

Abstract
Background. Natural killer (NK) cells are an important component of the innate immune defense against viruses, including hepatitis C virus (HCV). The cell culture system using HCV-permissive Huh-7.5 cells make studies on interaction of NK cells and HCV-infected target cells possible. We used this system to characterize interactions of HCV-infected Huh-7.5 cells and NK cells from healthy controls and patients with acute HCV infection.
Methods. IFNα- and IL-2 stimulated NK cells were cultured with HCV-infected hepatoma cells and subsequently analyzed (for degranulation and cytokine production) via multicolour flow cytometry. Luciferase assyas have been used to study inhibition of HCV replication. Further, PBMC from patients with acute hepatitis C as well as HCV-infected Huh7.5 cells have been analyzed via flow cytometry for expression of NK cell receptors and ligands, respectively.
Results. After interferon (IFN) α stimulation, NK cells from healthy controls and patients with acute hepatitis C efficiently recognized both HCV-infected and uninfected hepatoma cells. Subsequent dissection of receptor-ligand interaction revealed a dominant role for DNAM-1 and a complementary contribution of NKG2D for NK cell activation in this setting. Furthermore, IFN-α–stimulated NK cells effectively inhibited HCV replication in a DNAM-1–dependent manner.
Conclusions. Human NK cells recognize HCV-infected hepatoma cells after IFN-α stimulation in a DNAM-1–dependent manner. Furthermore, interaction of IFN-α–stimulated NK cells with HCV-infected hepatoma cells efficiently reduced HCV replication. This study opens up future studies of NK cell interaction with HCV-infected hepatocytes to gain further insight into the pathogenesis of human HCV infection and the therapeutic effects of IFN-α.
Hepatitis C virus (HCV) infection is a major global health burden with an estimated 130 million people chronically infected worldwide [1]. Hepatitis C virus is an enveloped RNA virus belonging to the family of Flaviviridae; it has a 9.6-kb genome encoding for a large polyprotein that is processed into structural and nonstructural viral proteins [2]. Since the discovery of HCV, its properties have undergone intense studies [2]. However, until a few years ago, it was not possible to reproduce the complete life cycle of HCV in vitro. This hampered investigations of immune cell interactions with HCV-infected cells. The development of a HCV cell culture system that allows complete replication of the virus in vitro [3–5] has opened up new possibilities for studies of the virus and interactions between HCV-infected cells and the host immune system [6]. Here, we have used this system to study natural killer (NK) cell interactions with HCV-infected hepatoma cells and their ability to inhibit HCV-replication.
Natural killer cells are CD56+CD3− lymphocytes belonging to the innate immune system [7]. They represent 5%–15% of peripheral blood mononuclear cells (PBMCs). Interestingly, NK cells are highly enriched in the liver where they make up 30%–40% of intrahepatic lymphocytes [8]. They can directly eliminate virus-infected and transformed cells [7, 9]. They recognize target cells via a germ-line–encoded repertoire of activating and inhibitory receptors [10]. Numerous activating receptors, such as NKG2D, NKG2C, the natural cytotoxicity receptors NKp30, NKp44, and NKp46, and DNAM-1, have been described [11–13]. Inhibition is mediated mainly via the inhibitory killer immunoglobulinlike receptors (KIR) and NKG2A, which recognize classical and nonclassical major histocompatibility complex class 1 molecules, respectively, on target cells [10, 11].
Although the role of adaptive immunity in spontaneous clearance of acute hepatitis C is well established [14], less is known about the direct role of NK cells in controlling HCV infection. Genetic association studies comparing patients who spontaneously cleared HCV infection with those who developed chronic hepatitis C have identified a link between HLA-C1:KIR2DL3 homozygosity and the resolution of infection [15, 16]. Furthermore, a number of studies, which have investigated phenotype and function of NK cells in patients with chronic hepatitis C, have demonstrated changes in the expression levels of individual inhibitory and activating receptors and consequently alterations in NK cell functionality [17–20]. However, few studies have more directly attempted to dissect the direct interaction between NK cells and HCV-infected target cells [21, 22].
We recently showed that interferon (IFN) α–stimulated NK cells up-regulate tumor necrosis factor (TNF)–related apoptosis-inducing ligand (TRAIL) to enable lysis of both HCV-infected and uninfected hepatoma cells [23]. From that point, the present study was designed to further characterize the interaction between NK cells and HCV-infected hepatoma cells with respect to target cell molecules involved in NK cell activation and the ability of NK cells from healthy controls and patients with acute hepatitis C to reduce HCV replication. We demonstrate that IFN-α–activated NK cells require DNAM-1 for recognition of HCV-infected hepatoma cells, and that this recognition efficiently reduces HCV replication in the infected cells.
MATERIALS AND METHODS
HCV Huh-7.5 Hepatoma Cell Culture System
Hepatitis C virus (strain Jc1G) was produced as described elsewhere [24, 25]. Huh-7.5 cells were cultured in Dulbecco’s modified Eagle medium with 10% fetal calf serum (Life Technologies), 100 U/mL penicillin G, 100 μg/mL streptomycin, and 1x nonessential amino acids (Invitrogen). For infection, filtered HCV-containing culture supernatant was added to Huh-7.5 cells and incubated for 1 week; additional fresh media was added on day 3 or 4. The rate of infectivity was verified by flow cytometry with intracellular staining using a monoclonal antibody for HCV-NS5A protein (clone 9E10) [3]. The infection rate was >50% in all experiments culturing HCV-infected Huh-7.5 cells with effector cells.
Patient Material
Six patients with acute hepatitis C were recruited at Hannover Medical School, Hannover, Germany. All patients gave informed consent for participation in this study. The study was part of a protocol that had been approved by the ethics committee of the Hannover Medical School. Patient characteristics are given in Supplementary Table 1.
Healthy donors were recruited at the Karolinska University Hospital, Stockholm, Sweden, and the Hannover Medical School. Peripheral blood mononuclear cells were isolated through standard density-gradient separation and either cryopreserved or used fresh in experiments.
Antibodies Used for Flow Cytometry
The Supplementary information includes details on antibodies used for flow cytometry, phenotyping of HCV-infected and uninfected Huh-7.5 cells and NK cells from healthy controls and patients with acute hepatitis C, and flow cytometry staining details.
Functional NK Cell Assays
In some experiments, NK cells were purified using the NK Cell Isolation Kit (Miltenyi). Purified NK cells were used in parallel with whole PBMCs in coculture experiments with target cells to verify similar levels of responsiveness comparing purified NK cells and whole PBMCs. In more detail, whole PBMCs or purified NK cells were stimulated overnight with 100 ng/mL IFN-α-2a (PBL InterferonSource) or 1000 U/mL interleukin 2 (IL-2) (Proleukin; Chiron) or incubated in medium alone without stimulation. After stimulation, PBMCs or NK cells were washed once with phosphate-buffered saline (PBS) and resuspended in complete Roswell Park Memorial Institute 1640 medium.
Effector cells were cocultured with Jurkat, K562, HepG2, Hep3B, Huh-7, and Huh-7.5 cells in a ratio of 1:1 for isolated NK cells and 10:1 for whole PBMCs, as described elsewhere [26]. Briefly, to measure degranulation and cytokine production by the effector cells, GolgiPlug (BD Bioscience) was added after a 1-hour culture. After an additional 5 hours of incubation, cells were stained with anti-CD107a–fluorescein isothiocyanate, anti-CD56–phycoerythrin (PE)–cyanine 7 (Cy7), anti-CD3–Pacific Blue, anti-CD14–electron coupled dye (ECD), and Live/Dead Fixable Aqua Dead Cell Stain Kit or with anti-CD107a–PE, anti-CD56–PE-Cy7, anti-CD3–Pacific Blue, anti-CD14–Horizon V500, anti-CD19–Horizon V500, and Live/Dead Fixable Aqua Dead Cell Stain Kit for 20 minutes at 4°C in the dark. Cells were washed twice with PBS containing 5% fetal calf serum; cells were incubated in BD Cytofix/Cytoperm for 20 minutes at 4°C. Intracellular staining for IFN-γ and TNF-α was performed as described in the Supplementary information.
Blocking of NK Cell–Target Cell Interaction
Purified NK cells or PBMCs were either rested or stimulated with 100 ng/mL IFN-α-2a overnight. After washing, effector and target cells were mixed at a ratio of 1:1 for NK cells and 10:1 for PBMCs. To identify ligand-receptor interactions, 5 μg/mL blocking antibodies (anti–DNAM-1 [clone DX11; BD Bioscience], anti-NKG2D [clone M585; Amgen], anti-NKp30 [clone Z25; Beckman Coulter], anti-NKp46 [clone BAB281; Beckman Coulter], or immunoglobulin G1 (IgG1) isotype control [clone MOPC21; BD Bioscience]) were added to the effector cells 30 minutes before and during the coculture. The level of blocking was quantified by determining differences in degranulation and production of IFN-γ and TNF-α by flow cytometry, as outlined above.
Luciferase Assay to Measure HCV Replication
Huh-7.5 cells were transfected by electroporation with a reporter virus genome carrying a luciferase-reporter gene (Jc1-Luc [27]), as shown elsewhere [27]. Forty-eight hours after transfection, Huh-7.5 cells were trypsinized and 40 000 cells were seeded per well in a 24-well plate. After 5–6 hours, 0.2 × 106 NK cells were added for 8 hours and then washed away. Target cells were lysed to determine HCV replication via firefly luciferase assays, as described elsewhere [28]. All luciferase assays were performed in duplicate. For the coculture, purified NK cells stimulated overnight with 100 ng/mL of IFN-α-2b (Intron A; Essex Pharma) or incubated in medium without stimulation were used as effector cells. Before their addition to transfected Huh-7.5 cells, NK cells were washed once with PBS. During the coculture, 10 μg/mL anti–DNAM-1 antibody was added to block interaction of NK cells and transfected Huh-7.5 cells. As a control, IgG1 antibody was used.
For assessment of noncytolytic effector mechanisms, supernatants obtained from coculture of Huh-7.5 cells and previously IFN-α–stimulated purified NK cells were added to HCV-infected (Jc1-F-Luc) Huh-7.5 cells. Replication of HCV replication was determined after 48 hours via luciferase assay. During culturing of HCV-infected Huh-7.5 cells, 10 μg/mL anti-IFN-γ (clone NIB42) or anti-TNF-α antibody (clone MAb1) was added. As a control, IgG1 antibody (clone P3.6.2.8.; all eBioscience) was used.
Statistical Analyses
Data are presented as means ± standard errors of the mean. For preparation of diagrams, Prism 5 software (GraphPad Software) was used. For statistical analyses of coculture assays, nonparametrical paired t tests (Wilcoxon) were used.
RESULTS
Recognition of HCV-Infected and Uninfected Huh-7.5 Cells by NK Cells From Healthy Controls and Patients With Hepatitis C
To investigate how NK cells from healthy controls and patients with acute hepatitis C recognize HCV-infected target cells, we used the recently developed HCV cell culture system [4]. Thawed PBMCs from healthy donors, either kept without stimulation or stimulated overnight with IFN-α or IL-2, were exposed to HCV-infected or uninfected Huh-7.5 cells. CD56dim (low expression levels of CD56) NK cells were subsequently analyzed by flow cytometry for degranulation and production of IFN-γ and TNF-α. Unstimulated CD56dim NK cells did not respond to HCV-infected or uninfected Huh-7.5 cells, whereas those stimulated with IFN-α and, to a lesser extent, IL-2, responded with degranulation and cytokine production (Figure 1A). Stimulation with IFN-α induced multifunctional NK cell responses (ie, simultaneous degranulation and production of IFN-γ and TNF-α) (Figure 1C). No difference in NK cell responses could be detected when comparing HCV-infected and uninfected Huh-7.5 cells (Figure 1A). Next, we investigated whether NK cells from patients with acute hepatitis C also were able to recognize HCV-infected or uninfected Huh-7.5 cells (Figure 1B). Phenotypically, NK cells from patients with acute hepatitis C displayed an overall activated phenotype expressing higher levels of activation receptors (Figure 3D–F). With respect to function, CD56dim NK cells from these patients showed a pattern of responsiveness similar to that of CD56dim NK cells from healthy controls (Figure 1A and 1B).
Figure 1.

Interferon (IFN) α–stimulated natural killer (NK) cells recognize hepatitis C virus (HCV)–infected and uninfected hepatoma cells. A, B, Frozen peripheral blood mononuclear cells (PBMCs) were thawed and either rested or stimulated with interleukin 2 (IL-2) or IFN-α overnight before coculturing for 5 hours with HCV-infected or uninfected Huh-7.5 cells. A, Degranulation (upper diagram), IFN-γ (middle diagram), and tumor necrosis factor (TNF) α (lower diagram) production of CD56dim NK cells from 4 healthy controls. Wilcoxon signed rank test did not reach significant values owing to the small number of samples. B, Degranulation (upper diagram), IFN-γ (middle diagram), and TNF-α (lower diagram) production by CD56dim NK cells compiled for 6 patients with acute hepatitis C. P < .05 for IFN-α–stimulated cells cultured without target cells vs HCV-infected and uninfected Huh-7.5 cells, for degranulation as well as IFN-γ and TNF-α production (Wilcoxon signed rank test); P < .05 for resting cells cultured without target cells vs HCV-infected and uninfected Huh-7.5 cells for degranulation. C, Mean frequency of CD56dim NK cells with 1 functional activity (upper diagram), 2 functional activities (middle diagram), and all 3 functional activities (lower diagram) investigated (degranulation, IFN-γ, and TNF-α production) for thawed PBMCs from 4 healthy controls.
Figure 3.

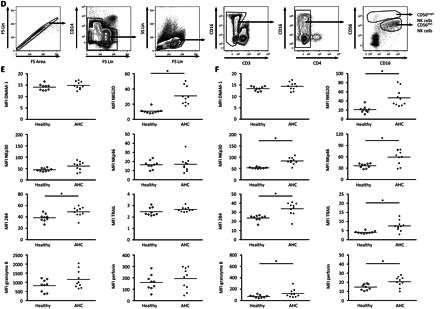
Natural killer (NK) cell receptor ligand expression on Huh-7.5 cells and expression of NK cell receptor in acute hepatitis C virus (HCV) infection. A, Representative stainings for the HCV protein NS5A of HCV-infected (left) and uninfected (right) Huh-7.5 cells. B, Histograms of HCV-uninfected (NS5A−) (left) and HCV-infected Huh-7.5 (NS5A+) (middle) cells from the same sample and control Huh-7.5 cells (uninfected) (right), showing expression of different NK cell receptor ligands (black) and the respective isotype control (gray). One representative experiment of 3 is shown. (TRAIL, tumor necrosis factor–related apoptosis-inducing ligand). C, Mean relative expression levels of the indicated NK cell receptor ligands are summarized for 3 independent experiments. Mean relative expression was calculated as the fold increase compared with the isotype control. D, Plots from 1 representative individual demonstrating the gating strategy to identify CD56bright and CD56dim NK cells used for the phenotypical characterization. E, Individual data of patients with acute hepatitis C (AHC) (n = 10) (filled rhombs) as compared with healthy controls (n = 9) (open rhombs) for the expression level of the different receptors investigated on CD56dim NK cells. *P < .05. F, Individual data from patients with AHC (n = 10) (filled rhombs), as compared with healthy controls (n = 9) (open rhombs), for the expression level of the different receptors investigated on CD56bright NK cells. *P < .05. Abbreviations: FS, Forward scatter; LFA-1, lymphocyte function associated antigen 1; Lin, linear; MFI, Mean Fluorescence Intensity; MICA, MHC class I chain-related genes A; MICB, MHC class I chain-related genes B; SS, side scatter; ULBP, UL16 binding protein.
Because IFN-α stimulation of NK cells seemed to be a prerequisite for strong NK cell responses against HCV-infected and uninfected Huh-7.5 cells, and because IL-2 caused only a minor increase in degranulation and cytokine production, we next addressed whether the observed IFN-α dependency on eliciting strong responses against hepatoma cells was specific for this type of target cell. To do this, we evaluated NK cell responses to the lymphoma cell lines K562 and Jurkat as well as the hepatoma cells lines Huh-7, Huh-7.5, HepG2, and Hep3B (Figure 2). Taken together, the data convey that resting NK cells respond poorly against hepatoma cell lines, with low levels of degranulation and cytokine production, yet exhibit strong responses against K562 and Jurkat cells. On the other hand, IFN-α stimulation was associated with a more efficient potentiation of NK cell responses against hepatoma cells, as compared with stimulation by IL-2. However, this was not the case for NK cell responses against K562 and Jurkat cells, in which IFN-α and IL-2 increased NK cell function to a similar level (Figure 2).
Figure 2.
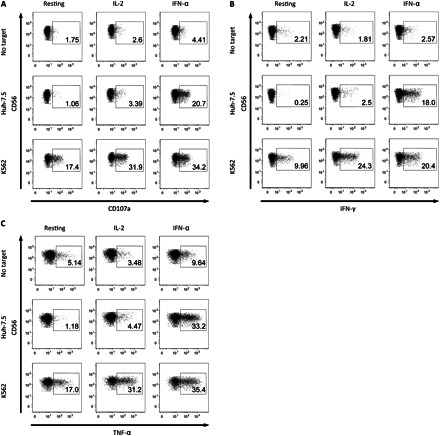

Interferon (IFN) α is necessary for natural killer (NK) cell recognition of Huh-7.5 cells. A–C, Representative stainings for degranulation (A), IFN-γ (B), and tumor necrosis factor (TNF) α (C) production of CD56dim NK cells from 1 healthy donor. Peripheral blood mononuclear cells (PBMCs) were either rested or stimulated with interleukin 2 (IL-2) or IFN-α overnight before coculturing for 6 hours either without target cells or with K562 or Huh-7.5 cells. D, Frequency of degranulation (upper diagrams), IFN-γ (middle diagrams), and TNF-α (lower diagrams) production by CD56dim NK cells from healthy controls; fold increases are normalized to the respective resting sample to each cell line. Fresh isolated PBMCs, either rested or stimulated with IL-2 or IFNα overnight, were cocultured with different target cell lines. *P < .05, **P < .01, and ***P < .001 (Wilcoxon signed rank test).
To conclude, NK cells from healthy controls and patients with acute hepatitis C recognized Huh-7.5 cells. This recognition depended on activation of NK cells by IFN-α. Unlike its effect on other target cells, IL-2 was not as effective as IFN-α at activating NK cells against hepatoma cell lines, including HCV-infected or uninfected Huh-7.5 cells.
Effect of HCV Infection on NK Cell–Receptor Ligand Expression in Huh-7.5 Cells
Because NK cells responded equally well with respect to degranulation and cytokine production when cocultured with HCV-infected and uninfected Huh-7.5 cells, it was reasonable to assume that HCV does not cause any major changes in the hepatoma cells that might affect recognition by NK cells. To confirm this, we evaluated the expression patterns of multiple ligands for activating and inhibitory NK cell receptors on HCV-infected and uninfected Huh-7.5 cells (Figure 3A–C). An infection rate of 25%–75% was used, allowing a direct comparison of differences in NK cell receptor ligand expression on HCV-positive and HCV-negative Huh-7.5 cells within the same sample (Figure 3A). Uninfected Huh-7.5 cells stained positive for the DNAM-1 ligands CD112 and CD155 for 2 of 6 investigated NKG2D ligands as well as for CD102 and CD54, both ligands for lymphocyte function associated antigen 1 (LFA-1) (Figure 3B). As hypothesized, infection with HCV did not cause any major changes in the expression level of the investigated NK cell receptor ligands on Huh-7.5 (Figure 3B and 3C).
Recognition of HCV-Infected and Uninfected Huh-7.5 Cells by IFN-α–Stimulated NK Cells via DNAM-1
We next aimed to identify the specific receptor-ligand interaction responsible for NK cell recognition of HCV-infected and uninfected Huh-7.5 cells. Because Huh-7.5 cells expressed ligands to the activating receptors NKG2D and DNAM-1, we blocked these 2 interactions in coculture experiments. We also assessed involvement of the activating receptors NKp30 and NKp46. Initial studies involved NK cells from healthy controls. For both HCV-infected and uninfected Huh-7.5 cells, blocking of DNAM-1 resulted in a sharp decrease in IFN-α–stimulated CD56dim NK cell functions (Figure 4A and 4B). In addition, a minor role was identified for NKG2D in NK cell-recognition of Huh-7.5 cells (Figure 4A and 4B). Of note, NKp30 and NKp46 blocking did not affect CD56dim NK cell responses (Figure 4B). Moreover, the combination of several blocking antibodies revealed an additive blocking effect of DNAM-1 and NKG2D (Figure 4C). Combinations of blocking antibodies not involving anti–DNAM-1 antibody had only minor effects on recognition of Huh-7.5 cells by IFN-α–stimulated CD56dim NK cells (Figure 4C). Subsequently, we investigated whether NK cells from patients with acute hepatitis C also required DNAM-1 to respond against HCV-infected Huh-7.5 cells. Similarly to findings in healthy controls, degranulation and cytokine production of CD56dim NK cells from patients with acute hepatitis C were efficiently inhibited by DNAM-1 blocking. Infection of the target cells by HCV did not change the DNAM-1 dependency for NK cell recognition (Figure 4D). The results are further supported by the finding that DNAM-1 expression levels were not altered by IFN-α stimulation (Figure 4E) and did also not differ between healthy controls and patients with acute hepatitis C (Figure 3E and 3F). Together, these results suggest that IFN-α–stimulated CD56dim NK cells from patients with acute hepatitis C and healthy controls recognize HCV-infected and uninfected Huh-7.5 cells in a DNAM-1–dependent manner.
Figure 4.
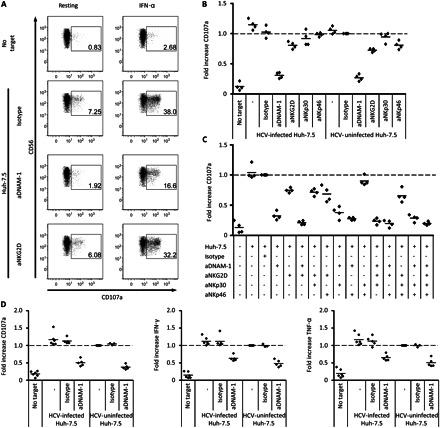
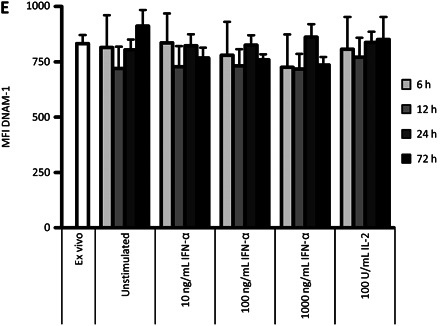
DNAM-1 is necessary for natural killer (NK) cell recognition of hepatitis virus C (HCV)–infected and uninfected Huh-7.5 cells. A, Representative stainings for degranulation of rested or interferon (IFN) α–stimulated CD56dim NK cells from 1 healthy individual after coculture with Huh-7.5 cells plus the addition of the indicated blocking antibodies. B, Summarized data for IFN-α–stimulated CD56dim cells from 4 healthy donors. Fresh isolated peripheral blood mononuclear cells (PBMCs) were cocultured with HCV-infected or uninfected Huh-7.5 cells and blocking antibodies against the indicated activating NK cell receptors. B–D, Fold increases were normalized to IFN-α–stimulated CD56dim NK cells cocultured with uninfected Huh-7.5 cells in the presence of an isotype control antibody. C, Summarized data of IFN-α–stimulated CD56dim NK cells from 4 healthy donors. Fresh isolated PBMCs were cocultured with Huh-7.5 cells and different combinations of blocking antibodies against activating NK cell receptors. Wilcoxon signed rank test could not be performed owing to normalization of the samples and the small sample number. D, Summarized data from 6 patients with acute hepatitis C for degranulation (left), IFN-γ (middle), and tumor necrosis factor (TNF) α (right) production of CD56dim NK cells after coculture of thawed and IFN-α–stimulated PBMCs with HCV-infected and uninfected Huh-7.5 cells; blocking anti–DNAM-1 antibody and isotype control antibody was added as indicated. E, DNAM-1 expression levels on CD56dim NK cells after stimulation with IFN-α and interleukin 2 (IL-2) in the indicated doses at 6, 12, 24, and 72 hours. Mean values for 3 healthy individuals are shown. Abbreviations: aDNAM-1, anti-DNAM-1 antibody; MFI, Mean Fluorescence Intensity.
Inhibition of HCV Replication by NK Cells via DNAM-1
One of the advantages of implementing cellular assays with HCV-infected Huh-7.5 cells is the opportunity to evaluate the specific ability of NK cells to exhibit antiviral effects. We used this option to test NK cells for the direct inhibition of HCV replication and, if inhibition was observed, to determine whether it was DNAM-1 dependent. To do this, NK cells were cocultured with Huh-7.5 cells that had been transfected with an HCV genome expressing a luciferase-reporter gene [27]. Interestingly, HCV replication, as measured by luciferase activity, was only slightly affected by the addition of IFN-α alone. Resting nonactivated NK cells had a modest effect on HCV replication, whereas HCV replication was greatly reduced when NK cells had been prestimulated with IFN-α (Figure 5A). Here, blocking of DNAM-1 almost completely restored HCV replication (Figure 5A).
Figure 5.
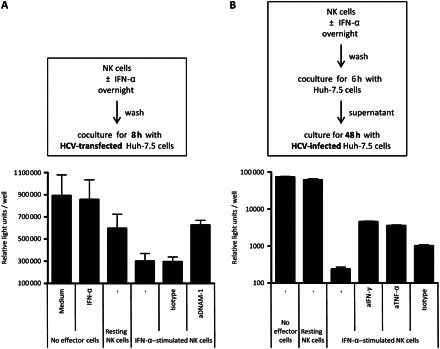
Interferon (IFN) α–stimulated natural killer (NK) cells inhibit hepatitis C virus (HCV) replication in a DNAM-1–dependent manner. A, HCV-Luc–transfected Huh-7.5 cells were cocultured for 8 hours with rested or IFN-α–stimulated NK cells. Replication of HCV was determined by luciferase assay. As indicated, 10 μg/mL blocking anti–DNAM-1 or isotype antibody were added. Mean values for 7 healthy donors are shown. B, HCV-Luc–infected Huh-7.5 cells were cultured for 48 hours with supernatant obtained after coculture of previously IFN-α–stimulated purified NK cells and Huh-7.5 cells. As indicated, 10 μg/mL of blocking anti–IFN-γ, anti–tumor necrosis factor (TNF) α, or isotype antibody were added. Mean values for experiments with supernatants from 3 individuals are shown.
Except for contact-dependent cytotoxic mechanisms for inhibition of HCV replication, it is plausible to assume that soluble factors such as cytokines might also have the capacity to reduce HCV-replication. To further investigate the effect of soluble factors on HCV replication, supernatants of Huh-7.5 cells cocultured with previously IFN-α–stimulated purified NK cells were added to HCV-infected Huh-7.5 cells, leading to reduced HCV replication (Figure 5B). HCV replication could be partially restored by addition of blocking antibodies against IFN-γ and TNF-α (Figure 5B). We have shown elsewhere that TRAIL is involved in killing of HCV-infected Huh-7.5 cells [23]; thus, it is plausible that TRAIL, found in the supernatant, is also responsible in part for the inhibition of HCV replication. To summarize, these data suggest that IFN-α–stimulated NK cells can efficiently reduce HCV replication in hepatoma cells. This effect is mediated primarily via DNAM-1.
DISCUSSION
Cumulative evidence suggests that NK cells contribute to the control of HCV infection. In this article we show the following 3 things: (1) NK cells from healthy controls as well as patients with acute hepatitis C can efficiently recognize HCV-infected hepatoma cells when stimulated with IFN-α and suppress replication of the virus; (2) HCV infection does not alter the expression of NK cell receptor ligands on hepatoma cells; and (3) the induction of effector functions in IFN-α–stimulated NK cells, following coculture with hepatoma cells, depends on the activation receptor DNAM-1.
We show here that efficient recognition of hepatoma cells by NK cells is dependent on IFN-α stimulation. This finding is rather surprising, because previous studies investigating the interaction between NK cells and various malignant cell types have indicated that stimulation of NK cells with IL-2 is sufficient [29, 30]. Thus, we conclude that immunomodulatory effects of IFN-α in the treatment of HCV infection can be attributed, at least partly, to induction of functional changes of NK cells enabling them to recognize hepatoma cells. Of note, a significant proportion of IFN-α–stimulated NK cells cocultured with hepatoma cells was polyfunctional. The reduction of HCV replication could be due to lysis of infected cells [23] as well as noncytotoxic mechanisms [21].
Overall, HCV infection did not induce any obvious changes in expression levels of various NK cell receptor ligands. This could suggest that functional impairment of NK cells in HCV infection is mediated mainly by direct effects on the NK cells rather than by interference of HCV with NK cell receptor ligands on the target cells. Support for this probability comes from earlier descriptions of the inhibitory effects of HCV-E2 protein on NK cell function [31, 32]. However, a more recent study did not confirm these findings using full infectious particles instead of HCV-E2 protein [33]. In our hands, NK cells from patients with acute hepatitis C were functional and responded to IFN-α stimulation in vitro equally well as NK cells from healthy controls. However, we did not have the possibility to directly compare the functional characteristics of NK cells from patients with acute hepatitis C and healthy controls. There might be interindividual differences in the intrinsic capacity to respond to IFN-α, because PBMCs from patients who respond to treatment with IFN-α show higher up-regulation of TRAIL upon IFN-α stimulation in vitro than do PBMCs from patients who do not respond to such treatment [23].
NKp30 expression may be altered in chronic hepatitis C [17, 22, 34], and the molecule has been designated as a mediator of interactions between NK cells and antigen-presenting cells [35]. Recently, it was shown that the inhibitory effects of myeloid-derived suppressor cells on NK cells are mediated via NKp30 [36]. The role of NKp46 in hepatitis C remains controversial [17, 19, 34, 37]; nevertheless, this receptor is involved in interactions between NK cells and antigen-presenting cells and tumor cells [35, 38]. However, neither NKp30 nor NKp46 seemed to be involved in NK cell recognition of Huh-7.5 cells. NKG2D partially contributed to the recognition of hepatoma cells by IFN-α–stimulated NK cells, yet the most dominant effect was evident in the presence of DNAM-1. DNAM-1 is expressed by almost all human NK cells, T cells, and monocytes [39]. To the best of our knowledge, no data are available on the role of DNAM-1 on NK cells in either acute or chronic hepatitis C. However, the DNAM-1 is involved in the interactions between NK cells and numerous tumor cells, including melanoma, ovarian carcinoma, leukemia, and neuroblastoma cells [40–43]. Moreover, DNAM-1 is involved in the killing of dendritic cells [44]. The ligands for DNAM-1, CD112, and CD155 are 2 related molecules belonging to the nectin family [45]. Importantly, CD155 is highly expressed on Huh-7.5 cells, indicating that hepatoma cells are indeed delivering signals to NK cells via CD155:DNAM-1 interaction. However, our results cannot exclude the possibility that ligation of additional NK cell receptors is required to induce full NK cell effector functions against HCV-infected hepatoma cells.
Because the recognition of hepatoma cells by NK cells required stimulation with IFN-α, and because this recognition was mediated by DNAM-1, one could speculate that the effect of IFN-α was mediated via changes in DNAM-1 receptor expression. However, this was not the case. Stimulation of NK cells with IFN-α in vitro did not affect DNAM-1 expression on NK cells. Although NK cells from patients with acute hepatitis C exhibit an activated phenotype, DNAM-1 expression was not altered, as compared with healthy controls. Thus, these data suggest that IFN-α may alter the intracellular signaling of DNAM-1. Alternatively, IFN-α may regulate another yet unknown receptor on NK cells, with DNAM-1 acting as a coreceptor.
DNAM-1 has been shown to amplify effector functions of immune cells as a stimulatory molecule in various contexts, including allergic inflammation [46], autoimmune diseases [47], and viral infections such as human immunodeficiency virus infection [48]. Thus, DNAM-1 has already been suggested as a therapeutic target for modulating immune responses [12]. For example, NK cell responses against tumors and pathogens might be augmented by DNAM-1 activation. Therefore, understanding in detail how IFN-α enhances DNAM-1–dependent recognition of hepatoma cells might lead to the identification of potential therapeutic targets to enhance innate immunity against not only HCV infection but also other hepatotropic pathogens and primary hepatocellular carcinomas. Conversely, blocking DNAM-1 could down-regulate immune responses against hepatocytes, which may have implications in the context of autoimmune hepatitis.
It is important to bear in mind that results reported here were obtained using a tumor cell line as a model system and not primary hepatocytes. Recently, however, persistent infection with HCV has been demonstrated in primary human hepatocytes [49], which will provide an interesting ground to substantiate the present results.
In conclusion, IFN-α increases recognition of HCV-infected and uninfected hepatoma cells by NK cells from patients with acute hepatitis C, potentially contributing to the control of HCV infection. This effect is mediated via DNAM-1. Understanding the detailed molecular mechanism by which IFN-α modulates DNAM-1–dependent interactions of NK cells with hepatoma cells may help identify novel immunomodulatory treatment approaches for hepatitis C.
Supplementary Data
Supplementary materials are available at The Journal of Infectious Diseases online (http://www.oxfordjournals.org/our_journals/jid/). Supplementary materials consist of data provided by the author that are published to benefit the reader. The posted materials are not copyedited. The contents of all supplementary data are the sole responsibility of the authors. Questions or messages regarding errors should be addressed to the author.
Notes
Acknowledgments.
We thank patients and blood donors who have contributed clinical material to this study.
K. A. S., N. K. B., H. G. L., and H. W. designed the study. K. A. S., N. K. B., S. C., and S. L. acquired the data. K. A. S., N. K. B., S. C., S. L., J. J., J. W., P. M., T. P., M. C., H. G. L., and H. W. analyzed and interpreted the data. L. B. D. and C. M. R. contributed new reagents. K. A. S., N. K. B., H. G. L. and H. W. wrote the manuscript. K. A. S., N. K. B., S. C., S. L., J. J., J. W., P. M., L. B. D., C. M. R., M. P. M., T. P., M. C., H. G. L., and H. W. reviewed and edited the manuscript.
Financial support.
This work was funded by the International Research Training Group 1273 funded by the German Research Foundation (DFG), the Federal Ministry of Education and Research (BMBF) (grant 01Kl0788 to H. W. and M. C.), the Swedish Medical Research Council, the Swedish Cancer Society, the Swedish Fund for Research without Animal Experiments, the Greenberg Medical Research Institute, and the Starr Foundation. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Potential conflicts of interest.
All authors: No reported conflicts.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.Shepard CW, Finelli L, Alter MJ. Global epidemiology of hepatitis C virus infection. Lancet Infect Dis. 2005;5:558–67. doi: 10.1016/S1473-3099(05)70216-4. [DOI] [PubMed] [Google Scholar]
- 2.Moradpour D, Penin F, Rice CM. Replication of hepatitis C virus. Nat Rev Microbiol. 2007;5:453–63. doi: 10.1038/nrmicro1645. [DOI] [PubMed] [Google Scholar]
- 3.Lindenbach BD, Evans MJ, Syder AJ, et al. Complete replication of hepatitis C virus in cell culture. Science. 2005;309:623–6. doi: 10.1126/science.1114016. [DOI] [PubMed] [Google Scholar]
- 4.Wakita T, Pietschmann T, Kato T, et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med. 2005;11:791–6. doi: 10.1038/nm1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhong J, Gastaminza P, Cheng G, et al. Robust hepatitis C virus infection in vitro. Proc Natl Acad Sci U S A. 2005;102:9294–9. doi: 10.1073/pnas.0503596102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jo J, Lohmann V, Bartenschlager R, Thimme R. Experimental models to study the immunobiology of hepatitis C virus. J Gen Virol. 2011;92:477–93. doi: 10.1099/vir.0.027987-0. [DOI] [PubMed] [Google Scholar]
- 7.Cooper MA, Fehniger TA, Caligiuri MA. The biology of human natural killer-cell subsets. Trends Immunol. 2001;22:633–40. doi: 10.1016/s1471-4906(01)02060-9. [DOI] [PubMed] [Google Scholar]
- 8.Norris S, Collins C, Doherty DG, et al. Resident human hepatic lymphocytes are phenotypically different from circulating lymphocytes. J Hepatol. 1998;28:84–90. doi: 10.1016/s0168-8278(98)80206-7. [DOI] [PubMed] [Google Scholar]
- 9.Smyth MJ, Cretney E, Kelly JM, et al. Activation of NK cell cytotoxicity. Mol Immunol. 2005;42:501–10. doi: 10.1016/j.molimm.2004.07.034. [DOI] [PubMed] [Google Scholar]
- 10.Bryceson YT, March ME, Ljunggren HG, Long EO. Activation, coactivation, and costimulation of resting human natural killer cells. Immunol Rev. 2006;214:73–91. doi: 10.1111/j.1600-065X.2006.00457.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Biassoni R, Cantoni C, Pende D, et al. Human natural killer cell receptors and co-receptors. Immunol Rev. 2001;181:203–14. doi: 10.1034/j.1600-065x.2001.1810117.x. [DOI] [PubMed] [Google Scholar]
- 12.Elishmereni M, Bachelet I, Levi-Schaffer F. DNAM-1: an amplifier of immune responses as a therapeutic target in various disorders. Curr Opin Investig Drugs. 2008;9:491–6. [PubMed] [Google Scholar]
- 13.Bryceson YT, March ME, Ljunggren HG, Long EO. Synergy among receptors on resting NK cells for the activation of natural cytotoxicity and cytokine secretion. Blood. 2006;107:159–66. doi: 10.1182/blood-2005-04-1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rehermann B. Hepatitis C virus versus innate and adaptive immune responses: a tale of coevolution and coexistence. J Clin Invest. 2009;119:1745–54. doi: 10.1172/JCI39133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khakoo SI, Thio CL, Martin MP, et al. HLA and NK cell inhibitory receptor genes in resolving hepatitis C virus infection. Science. 2004;305:872–4. doi: 10.1126/science.1097670. [DOI] [PubMed] [Google Scholar]
- 16.Romero V, Azocar J, Zuniga J, et al. Interaction of NK inhibitory receptor genes with HLA-C and MHC class II alleles in hepatitis C virus infection outcome. Mol Immunol. 2008;45:2429–36. doi: 10.1016/j.molimm.2008.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nattermann J, Feldmann G, Ahlenstiel G, Langhans B, Sauerbruch T, Spengler U. Surface expression and cytolytic function of natural killer cell receptors is altered in chronic hepatitis C. Gut. 2006;55:869–77. doi: 10.1136/gut.2005.076463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oliviero B, Varchetta S, Paudice E, et al. Natural killer cell functional dichotomy in chronic hepatitis B and chronic hepatitis C virus infections. Gastroenterology. 2009;137:1151–60. doi: 10.1053/j.gastro.2009.05.047. 1160 e1–7. [DOI] [PubMed] [Google Scholar]
- 19.Ahlenstiel G, Titerence RH, Koh C, et al. Natural killer cells are polarized toward cytotoxicity in chronic hepatitis C in an interferon-alfa–dependent manner. Gastroenterology. 2010;138:325–35. doi: 10.1053/j.gastro.2009.08.066. e1–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Golden-Mason L, Madrigal-Estebas L, McGrath E, et al. Altered natural killer cell subset distributions in resolved and persistent hepatitis C virus infection following single source exposure. Gut. 2008;57:1121–8. doi: 10.1136/gut.2007.130963. [DOI] [PubMed] [Google Scholar]
- 21.Wang SH, Huang CX, Ye L, et al. Natural killer cells suppress full cycle HCV infection of human hepatocytes. J Viral Hepat. 2008;15:855–64. doi: 10.1111/j.1365-2893.2008.01014.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Golden-Mason L, Cox AL, Randall JA, Cheng L, Rosen HR. Increased natural killer cell cytotoxicity and NKp30 expression protects against hepatitis C virus infection in high-risk individuals and inhibits replication in vitro. Hepatology. 2010;52:1581–9. doi: 10.1002/hep.23896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stegmann KA, Bjorkstrom NK, Veber H, et al. Interferon-alpha–induced TRAIL on natural killer cells is associated with control of hepatitis C virus infection. Gastroenterology. 2010;138:1885–97. doi: 10.1053/j.gastro.2010.01.051. [DOI] [PubMed] [Google Scholar]
- 24.Pietschmann T, Kaul A, Koutsoudakis G, et al. Construction and characterization of infectious intragenotypic and intergenotypic hepatitis C virus chimeras. Proc Natl Acad Sci U S A. 2006;103:7408–13. doi: 10.1073/pnas.0504877103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kato T, Date T, Murayama A, Morikawa K, Akazawa D, Wakita T. Cell culture and infection system for hepatitis C virus. Nat Protoc. 2006;1:2334–9. doi: 10.1038/nprot.2006.395. [DOI] [PubMed] [Google Scholar]
- 26.Bryceson YT, Fauriat C, Nunes JM, et al. Functional analysis of human NK cells by flow cytometry. Methods Mol Biol. 2010;612:335–52. doi: 10.1007/978-1-60761-362-6_23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koutsoudakis G, Kaul A, Steinmann E, et al. Characterization of the early steps of hepatitis C virus infection by using luciferase reporter viruses. J Virol. 2006;80:5308–20. doi: 10.1128/JVI.02460-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Steinmann E, Brohm C, Kallis S, Bartenschlager R, Pietschmann T. Efficient trans-encapsidation of hepatitis C virus RNAs into infectious virus-like particles. J Virol. 2008;82:7034–46. doi: 10.1128/JVI.00118-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang B, Zhang J, Tian Z. Comparison in the effects of IL-2, IL-12, IL-15 and IFN alpha on gene regulation of granzymes of human NK cell line NK-92. Int Immunopharmacol. 2008;8:989–96. doi: 10.1016/j.intimp.2008.03.001. [DOI] [PubMed] [Google Scholar]
- 30.Frederick M, Grimm E, Krohn E, Smid C, Yu TK. Cytokine-induced cytotoxic function expressed by lymphocytes of the innate immune system: distinguishing characteristics of NK and LAK based on functional and molecular markers. J Interferon Cytokine Res. 1997;17:435–47. doi: 10.1089/jir.1997.17.435. [DOI] [PubMed] [Google Scholar]
- 31.Tseng CT, Klimpel GR. Binding of the hepatitis C virus envelope protein E2 to CD81 inhibits natural killer cell functions. J Exp Med. 2002;195:43–9. doi: 10.1084/jem.20011145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Crotta S, Stilla A, Wack A, et al. Inhibition of natural killer cells through engagement of CD81 by the major hepatitis C virus envelope protein. J Exp Med. 2002;195:35–41. doi: 10.1084/jem.20011124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yoon JC, Shiina M, Ahlenstiel G, Rehermann B. Natural killer cell function is intact after direct exposure to infectious hepatitis C virions. Hepatology. 2009;49:12–21. doi: 10.1002/hep.22624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.De Maria A, Fogli M, Mazza S, et al. Increased natural cytotoxicity receptor expression and relevant IL-10 production in NK cells from chronically infected viremic HCV patients. Eur J Immunol. 2007;37:445–55. doi: 10.1002/eji.200635989. [DOI] [PubMed] [Google Scholar]
- 35.Spaggiari GM, Carosio R, Pende D, et al. NK cell-mediated lysis of autologous antigen-presenting cells is triggered by the engagement of the phosphatidylinositol 3-kinase upon ligation of the natural cytotoxicity receptors NKp30 and NKp46. Eur J Immunol. 2001;31:1656–65. doi: 10.1002/1521-4141(200106)31:6<1656::aid-immu1656>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 36.Hoechst B, Voigtlaender T, Ormandy L, et al. Myeloid derived suppressor cells inhibit natural killer cells in patients with hepatocellular carcinoma via the NKp30 receptor. Hepatology. 2009;50:799–807. doi: 10.1002/hep.23054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bozzano F, Picciotto A, Costa P, et al. Activating NK cell receptor expression/function (NKp30, NKp46, DNAM-1) during chronic viraemic HCV infection is associated with the outcome of combined treatment. Eur J Immunol. 2011;41:2905–15. doi: 10.1002/eji.201041361. [DOI] [PubMed] [Google Scholar]
- 38.Pessino A, Sivori S, Bottino C, et al. Molecular cloning of NKp46: a novel member of the immunoglobulin superfamily involved in triggering of natural cytotoxicity. J Exp Med. 1998;188:953–60. doi: 10.1084/jem.188.5.953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shibuya A, Campbell D, Hannum C, et al. DNAM-1, a novel adhesion molecule involved in the cytolytic function of T lymphocytes. Immunity. 1996;4:573–81. doi: 10.1016/s1074-7613(00)70060-4. [DOI] [PubMed] [Google Scholar]
- 40.Lakshmikanth T, Burke S, Ali TH, et al. NCRs and DNAM-1 mediate NK cell recognition and lysis of human and mouse melanoma cell lines in vitro and in vivo. J Clin Invest. 2009;119:1251–63. doi: 10.1172/JCI36022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Carlsten M, Bjorkstrom NK, Norell H, et al. DNAX accessory molecule-1 mediated recognition of freshly isolated ovarian carcinoma by resting natural killer cells. Cancer Res. 2007;67:1317–25. doi: 10.1158/0008-5472.CAN-06-2264. [DOI] [PubMed] [Google Scholar]
- 42.Pende D, Spaggiari GM, Marcenaro S, et al. Analysis of the receptor–ligand interactions in the natural killer-mediated lysis of freshly isolated myeloid or lymphoblastic leukemias: evidence for the involvement of the poliovirus receptor (CD155) and nectin-2 (CD112) Blood. 2005;105:2066–73. doi: 10.1182/blood-2004-09-3548. [DOI] [PubMed] [Google Scholar]
- 43.Castriconi R, Dondero A, Corrias MV, et al. Natural killer cell-mediated killing of freshly isolated neuroblastoma cells: critical role of DNAX accessory molecule-1-poliovirus receptor interaction. Cancer Res. 2004;64:9180–4. doi: 10.1158/0008-5472.CAN-04-2682. [DOI] [PubMed] [Google Scholar]
- 44.Pende D, Castriconi R, Romagnani P, et al. Expression of the DNAM-1 ligands, nectin-2 (CD112) and poliovirus receptor (CD155), on dendritic cells: relevance for natural killer-dendritic cell interaction. Blood. 2006;107:2030–6. doi: 10.1182/blood-2005-07-2696. [DOI] [PubMed] [Google Scholar]
- 45.Bottino C, Castriconi R, Pende D, et al. Identification of PVR (CD155) and nectin-2 (CD112) as cell surface ligands for the human DNAM-1 (CD226) activating molecule. J Exp Med. 2003;198:557–67. doi: 10.1084/jem.20030788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bachelet I, Levi-Schaffer F. Mast cells as effector cells: a co-stimulating question. Trends Immunol. 2007;28:360–5. doi: 10.1016/j.it.2007.06.007. [DOI] [PubMed] [Google Scholar]
- 47.Tao D, Shangwu L, Qun W, et al. CD226 expression deficiency causes high sensitivity to apoptosis in NK T cells from patients with systemic lupus erythematosus. J Immunol. 2005;174:1281–90. doi: 10.4049/jimmunol.174.3.1281. [DOI] [PubMed] [Google Scholar]
- 48.Mavilio D, Lombardo G, Kinter A, et al. Characterization of the defective interaction between a subset of natural killer cells and dendritic cells in HIV-1 infection. J Exp Med. 2006;203:2339–50. doi: 10.1084/jem.20060894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ploss A, Khetani SR, Jones CT, et al. Persistent hepatitis C virus infection in microscale primary human hepatocyte cultures. Proc Natl Acad Sci U S A. 2010;107:3141–5. doi: 10.1073/pnas.0915130107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.