

Abstract
Purpose
To review the current understanding of the pathophysiology of dilated cardiomyopathy (DCM) in patients with Duchenne and Becker muscular dystrophies, assessment of cardiac dysfunction for these patients, and the recommended pharmacological treatment options and ongoing research directions.
Data sources
Reviews and original research articles from scholarly journals and books.
Conclusions
Duchenne and Becker muscular dystrophies are debilitating neuromuscular disorders, both caused by mutations in the dystrophin gene. Most patients develop DCM as part of the disease course; in fact, DCM is the leading cause of death among these patients. Cardiac surveillance, including routine monitoring of electrocardiograms, echocardiograms, and appropriate blood biomarkers, may detect early DCM development. Although previous studies have shown that early administration of cardiac medications may delay the development of DCM, current standard of care does not emphasize cardiac surveillance and timely treatment. This, in turn, limits clinicians, including advanced practice nurses, to be optimally engaged in providing the most aggressive and proactive patient care.
Implications for practice
Implementing a routine cardiac assessment and timely pharmacological treatment in primary or specialty care settings is high-lighted as an important step to optimize cardiac health among patients with Duchenne and Becker muscular dystrophies.
Keywords: Cardiomyopathy, muscular dystrophy, dystrophin, genetics
Introduction
Duchenne muscular dystrophy (DMD) is an X-linked, neuromuscular disorder with an incidence of 1 in 3500 live newborn males (Emery, 1991). It is characterized by progressive, generalized weakness and wasting of muscle. The disease was first described by Meryon (1851) at a meeting of the Royal Medical and Chirurgical Society. He described a familial pattern with a male predilection that ultimately resulted in early death. Twenty years later, Duchenne, a French neurologist, published a complete histological description of muscle biopsies from 13 patients. He gave the condition the name “paralysie musculaire pseudohypertrophique” (pseudohypertrophic muscle paralysis), which accurately described the muscle paralysis and muscle hypertrophy characteristics of DMD patients (Duchenne, 1872). The term muscular dystrophy was adopted by Erb (1884), who defined the disease as a primary degeneration of the muscle. To date, there is still no effective treatment or cure for muscular dystrophy, despite extensive research for the past 100 years.
Clinically, DMD patients are diagnosed between 3 and 6 years of age with a delay in motor development and skeletal muscle weakness. To get up from the floor, DMD patients face the floor, plant their feet widely apart, raise their buttocks first, and use their hands to “walk” up the legs. This set of movements is termed the Gowers’ maneuver. As the disease progresses, the child reports difficulty or weakness in performing normal tasks such as walking, running, and climbing stairs. Underlying these challenges, these patients experience musculoskeletal wasting, muscle contractures, and severe scoliosis. These children usually lose ambulatory ability and require a wheelchair at as early as 10 to 12 years of age. The majority of DMD patients eventually develop dilated cardiomyopathy (DCM) in their midteens (English & Gibbs, 2006; Jefferies et al., 2005), characterized by enlarged heart chambers and reduced cardiac wall thickness. The cardiomyopathy accentuates respiratory problems caused by intercostal and diaphragmatic respiratory muscle weakness and scoliosis. In most cases, the DMD patient dies from cardiac or respiratory failure by 30 years of age.
Koenig and colleagues (1987) reported the important genetic aspect of this lethal disease in 1987 with their discovery that mutations in the dystrophin gene give rise to abnormal expression of the dystrophin protein within the myofiber, resulting in the muscle damage noted in DMD. The dystrophin gene is localized at the short arm of the X chromosome at cytogenetic band 21 (Xp21) and is expressed in all muscle types, including skeletal, smooth, and cardiac muscles, as well as in the brain and retina (Hoffman, Brown, & Kunkel, 1987; Hoffman, Hudecki, Rosenberg, Pollina, & Kunkel, 1988; Koenig et al.; Pillers et al., 1993). This seminal discovery and recent advances in genetic diagnostic techniques have revealed that mutations in the dystrophin gene can also result in several other disorders, including Becker muscular dystrophy (BMD). The reported incidence for BMD is at least 1 in 18,450 male live births (Bushby, Thambyayah, & Gardner-Medwin, 1991).
The spectrum of clinical presentation and severity of BMD is much broader than that of DMD. In general, most patients with BMD develop musculoskeletal symptoms at a much slower pace than DMD. The natural history of the disease varies considerably depending on the dystrophin gene mutation and the amounts of dystrophin protein expressed in muscle. Many BMD patients remain ambulatory until the third or fourth decade or later. Despite the milder skeletal muscle involvement in BMD, the majority of BMD patients still develop DCM (Nigro et al., 2006). Moreover, the severity and onset of cardiac symptoms in BMD patients are unrelated to the skeletal myopathy (Nigro et al., 1995; Kirchmann, Kececioglu, Korinthenberg, & Dittrich, 2005).
In agreement with this discordance between skeletal and cardiac involvement, recent genetic testing has revealed that mutations in the dystrophin gene are also the cause for some familial X-linked dilated cardiomyopathy (XLDCM), in which the phenotype is cardiac-specific, generally sparing the skeletal muscle. In addition to DMD, BMD, and XLDCM, dystrophin gene mutations can also cause other conditions as listed in Table 1, indicating the gene’s essential function in the biology of the muscles and the brain (Davies & Nowak, 2006).
Table 1.
Clinical phenotypes resulting from mutations in the dystrophin gene
| • Duchenne muscular dystrophy |
| • Becker muscular dystrophy |
| • X-linked dilated cardiomyopathya,b |
| • Retinal neurotransmission defectb |
| • Mental retardationb |
| • Psychiatric disturbancesb |
Can manifest without any musculoskeletal manifestation.
Usually present with Duchenne or Becker muscular dystrophy or both.
In summary, damage to respiratory and cardiac muscles significantly shortens the life expectancy of DMD and BMD patients. Advances in technology have greatly improved respiratory care for these patients, making DCM the leading cause of death among DMD and BMD patients (Eagle et al., 2002). The purpose of this article is to provide an overview of the current understanding in the pathophysiology of DCM in DMD and BMD, and to discuss the assessment and pharmacological treatments for DCM in this patient population. Finally, ongoing and future research directions are discussed.
Pathophysiology
Dystrophin
The dystrophin gene is the largest gene in the human genome, containing 79 exons and encompassing 2.6 million base pairs of the genomic sequence, which is about 1.5% of the entire X chromosome. Mutations in the dystrophin gene lead to errors in coding for the dystrophin protein. The rod-shaped dystrophin protein is localized under the muscle fiber membrane, or the sarcolemma. Dystrophin consists of four major domains: an amino terminal domain that anchors it to the intracellular actin cytoskeleton, a long flexible rod domain that absorbs the impact of muscle contraction, a cysteine-rich domain that links the intracellular cytoskeleton to the extracellular matrix, and the carboxyl terminal, which is less critical for preserving muscle function. Additionally, dystrophin is an essential member of the dystrophin-associated protein complex (DAPC), which protects the sarcolemma from mechanical stress during repeated cycles of muscle contraction and relaxation. Mutations that permit some gene expression are usually associated with the milder BMD clinical phenotype. The mutations of BMD patients are commonly in the rod domain but do occur elsewhere in the gene. In contrast, mutations that interrupt the linkage between the actin cytoskeleton (amino terminal) and extracellular matrix (cysteine-rich domain) or result in very low levels of dystrophin protein expression typically give rise to the more severe DMD phenotype.
When the function of dystrophin is compromised as a result of mutations, the sarcolemma is more fragile and susceptible to damage from muscle contractions leading to small tears in the membrane. Extracellular calcium enters the muscle fiber through these small tears, which in turn activates calcium-activated proteases leading to protein degradation (Constantin, Sebille, & Cognard, 2006). Evidence from DMD skeletal muscle biopsies has shown that the level of dystrophin protein in muscle is ≤ 5%, much less than the amount of dystrophin found in BMD patients. The increased permeability of the sarcolemma also leads to the release of intracellular muscle proteins from the myofiber, such as creatine kinase (CK). Increased level of CK in the serum above normal (35–174 U/L) is the hallmark of muscle damage and is a diagnostic tool of these two types of muscular dystrophy. The value of serum CK varies with the age and clinical progression in DMD and BMD patients, and it can reach values 200 to 300 times higher than normal (Zatz et al., 1991).
Dystrophin in the cardiomyocyte
Defects in or absence of dystrophin protein in the cardiomyocyte result in DCM through a similar pathway as described. Specifically, recent studies have suggested that absence or mutation of dystrophin disrupts the function of membrane ion channels, particularly the sarcolemmal stretch-activated channels, which respond to mechanical stress (Woolf et al., 2006). When cardiomyocytes with deficient or mutant dystrophin stretch during ventricular filling, the stretch-activated channels do not open appropriately, leading to increased influx of calcium (Williams & Allen, 2007). The high intracellular calcium activates calcium-induced calpains, a group of proteases, which will degrade troponin I and compromise contraction of the cardiomyocyte (Feng, Schaus, Fallavollita, Lee, & Canty, 2001; Gao et al., 1997). Calpain-mediated destruction of membrane protein allows more calcium entry. Eventually, the chronic calcium overload leads to the death of the cardiomyocyte (Raymackers et al., 2003). Figure 1 is a graphic representation of the cardiomyocyte pathophysiology pathways.
Figure 1.

Known pathways through which abnormal dystrophin leads to cardiomyocyte death.
Cardiac mechanical factors
There are several distinctive characteristics of the cardiomyocyte that contribute to the deterioration of the heart when dystrophin is dysfunctional or absent. First, unlike skeletal muscle, cardiac muscle repeatedly contracts at least 86,400 times per day. The calcium fluxes associated with each excitation-contraction cycle undoubtedly accelerate the deterioration process within cardiomyocytes compared to skeletal myofibers. Second, the cardiomyocyte contracts in the calcium-induced calcium release manner; small amounts of extracellular calcium enter the cell through the voltage-gated L-type calcium channels and induce larger amounts of intracellular calcium to be released into the cytoplasm. As described earlier, when extracellular calcium leaks into the cardiomyocyte because of dystrophin defect or deficiency, the L-type channels can be activated unnecessarily, causing the release of intracellular calcium and initiating a contraction. Ultimately, the elevated intracellular calcium concentration activates the cascade of protein degradation and cardiomyocyte death.
Cardiomyocyte fibrosis and dilation of the heart
The death of cardiomyocytes initiates an inflammatory cascade during which macrophages migrate to clear the damaged cells and debris. Following macrophage recruitment, fibroblasts invade the damaged area and form scar tissue or fibrosis in the heart. Fibrotic tissue is very inflexible compared to the normal cardiac tissue and thus restricts the efficiency of myocardial contraction. The fibrosis begins in the left ventricular wall in DMD and in the right ventricular wall in BMD, starting in the epicardium and advancing into the endocardium (Frankel & Rosser, 1976). It progressively spreads throughout most of the outer half of the ventricular wall. This pattern of fibrosis is unique to dystrophinopathy. The fibrotic region will gradually stretch, become thinner, lose contractility, and result in DCM. The dilation of the heart increases left ventricular volume, decreases systolic function, and often leads to mitral valve regurgitation, all of which result in decreased cardiac output and hemodynamic decompensation. The cardiac phenotype in each DMD or BMD patient results from the patient’s particular type of dystrophin gene mutation; however, the relationship between genotype and phenotype remains elusive. Figure 2 depicts the pathologic progression of DCM.
Figure 2.
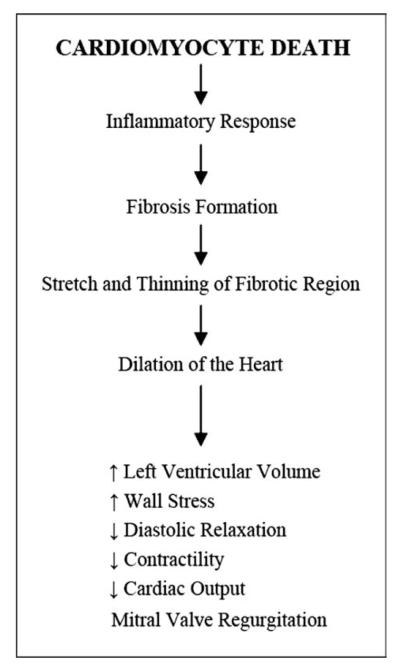
Progression from cardiomyocyte death to dilated cardiomyopathy.
Assessments
In the early stages of muscular dystrophies, cardiomyopathy is usually asymptomatic because of some compensatory mechanisms, including activation of the sympathetic nervous system (Lu & Hoey, 2000). Eventually, functional reserve of the heart is exhausted and the patient develops classic signs of DCM. Because DMD patients often have limited physical abilities, pathologic changes of the heart are seldom detected until the disease is quite severe; however, because the physical ability of BMD patients varies, cardiac manifestations for BMD patients are much more variable. In BMD patients who live a more active lifestyle, cardiac symptoms may lead the patient to the cardiologist before skeletal muscle weakness is noticed and the BMD diagnosis is made. In BMD patients who have minor physical limitation, the cardiac disease may negatively affect quality of life more than the skeletal symptoms. Therefore, early diagnosis and treatment of cardiomyopathy will greatly benefit patients with muscular dystrophy (English & Gibbs, 2006).
Cardiac condition can be assessed via 12-lead or Holter electrocardiogram (EKG) and echocardiogram. Cardiac magnetic resonance imaging (MRI) allows functional assessment, as do computerized tomography imaging, radionuclide imaging, and positron emission testing. Additionally, MRI can show the presence and degree of fibrosis. Clinicians have different preferences in setting the time intervals between cardiac follow-up visits. Emerging data have shown that assessment of cardiac function should begin on diagnosis of the muscular dystrophy, and then every year until an abnormality is identified. Once evidence of cardiac abnormality has been identified, the patient may be seen by the cardiologist every three months, or more frequently as necessary depending on the need to assess responses to treatment.
DCM pathology specific to DMD
Many DMD patients develop sinus tachycardia by 5 years of age and conduction changes by 10 years of age. Irregular conduction patterns seen in a 12-lead EKG include deep Q waves in leads I, aVL, V5, and V6, tall R waves in V1, and the R/S ratio of greater than 1 (Emery & Muntoni, 2003). Shortened PR intervals are seen in about 50% of patients, and QT prolongation is rare but can also be noted. From Holter EKG monitoring, resting sinus tachycardia, loss of circadian rhythm, and reduced heart rate variability caused by increased sympathetic activity can be observed (Kirchmann et al., 2005). With an advanced fibrosis, more serious arrhythmias may be seen, including atrial fibrillation, atrioventricular block, ventricular tachycardia, and ventricular fibrillation (Corrado et al., 2002).
With DMD, myocardial fibrosis and dilation first appear in the left ventricular wall behind the posterior mitral valve leaflet, which over time progress inferiorly toward the apex and into the septum, ultimately affecting the entire left ventricle. Visualization of posterior epicardial thinning, areas of dyskinesis and akinesis, as well as direct measurement of systolic and diastolic dysfunction are possible with echocardiogram or other imaging methods such as MRI. The E point-to-septal separation (EPSS) measures the minimal separation between the mitral valve anterior leaflet and the ventricular septum during early diastole. An EPSS of greater than 5–5.5 mm suggests that the left ventricle has become more spherical and can provide a clue for cardiac anatomical changes (Anderson, 2007). Likewise, the sphericity index, calculated by dividing the length of the left ventricle by the width of the left ventricle, is very useful in evaluating the shape of the left ventricular chamber. A sphericity index of greater than 0.66 suggests the presence of dilated cardiomyopathy (Tani, Minich, Williams, & Shaddy, 2005). Other signs of dilation include increased left ventricular diameter and volume, decreased shortening and ejection fractions, and development of mitral valve regurgitation.
DCM pathology specific to BMD
Becker-type muscular dystrophy patients have EKG presentations of DCM similar to those seen in DMD, including deep Q waves in leads I, aVL (or II, III aVF) and V6, tall R waves in V1 and shortened PR intervals. Bundle-branch block, nonspecific ST changes, and negative T waves have also been reported (Melacini et al., 1996), as have atrial or ventricular arrhythmias such as tachycardia, fibrillation, and flutter (Suselbeck, Haghi, Neff, Borggrefe, & Papavassiliu, 2005). Different from DMD, however, the Holter EKG in Becker patients does not show the increased automaticity of sympathetic activation (Kirchmann et al., 2005; Vita et al., 2001). Figure 3 shows the EKG of a BMD patient. The fibrosis in BMD has a similar pattern of progression; however, it often begins in the right ventricular wall. The echocardiographic measurement standards are the same as in DMD. In addition, measurement of the peak systolic strain rate and early diastolic strain rate may detect abnormal myocardial function not revealed by the measurement of the ejection fraction (Meune et al., 2004).
Figure 3.

Electrocardiogram of a patient with Becker muscular dystrophy. Note the short PR interval (98 ms, black horizontal bar, normal range: 120–200 ms), Q waves in leads 2, 3, aVF, V5, V6 (arrows), and tall R wave in V1 (asterisks). The EKG finding in patients with Duchenne muscular dystrophy is similar.
Other assessment tools
Chest X-ray is another tool used to reveal cardiomegaly, pleural effusion, and pulmonary congestion, supporting the diagnosis of DCM (Finsterer & Stollberger, 2003). Although it is common to measure blood levels of certain enzymes to evaluate cardiac pathology, this is not always helpful in the diagnosis of DCM of muscular dystrophy. The leakage of muscle proteins from skeletal muscle fibers into the serum because of the nature of the disease persistently maintains a high level of CK, which declines only with reduction in muscle mass. For example, the typical “cardiac-specific” protein, CK-MB, can also be detected during ongoing skeletal muscle regeneration; therefore, it is not cardiac-specific for DMD and BMD patients (Bodor et al., 1997; Nakada, Miyazaki, & Hirabayashi, 2002). On the other hand, the use of cardiac troponin I as a marker for myocardial damage among DMD and BMD patients has been reported (Matsumura, Saito, Fujimura & Shinno, 2007). Alternatively, brain natriuretic peptide (BNP), a cardiac hormone originally isolated from the brain, is used to monitor cardiac failure. BNP is synthesized by the cells in the atria and is released following left ventricular overloading and increased wall stress. Therefore, BNP level serves as a valuable clinical tool for monitoring the progression of DCM (Mori et al., 2002). BNP has been proposed as a biomarker for left ventricular dysfunction as well as the most accurate predictor of cardiac prognosis (Doust, Pietrzak, Dobson, & Glasziou, 2005). In practice, a diagnostic BNP value greater than 80–100 pg/mL is indicative of heart failure.
Pharmacological treatments
Pharmacological treatment should be offered to DMD and BMD patients with cardiac complications. Based on the fundamental pathogenesis of calcium accumulation in DMD and BMD, clinical trials have been conducted to test the rescuing effect of calcium channel blockers, including diltiazem, flunarizine, and nifedipine (Dick et al., 1986; Moxley et al., 1987; Pernice et al., 1988; Toifl, Presterl, & Graninger, 1991). These trials did not, however, demonstrate benefits; therefore, calcium blockers are not used. The progressive cardiac fibrosis eventually leads to reduction of the functional myocardial mass, increased wall stress, and decreased cardiac output. The decrease in cardiac output leads to activation of the renin-angiotensin-aldosterone system (RAAS), which is crucial in the regulation of sodium and water. The first RAAS component activated is renin, an enzyme that cleaves angiotensinogen to form angiotensin I. Subsequently, the angiotensin-converting enzyme (ACE), which is produced by endothelial cells in the lungs, transforms angiotensin I to angiotensin II. Angiotensin II stimulates the adrenal cortex to secrete aldosterone, promoting fluid and sodium retention.
Both angiotensin II and aldosterone contribute to the formation of fibrosis and overgrowth of connective tissue within the heart. Specifically, angiotensin II acts as a growth factor at sites of tissue repair and enhances the activity of a fibrogenic cytokine, TGF-β1 (Weber, 1999). Likewise, aldosterone is involved in the synthesis of fibrosis-forming collagen and reduces compliance of the heart (Bernal, Pitta, & Thatai, 2006). These harmful consequences of RAAS hyperactivity secondary to decreased cardiac output further complicate the myocardial fibrosis resulting from dystrophin deficiency in DMD and BMD patients. Therefore, the use of ACE inhibitors, angiotensin receptor blockers (ARBs), and aldosterone antagonists in DMD and BMD patients with cardiomyopathy is indicated. ACE inhibitors have been more extensively studied and therefore are prescribed more frequently. In 2003, a European group first recommended treating the progressive cardiomyopathy in DMD and BMD patients with ACE inhibitors and, if necessary, beta blockers. This group also emphasized that their recommendation was not evidence-based and encouraged large-scale trials (Bushby, Muntoni, & Bourke, 2003). Since then, several studies have been initiated.
An uncontrolled retrospective study showed that echocardiographic parameters, including left ventricular ejection fraction, fractional shortening, and sphericity index, in DMD and BMD patients were improved 3 years after administration of either ACE inhibitors alone or the combination of both ACE inhibitors and beta blockers (Jefferies et al., 2005). Around the same time, a double-blind multicenter study was conducted to evaluate the effect of preventive afterload reduction in muscular dystrophy patients. In this study, DMD patients between 9.5 and 13 years old with normal ventricular function were randomly assigned to receive 3 years of placebo or perindopril, an ACE inhibitor. After this 3-year period, every participant received 2 years of perindopril. At the conclusion of the study, a lower left ventricular ejection fraction was found in those subjects who did not take perindopril for the first 3 years (Duboc et al., 2005). The findings of this study not only suggested the beneficial effects of perindopril but also encouraged early pharmacological interventions to preserve cardiac function. A later study in 2006 tested another ACE inhibitor, enalapril, and monitored left ventricular systolic dysfunction in DMD patients with DCM, diagnosed based on left ventricular fractional shortening. Normalization of fractional shortening occurred in 43% of the patients on enalapril (Ramaciotti et al., 2006). This study provided more support for the use of ACE inhibitors in DMD and BMD patients with DCM. Although there is no universally agreed upon guideline for the best time to begin ACE inhibition therapy for these patients, current evidence suggests that an ACE inhibitor should be prescribed to the DMD or BMD patient with a left ventricular ejection fraction less than 55%, a sphericity index less than 0.66, or a myocardial performance index less than 0.35 (Bosser et al., 2004; Duboc et al., 2005; Jefferies et al., 2005; Naruse et al., 2004). Prescribing an ACE inhibitor dosed once a day will add to patients’ convenience and likelihood of compliance.
Given the beneficial effects of beta blockers used in patients with DCM and heart failure attributable to other causes, this class of drugs is frequently prescribed in addition to ACE inhibitors for DCM in DMD or BMD patients (Doing, Renlund, & Smith, 2002; Ishikawa, Bach, Ishikawa, & Minami, 1995). In a more recent Japanese study, patients with different types of muscular dystrophies were assigned to receive either an ACE inhibitor alone (cilazapril or enalapril) for at least 3 years or an ACE inhibitor plus a beta blocker (carvedilol) for at least 2 years. Interestingly, the combination therapy provided a significant improvement on left ventricular fractional shortening, whereas ACE inhibitor treatment alone maintained the fractional shortening at the same level (Kajimoto et al., 2006). These data support the combined use of ACE inhibitors and beta blockers. In clinical practice, beta blockers may be added to the treatment regimen when ventricular irritability (i.e., ventricular arrhythmias) is noted or when the ACE inhibitor does not improve echocardiographic indices of ventricular remodeling for 3 months (Jefferies et al., 2005).
Although ARBs are not currently used to treat DMD and BMD patients with dilated cardiomyopathy as often, their effects are being actively investigated in animal and human studies. One ARB, valsartan, together with enalapril was tested in an animal model of DCM. The results showed that animals that received combination therapy showed significant improvement in cardiac function as observed from echocardiogram and cardiac catheterization (Shimizu et al., 2002). Other studies have shown other ARBs, including candesartan, cilexetil, and losartan, to have therapeutic potential in animal models of DCM (De Mello & Specht, 2006; Shirai et al., 2005). In another study, the ARB losartan was administered to animal models of myopathy as a blockade of TFG-β signaling in skeletal muscle. This antagonism restored the regenerative capacity of the skeletal muscle, which is lost in muscular dystrophy. The results suggested that in addition to their cardiac therapeutic benefits, ARBs may also be useful in treating the skeletal muscle degeneration in muscular dystrophy (Cohn et al., 2007). Currently, the efficacy of ARBs has not been studied in large trials of DMD and BMD patients with DCM. Overall, pharmacological intervention has proven to be partially effective in managing the DCM of muscular dystrophy; however, DCM inevitably worsens as the patient becomes older and as the disease progresses, demonstrating the need for further research and development of new therapies.
There are many ongoing investigations to find a cure for DCM of muscular dystrophy. Several studies have explored the introduction of a modified, functional dystrophin gene via gene transfer (Townsend et al., 2007; Yue et al., 2003), as well as molecular correction of the mutated dystrophin gene in situ (Lu et al., 2003). A therapy involving intravenous delivery of a synthetic surfactant that functions as a cell membrane sealant has shown promise in strengthening the fragile and leaky sarcolemmal membrane responsible for the excessive calcium influx (Yasuda et al., 2005). To date, these approaches have been studied in only animals or a small number of humans. Although demonstrating significant promise, these studies are still in proof-of-concept or early clinical development.
Summary
In the not so distant past, the cardiac features of muscular dystrophy were often explained as being secondary to the respiratory abnormalities and musculoskeletal problems. It is now speculated that certain mutations within the dystrophin gene may be more likely than others to lead to cardiac involvement. Improved clinical care of these patients depends on a better genetic understanding of the dystrophin gene mutations and the pathophysiological consequences. Novel strategies using existing or newly developed pharmacologic agents may slow the progression of cardiac dysfunction. Methods to correct mutations or to replace the dysfunctional or absent protein in skeletal and cardiac muscle, however, will have the greatest impact on this disease. Although the disease has eluded a cure for nearly two centuries, the next decade may prove to be the most fruitful for altering the clinical course of muscular dystrophy. The challenge for clinicians is to keep updated with the latest research to provide optimal patient care.
Acknowledgments
The first author was supported by Grant Number T32RR023260 from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH). The authors sincerely appreciate helpful comments during the preparation of the manuscript provided by Donna McCarthy, PhD, RN, Professor of Nursing from The Ohio State University College of Nursing, and Jerry R. Mendell, MD, Professor of Neurology from The Ohio State University College of Medicine and Director of Center for Gene Therapy, the Research Institute at Nationwide Children’s Hospital.
Footnotes
Conflict of interest disclosure statement No relationship exists between any of the authors and any commercial entity or product mentioned in this article that might represent a conflict of interest. No inducements have been made by any commercial entity to submit the manuscript for publication.
References
- Anderson B. Echocardiography: The normal examination and echocardiographic measurement. MGA Graphics; Manly, Queensland, Australia: 2007. [Google Scholar]
- Bernal J, Pitta SR, Thatai D. Role of the renin-angiotensin-aldosterone system in diastolic heart failure: Potential for pharmacologic intervention. American Journal of Cardiovascular Drugs. 2006;6(6):373–381. doi: 10.2165/00129784-200606060-00004. [DOI] [PubMed] [Google Scholar]
- Bodor GS, Survant L, Voss EM, Smith S, Porterfield D, Apple FS. Cardiac troponin T composition in normal and regenerating human skeletal muscle. Clinical Chemistry. 1997;43(3):476–484. [PubMed] [Google Scholar]
- Bosser G, Lucron H, Lethor JP, Burger G, Beltramo F, Marie PY, et al. Evidence of early impairments in both right and left ventricular inotropic reserves in children with Duchenne’s muscular dystrophy. American Journal of Cardiology. 2004;93(6):724–727. doi: 10.1016/j.amjcard.2003.12.005. [DOI] [PubMed] [Google Scholar]
- Bushby K, Muntoni F, Bourke JP. 107th ENMC international workshop: The management of cardiac involvement in muscular dystrophy and myotonic dystrophy. 7th-9th June 2002, Naarden, the Netherlands. Neuromuscular Disorders. 2003;13(2):166–172. doi: 10.1016/s0960-8966(02)00213-4. [DOI] [PubMed] [Google Scholar]
- Bushby K, Thambyayah M, Gardner-Medwin D. Prevalence and incidence of Becker muscular dystrophy. Lancet. 1991;337(8748):1022–1024. doi: 10.1016/0140-6736(91)92671-n. [DOI] [PubMed] [Google Scholar]
- Cohn RD, van Erp C, Habashi JP, Soleimani AA, Klein EC, Lisi MT, et al. Angiotensin II type 1 receptor blockade attenuates TGF-beta-induced failure of muscle regeneration in multiple myopathic states. Nature Medicine. 2007;13(2):204–210. doi: 10.1038/nm1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constantin B, Sebille S, Cognard C. New insights in the regulation of calcium transfers by muscle dystrophin-based cytoskeleton: Implications in DMD. Journal of Muscle Research and Cell Motility. 2006;27(5-7):375–386. doi: 10.1007/s10974-006-9085-2. [DOI] [PubMed] [Google Scholar]
- Corrado G, Lissoni A, Beretta S, Terenghi L, Tadeo G, Foglia-Manzillo G, et al. Prognostic value of electrocardiograms, ventricular late potentials, ventricular arrhythmias, and left ventricular systolic dysfunction in patients with Duchenne muscular dystrophy. American Journal of Cardiology. 2002;89(7):838–841. doi: 10.1016/s0002-9149(02)02195-1. [DOI] [PubMed] [Google Scholar]
- Davies KE, Nowak KJ. Molecular mechanisms of muscular dystrophies: Old and new players. National Review of Molecular and Cell Biology. 2006;7(10):762–773. doi: 10.1038/nrm2024. [DOI] [PubMed] [Google Scholar]
- De Mello WC, Specht P. Chronic blockade of angiotensin II AT1-receptors increased cell-to-cell communication, reduced fibrosis and improved impulse propagation in the failing heart. Journal of the Renin-Angiotensin-Aldosterone System. 2006;7(4):201–205. doi: 10.3317/jraas.2006.038. [DOI] [PubMed] [Google Scholar]
- Dick DJ, Gardner-Medwin D, Gates PG, Gibson M, Simpson JM, Walls TJ. A trial of flunarizine in the treatment of Duchenne muscular dystrophy. Muscle & Nerve. 1986;9(4):349–354. doi: 10.1002/mus.880090412. [DOI] [PubMed] [Google Scholar]
- Doing AH, Renlund DG, Smith RA. Becker muscular dystrophy-related cardiomyopathy: A favorable response to medical therapy. Journal of Heart and Lung Transplantation. 2002;21(4):496–498. doi: 10.1016/s1053-2498(01)00316-3. [DOI] [PubMed] [Google Scholar]
- Doust J, Pietrzak E, Dobson A, Glasziou P. How well does B-type natriuretic peptide predict death and cardiac events in patients with heart failure: Systematic review. British Medical Journal. 2005;330(7492):625. doi: 10.1136/bmj.330.7492.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duboc D, Meune C, Lerebours G, Devaux JY, Vaksmann G, Becane HM. Effect of perindopril on the onset and progression of left ventricular dysfunction in Duchenne muscular dystrophy. Journal of the American College of Cardiology. 2005;45(6):855–857. doi: 10.1016/j.jacc.2004.09.078. [DOI] [PubMed] [Google Scholar]
- Duchenne GBA. Selections from the clinical works of dr Duchenne (de Boulogne) (G. Poore, Trans.) 3rd ed The New Sydenham Society; London: 1872. L’electricisation localisée; pp. 42–87. [Google Scholar]
- Eagle M, Baudouin SV, Chandler C, Giddings DR, Bullock R, Bushby K. Survival in Duchenne muscular dystrophy: Improvements in life expectancy since 1967 and the impact of home nocturnal ventilation. Neuromuscular Disorders. 2002;12(10):926–929. doi: 10.1016/s0960-8966(02)00140-2. [DOI] [PubMed] [Google Scholar]
- Emery A. Population frequencies of inherited neuromuscular diseases: A world survey. Neuromuscular Disorders. 1991;1(1):19–29. doi: 10.1016/0960-8966(91)90039-u. [DOI] [PubMed] [Google Scholar]
- Emery AE, Muntoni F. Duchenne muscular dystrophy. 3rd ed Oxford University Press; Oxford: 2003. [Google Scholar]
- English K, Gibbs J. Cardiac monitoring and treatment for children and adolescents with neuromuscular disorders. Developmental Medicine & Child Neurology. 2006;48(3):231–235. doi: 10.1017/S0012162206000491. [DOI] [PubMed] [Google Scholar]
- Erb WH. Über die “juvenile form” der progressiven muskelatrophie und ihre beziehungen zur sogenannten pseudohypertrophie der muskeln. Deutsches Archiv für klinische Medizin. 1884;34:467–519. [Google Scholar]
- Feng J, Schaus BJ, Fallavollita JA, Lee TC, Canty JM., Jr Preload induces troponin I degradation independently of myocardial ischemia. Circulation. 2001;103(16):2035–2037. doi: 10.1161/01.cir.103.16.2035. [DOI] [PubMed] [Google Scholar]
- Finsterer J, Stollberger C. The heart in human dystrophinopathies. Cardiology. 2003;99(1):1–19. doi: 10.1159/000068446. [DOI] [PubMed] [Google Scholar]
- Frankel K, Rosser R. The pathology of the heart in progressive muscular dystrophy: epimyocardial fibrosis. Human Pathology. 1976;7(4):375–386. doi: 10.1016/s0046-8177(76)80053-6. [DOI] [PubMed] [Google Scholar]
- Gao WD, Atar D, Liu Y, Perez NG, Murphy AM, Marban E. Role of troponin I proteolysis in the pathogenesis of stunned myocardium. Circulation Research. 1997;80(3):393–399. [PubMed] [Google Scholar]
- Hoffman EP, Brown RH, Kunkel LM. Dystrophin: The protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51:919–928. doi: 10.1016/0092-8674(87)90579-4. [DOI] [PubMed] [Google Scholar]
- Hoffman EP, Hudecki MS, Rosenberg PA, Pollina CM, Kunkel LM. Cell and fiber-type distribution of dystrophin. Neuron. 1988;1(5):411–420. doi: 10.1016/0896-6273(88)90191-2. [DOI] [PubMed] [Google Scholar]
- Ishikawa Y, Bach JR, Ishikawa Y, Minami R. A management trial for Duchenne cardiomyopathy. American Journal of Physical Medicine & Rehabilitation. 1995;74(5):345–350. [PubMed] [Google Scholar]
- Jefferies JL, Eidem BW, Belmont JW, Craigen WJ, Ware SM, Fernbach SD, et al. Genetic predictors and remodeling of dilated cardiomyopathy in muscular dystrophy. Circulation. 2005;112(18):2799–2804. doi: 10.1161/CIRCULATIONAHA.104.528281. [DOI] [PubMed] [Google Scholar]
- Kajimoto H, Ishigaki K, Okumura K, Tomimatsu H, Nakazawa M, Saito K, et al. Beta-blocker therapy for cardiac dysfunction in patients with muscular dystrophy. Circulation Journal. 2006;70(8):991–994. doi: 10.1253/circj.70.991. [DOI] [PubMed] [Google Scholar]
- Kirchmann C, Kececioglu D, Korinthenberg R, Dittrich S. Echocardiographic and electrocardiographic findings of cardiomyopathy in Duchenne and Becker-Kiener muscular dystrophies. Pediatric Cardiology. 2005;26(1):66–72. doi: 10.1007/s00246-004-0689-2. [DOI] [PubMed] [Google Scholar]
- Koenig M, Hoffman E, Bertelson C, Monaco A, Feener C, Kunkel L. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell (Cambridge) 1987;50(3):509–517. doi: 10.1016/0092-8674(87)90504-6. [DOI] [PubMed] [Google Scholar]
- Lu QL, Mann CJ, Lou F, Bou-Gharios G, Morris GE, Xue SA, et al. Functional amounts of dystrophin produced by skipping the mutated exon in the mdx dystrophic mouse. Nature Medicine. 2003;9(8):1009–1014. doi: 10.1038/nm897. [DOI] [PubMed] [Google Scholar]
- Lu S, Hoey A. Age- and sex-associated changes in cardiac beta(1)-adrenoceptors from the muscular dystrophy (mdx) mouse. Journal of Molecular and Cellular Cardiology. 2000;32(9):1661–1668. doi: 10.1006/jmcc.2000.1200. [DOI] [PubMed] [Google Scholar]
- Matsumura T, Saito T, Fujimura H, Shinno S. Cardiac troponin I for accurate evaluation of cardiac status in myopathic patients. Brain and Development. 2007;29(8):496–501. doi: 10.1016/j.braindev.2007.01.009. [DOI] [PubMed] [Google Scholar]
- Melacini P, Fanin M, Danieli GA, Villanova C, Martinello F, Miorin M, et al. Myocardial involvement is very frequent among patients affected with subclinical Becker’s muscular dystrophy. Circulation. 1996;94(12):3168–3175. doi: 10.1161/01.cir.94.12.3168. [DOI] [PubMed] [Google Scholar]
- Meryon E. On fatty degeneration of the voluntary muscles: Report of the Royal Medical and Chirurgical Society. Lancet. 1851;2:588–589. [Google Scholar]
- Meune C, Pascal O, Becane HM, Heloire F, Christoforou D, Laforet P, et al. Reliable detection of early myocardial dysfunction by tissue Doppler echocardiography in Becker muscular dystrophy. Heart (British Cardiac Society) 2004;90(8):947–948. doi: 10.1136/hrt.2003.021113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori K, Manabe T, Nii M, Hayabuchi Y, Kuroda Y, Tatara K. Plasma levels of natriuretic peptide and echocardiographic parameters in patients with Duchenne’s progressive muscular dystrophy. Pediatric Cardiology. 2002;23(2):160–166. doi: 10.1007/s00246-001-0040-0. [DOI] [PubMed] [Google Scholar]
- Moxley RT, 3rd, Brooke MH, Fenichel GM, Mendell JR, Griggs RC, Miller JP, et al. Clinical investigation in Duchenne dystrophy. VI. Double-blind controlled trial of nifedipine. Muscle & Nerve. 1987;10(1):22–33. doi: 10.1002/mus.880100106. [DOI] [PubMed] [Google Scholar]
- Nakada K, Miyazaki J, Hirabayashi T. Expression of multiple troponin T isoforms in chicken breast muscle regeneration induced by sub-serous implantation. Differentiation; Research in Biological Diversity. 2002;70(2–3):92–100. doi: 10.1046/j.1432-0436.2002.700204.x. [DOI] [PubMed] [Google Scholar]
- Naruse H, Miyagi J, Arii T, Ohyanagi M, Iwasaki T, Jinnai K. The relationship between clinical stage, prognosis and myocardial damage in patients with Duchenne-type muscular dystrophy: Five-year follow-up study. Annals of Nuclear Medicine. 2004;18(3):203–208. doi: 10.1007/BF02985001. [DOI] [PubMed] [Google Scholar]
- Nigro G, Comi L, Politano L, Limongelli F, Nigro V, De Rimini M, et al. Evaluation of the cardiomyopathy in Becker muscular dystrophy. Muscle and Nerve. 1995;18(3):283–291. doi: 10.1002/mus.880180304. [DOI] [PubMed] [Google Scholar]
- Nigro G, Politano L, Passamano L, Palladino A, De Luca F, Nigro G, et al. Cardiac treatment in neuro-muscular diseases. Acta Myologica. 2006;25(3):119–123. [PubMed] [Google Scholar]
- Pernice W, Beckmann R, Ketelsen UP, Frey M, Schmidt-Redemann B, Haap KP, et al. A double-blind placebo controlled trial of diltiazem in Duchenne dystrophy. Klinische Wochenschrift. 1988;66(13):565–570. doi: 10.1007/BF01720830. [DOI] [PubMed] [Google Scholar]
- Pillers DA, Bulman DE, Weleber RG, Sigesmund DA, Musarella MA, Powell BR, et al. Dystrophin expression in the human retina is required for normal function as defined by electroretinography. Nature Genetics. 1993;4(1):82–86. doi: 10.1038/ng0593-82. [DOI] [PubMed] [Google Scholar]
- Ramaciotti C, Heistein LC, Coursey M, Lemler MS, Eapen RS, Iannaccone ST, et al. Left ventricular function and response to enalapril in patients with Duchenne muscular dystrophy during the second decade of life. The American Journal of Cardiology. 2006;98(6):825–827. doi: 10.1016/j.amjcard.2006.04.020. [DOI] [PubMed] [Google Scholar]
- Raymackers JM, Debaix H, Colson-Van Schoor M, De Backer F, Tajeddine N, Schwaller B, et al. Consequence of parvalbumin deficiency in the mdx mouse: Histological, biochemical and mechanical phenotype of a new double mutant. Neuromuscular Disorders. 2003;13(5):376–387. doi: 10.1016/s0960-8966(03)00031-2. [DOI] [PubMed] [Google Scholar]
- Shimizu T, Okamoto H, Chiba S, Matsui Y, Sugawara T, Onozuka H, et al. Long-term combined therapy with an angiotensin type I receptor blocker and an angiotensin converting enzyme inhibitor prolongs survival in dilated cardiomyopathy. Japanese Heart Journal. 2002;43(5):531–543. doi: 10.1536/jhj.43.531. [DOI] [PubMed] [Google Scholar]
- Shirai K, Watanabe K, Ma M, Wahed MI, Inoue M, Saito Y, et al. Effects of angiotensin-II receptor blocker candesartan cilexetil in rats with dilated cardiomyopathy. Molecular and Cellular Biochemistry. 2005;269(1–2):137–142. doi: 10.1007/s11010-005-3446-9. [DOI] [PubMed] [Google Scholar]
- Suselbeck T, Haghi D, Neff W, Borggrefe M, Papavassiliu T. Midwall myocardial fibrosis in Becker-Kiener muscular dystrophy. Zeitschrift fur Kardiologie. 2005;94(7):465–468. doi: 10.1007/s00392-005-0249-7. [DOI] [PubMed] [Google Scholar]
- Tani LY, Minich LL, Williams RV, Shaddy RE. Ventricular remodeling in children with left ventricular dysfunction secondary to various cardiomyopathies. American Journal of Cardiology. 2005;96(8):1157–1161. doi: 10.1016/j.amjcard.2005.06.047. [DOI] [PubMed] [Google Scholar]
- Toifl K, Presterl E, Graninger W. Ineffectiveness of diltiazem in Duchenne muscular dystrophy: A placebo-controlled double-blind study. Wiener Klinische Wochenschrift. 1991;103(8):232–235. [PubMed] [Google Scholar]
- Townsend D, Blankinship MJ, Allen JM, Gregorevic P, Chamberlain JS, Metzger JM. Systemic administration of micro-dystrophin restores cardiac geometry and prevents dobutamine-induced cardiac pump failure. Molecular Therapy. 2007;15(6):1086–1092. doi: 10.1038/sj.mt.6300144. [DOI] [PubMed] [Google Scholar]
- Vita G, Di Leo R, De Gregorio C, Papalia A, Rodolico C, Coglitore S, et al. Cardiovascular autonomic control in Becker muscular dystrophy. Journal of the Neurological Sciences. 2001;186(1–2):45–49. doi: 10.1016/s0022-510x(01)00500-7. [DOI] [PubMed] [Google Scholar]
- Weber KT. Angiotensin II and connective tissue: Homeostasis and reciprocal regulation. Regulatory Peptides. 1999;82(1–3):1–17. doi: 10.1016/s0167-0115(99)00032-4. [DOI] [PubMed] [Google Scholar]
- Williams IA, Allen DG. Intracellular calcium handling in ventricular myocytes from mdx mice. American Journal of Physiology. 2007;292(2):H846–H855. doi: 10.1152/ajpheart.00688.2006. [DOI] [PubMed] [Google Scholar]
- Woolf PJ, Lu S, Cornford-Nairn R, Watson M, Xiao XH, Holroyd SM, et al. Alterations in dihydropyridine receptors in dystrophin-deficient cardiac muscle. American Journal of Physiology. 2006;290(6):H2439–H2445. doi: 10.1152/ajpheart.00844.2005. [DOI] [PubMed] [Google Scholar]
- Yasuda S, Townsend D, Michele DE, Favre EG, Day SM, Metzger JM. Dystrophic heart failure blocked by membrane sealant poloxamer. Nature. 2005;436(7053):1025–1029. doi: 10.1038/nature03844. [DOI] [PubMed] [Google Scholar]
- Yue Y, Li Z, Harper SQ, Davisson RL, Chamberlain JS, Duan D. Microdystrophin gene therapy of cardiomyopathy restores dystrophin-glycoprotein complex and improves sarcolemma integrity in the mdx mouse heart. Circulation. 2003;108(13):1626–1632. doi: 10.1161/01.CIR.0000089371.11664.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zatz M, Rapaport D, Vainzof M, Passos-Bueno M, Bortolini E, Pavanello Rde C, et al. Serum creatine-kinase (CK) and pyruvate-kinase (PK) activities in Duchenne (DMD) as compared with Becker (BMD) muscular dystrophy. Journal of the Neurological Sciences. 1991;102(2):190–196. doi: 10.1016/0022-510x(91)90068-i. [DOI] [PubMed] [Google Scholar]