

Abstract
The molecular mechanism underlying Ca2+/calmodulin (CaM)-dependent kinase II (CaMKII)-mediated regulation of the mouse transient receptor potential channel TRPC6 was explored by chimera, deletion and site-directed mutagenesis approaches. Induction of currents (ICCh) in TRPC6-expressing HEK293 cells by a muscarinic agonist carbachol (CCh; 100 μm) was strongly attenuated by a CaMKII-specific peptide, autocamtide-2-related inhibitory peptide (AIP; 10 μm). TRPC6/C7 chimera experiments showed that the TRPC6 C-terminal sequence is indispensable for ICCh to be sensitive to AIP-induced CaMKII inhibition. Further, deletion of a distal region (Gln855–Glu877) of the C-terminal CaM/inositol-1,4,5-trisphosphate receptor binding domain (CIRB) of TRPC6 was sufficient to abolish ICCh. Systematic alanine scanning for potential CaMKII phosphorylation sites revealed that Thr487 was solely responsible for the activation of the TRPC6 channel by receptor stimulation. The abrogating effect of the alanine mutation of Thr487 (T487A) was reproduced with other non-polar amino acids, namely glutamine or asparagine, while being partially rescued by phosphomimetic mutations with glutamate or aspartate. The cellular expression and distribution of TRPC6 channels did not significantly change with these mutations. Electrophysiological and immunocytochemical data with the Myc-tagged TRPC6 channel indicated that Thr487 is most likely located at the intracellular side of the cell membrane. Overexpression of T487A caused significant reduction of endogenous TRPC6-like current induced by Arg8-vasopressin in A7r5 aortic myocytes. Based on these results, we propose that the optimal spatial arrangement of a C-terminal domain (presumably the distal CIRB region) around a single CaMKII phosphorylation site Thr487 may be essential for CaMKII-mediated regulation of TRPC6 channels. This mechanism may be of physiological significance in a native environment such as in vascular smooth muscle cells.
Key points
Ca2+/calmodulin (CaM)-dependent kinase II (CaMKII) plays pivotal roles in diverse Ca2+-mediated cellular functions including the physiology/pathophysiology of the cardiovascular system, through modulation of a variety of Ca2+-permeable channels such as a non-voltage-gated Ca2+ channel TRPC6.
In this study, we investigated the molecular mechanism underlying its positive regulation by CaMKII with chimera, deletion and site-directed mutagenesis approaches.
The results indicate that two spatially separated sites of TRPC6 channel, i.e. a distal part of the C-terminal inositol-1,4,5-trisphosphate receptor/CaM binding domain and Thr487 located on the putative first intracellular loop, are crucial for the CaMKII-mediated regulation of TRPC6 channels.
This mechanism may serve as an effective positive feedback regulation of Ca2+ influx through TRPC6 channels, in concert with intracellular and transmembrane Ca2+ mobilization upon phospholipase C-coupled receptor stimulation by neurohormonal factors, thereby fine-tuning the cardiovascular functions.
Disruption of these could lead to pathological states such as cardiac hypertrophy and arrhythmia, hypertension and atherosclerosis.
Introduction
Ca2+/calmodulin (CaM)-dependent kinase II (CaMKII) is a serine/threonine kinase ubiquitously distributed in a broad range of tissues, and capable of phosphorylating a variety of protein substrates. CaMKII is activated by the binding of Ca2+/CaM in response to the elevation of the cytosolic Ca2+ concentration ([Ca2+]i), and can retain its activity beyond the [Ca2+]i signal by subsequent auto-phosphorylation (Hudmon & Schulman, 2002). As a result of these unique properties, CaMKII plays pivotal roles in diverse Ca2+-mediated cellular functions in a wide range of excitable and non-excitable tissues, including the cardiovascular system, in both homeostatic and activity-dependent manners (Schulman, 2004). For example, in the normal heart, duration- and frequency-dependent activation of CaMKII by Ca2+ influx during the action potential facilitates excitation–contraction coupling. In the failing heart, the excessive activation of CaMKII resulting from aberrant Ca2+ handling leads to myocardial dysfunction such as pumping failure and arrhythmia (Couchonnal & Anderson, 2008; Bers & Grandi, 2009). CaMKII is also involved in cardiac hypertrophy, where sustained stresses cause the translocation of CaMKII into the nucleus which in turn promotes the transcription of hypertrophic genes (Zhang & Brown, 2004). In blood vessels, CaMKII has been implicated in vascular remodelling, mobility, angiogenesis and enhanced reactivity associated with vascular dysfunction and diseases (Yousif et al. 2008; Banumathi et al. 2011; Kim et al. 2011; Li et al. 2011; Singer, 2012; Scott et al. 2012).
CaMKII not only acts as a key downstream effector of various Ca2+-mobilizing mechanisms, but by virtue of phosphorylation, regulates a variety of ion channels participating in Ca2+ homeostasis/dynamics in the cell. This is exemplified by voltage-gated Na+ and Ca2+ channels (Welsby et al. 2003; Yao et al. 2006; Wang et al. 2009; Aiba et al. 2010; Abiria & Colbran, 2010; Blaich et al. 2010), intracellular Ca2+ release channels (ryanodine receptor, inositol-1,4,5-trisphosphate receptor (IP3R) (Ai et al. 2005; Bare et al. 2005; Chelu et al. 2009) and transient receptor potential (TRP) channels (TRPV1, TRPC6) (Jung et al. 2004; Shi et al. 2004). Recent studies have disclosed that one of major cardiovascular isoforms of the TRP superfamily TRPC6 is subject to phosphorylation-mediated modification by several serine (Ser)/threonine (Thr) kinases including protein kinases A, C and G, CaMKII and extracellular signal-related kinase (ERK) 1/2 as well as non-receptor tyrosine kinases Lyn and Src (Yao et al. 2005; Nishioka et al. 2011; Shen et al. 2011). Activation of the TRPC6 channel investigated in a recombinant system showed its bell-shaped dependence on steady-state [Ca2+]i (peak activation: 200–300 nm), and was greatly decelerated by removal of external Ca2+ during receptor stimulation and markedly attenuated by the pharmacological inhibition of CaMKII (Shi et al. 2004). These observations suggest that, in response to both tonic and dynamic changes in [Ca2+]i, CaMKII may effectively tune TRPC6 channel activities to regulate associated cellular functions. There is, however, considerable paucity of knowledge about how CaMKII regulates TRPC6 channel activity especially at the molecular level. In this study, to address this question, we explored the molecular basis of CaMKII-mediated TRPC6 channel phosphorylation by employing chimera, deletion and site-directed mutation analyses. In the mutagenesis analysis, the fluorescence imaging method was first employed to screen a large number of mutants, the positive results of which were further confirmed by the patch clamp technique. The results show that, although a C-terminal CaM-binding domain is necessary for CaMKII-mediated regulation, the 487th threonine residue (Thr487), which is probably located on the first intracellular loop, is critical for CaMKII-mediated phosphorylation. Charge-neutralizing mutations of this amino acid residue caused almost total abolition of both expressed and endogenous TRPC6 channel activation by receptor stimulation.
Methods
Construction of chimeras and mutants
Full-length mouse TRPC6 and TRPC7 DNAs were cloned from the mouse brain cDNA library. Polymerase chain reaction (PCR) was used to construct the TRPC6/TRPC7 chimeras (TRPC6/C7) as previously described (Shi et al. 2004; see also Fig. 2A). For example, for Ch776, the mouse TRPC7 sequence containing its N-terminus (1–348) and transmembrane domain (349–672) was linked to the C-terminal sequence of mouse TRPC6 (727–930). For the construction of other chimeras such as Ch677, Ch676, Ch766, Ch667 and Ch767, see Fig. 2A. A two-step PCR method was employed to create deletions or an insertion in TRPC6 (△Lys824–Glu877, △Lys824–Tyr854, △Gln855–Glu877; Myc-tag ‘GGEQKLISEEDLGG’ insertion after Ser475; Figs 2E and 3A). Single amino acid mutations of mouse TRPC6 and TRPC7 as well as of chimeric Ch776 in Fig. 4 were introduced by PCR techniques using the QuickChange Site-directed Mutagenesis Kit (Stratagene, San Diego, CA, USA) according to the manufacturer's instructions. The wild-type, chimeric and mutant cDNAs were all subcloned into the pCI-neo expression vector and their authenticity and accuracy were verified by direct sequencing with a genetic analyser (ABI PRISM 3100, Applied Biosystems, Tokyo, Japan).
Figure 2. C-terminal domain of TRPC6 is essential for the action of calmodulin (CaM)-mediated regulation.
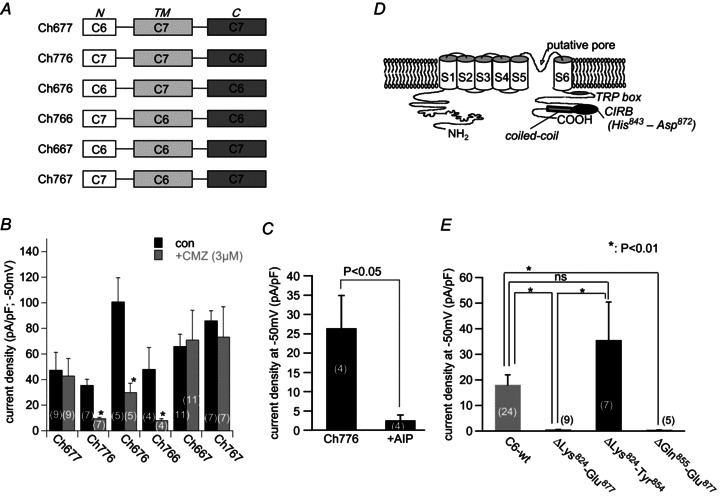
A, schematic illustration of the chimeric constructs of TRPC6 and TRPC7. B, comparison of the current density of 6 TRPC6/C7 chimeras before and after treatment with calmidazolium (CMZ, 3 μm). *P < 0.05 with unpaired t test. C, ICCh density (at −50 mV) with and without inclusion of AIP (10 μm) in the patch pipette. ICCh was evoked by 100 μm CCh 10 min after the establishment of whole-cell conditions. D, hypothetical TRPC6 channel structure with C-terminal CaM binding domain and coiled-coil region; CIRB, calmodulin, IP3 receptor binding domain. E, density of ICCh recorded from deletion mutants of TRPC6 CIRB domain. Deletion of the distal CIRB sequence caused severe inhibition of TRPC6 current density. *P < 0.01 with Tukey's multiple comparison test.
Figure 3. Screening of CaMKII phosphorylation sites by Fura-2 fluorescence imaging.
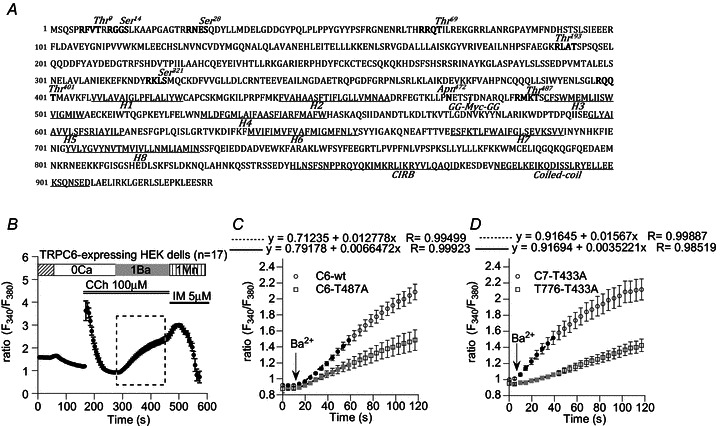
A, CaMKII phosphorylation motifs in murine TRPC6 sequence. Eight putative phosphorylation sites are highlighted in bold black. Seven of them are located in the N-terminus but Thr487 is between the second (H2) and third (H3) hydrophobic segments. B, the protocol used for the screening showing a typical receptor response recorded from HEK293 cells expressing wild-type (wt) TRPC6. Addition of Ba2+ (1 mm) after CCh (100 μm) application in the absence of Ca2+ induced a rapid fluorescence increase due to Ba2+ entry. A combination of ionomycin (IM) and Mn2+ was finally applied to quench Fura-2 to determine the background fluorescence. C, time-course plot of averaged Ba2+ fluorescence ratio in wild-type TRPC6 (C6-wt; magnified from the rectangle in B) and TRPC6-T487A mutant (C6-T487A)-expressing HEK cells. Neutralization of Thr487 by Ala substitution caused strong inhibition of CCh-induced Ba2+ entry. D, similar inhibition of the Ba2+ entry was also observed with the T776-T433A chimeric mutant. Ala substitution of an H2–H3 intersegmental CaMKII motif Thr433 (Supplemental Fig 1), which corresponds to Thr487 in TRPC6, resulted in a remarkable reduction in Ba2+ entry. Symbols and vertical bars denote mean ± SEM derived from 10–20 cells. In both C and D, linear regression of black or grey points were performed to calculate the rate of the ratio increase (dRBa/dt), and the results (‘x’ coefficients) are shown above the plots.
Figure 4. Summary of CCh-induced Ba2+ entry rate (dRBa/dt) compared among wild-type TRPC6 (C6-wt), TRPC7 (C7-wt), T776 chimera and their mutants carrying Ala substitution for CaMKII phosphorylation sites.
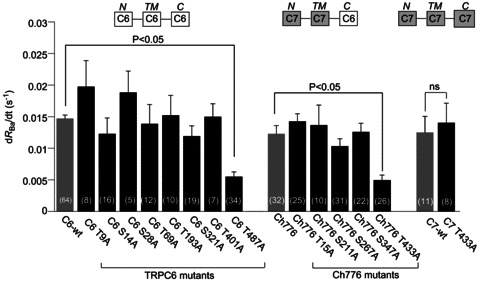
In most of the mutants, the rate of Ba2+ entry was unchanged compared with their parental TRPs, while greatly reduced in the two mutants, C6-T487A and Ch776-T433A. The Ala-substituted TRPC7 mutant (C7-T433A), however, showed comparable Ba2+ entry rate to wild-type TRPC7. The numbers in parentheses are numbers of independently performed experiments. P < 0.05, statistically significant, or ns, not significant, with Dunnett's multiple comparison test.
Cell culture and transfection
The human embryonic kidney cell line, HEK293, and an established aortic smooth muscle cell line, A7r5, were purchased from ATCC (Manassas, VA, USA) and used in the present study. The cells were maintained in Dulbecco's modified culture medium (DMEM) supplemented with 10% fetal bovine serum and a mixture of antibiotics (100 units ml−1 penicillin and streptomycin; Gibco, Invitrogen, USA) and passaged every 3–4 days.
For transfection, the cells were allowed to grow to 50–80% confluency in 35 mm culture dishes. DNAs for TRPC6 chimeras and their mutants were transfected into HEK293 cells together with pCI-neo-πH3-CD8 (cDNA of the T-cell antigen CD8) at the ratio of 5–10:1 using transfection reagent SuperFect (Qiagen, Germany). For the transfection of A7r5 myocytes, lipofectamine 2000 was used according to the manufacturer's instructions (Invitrogen, USA). Incubating for about 24 h after transfection, the cells were trypsinized and replated onto coverslips that had been pre-coated with poly-l-lysine (100 μg ml−1). Patch clamp and Ba2+ imaging experiments were performed within 24–48 h thereafter. A commercial polystyrene beads-conjugated anti-CD8 antibody (Dynabeads M-450 CD8, Dynal Biotech) was used to select cells which had successfully expressed exogenous proteins.
Electrophysiology
The whole-cell mode of the patch clamp technique was employed in this study. Patch electrodes were fabricated from 1.5 mm borosilicate glass capillaries (Sutter Instruments, USA) using an automated electrode puller (Sutter Instruments) and heat-polished in a microforge bearing a platinum/iridium wire (Narishige, Japan). Pipettes with a resistance of 3–5 MΩ (when filled with internal solution) were chosen for the patch clamp recording. Voltage generation and current signal acquisition were implemented through a high-impedance low-noise patch clamp amplifier (EPC7; HEKA Electronics, Germany) which was driven by Clampex v.6.2 software (Axon Instruments, Union City, CA, USA). Sampled data were stored on a hard disc of a PC after low-pass filtering at 1 kHz and digitization at 5 kHz. Data analysis was performed offline using a versatile analysis software ‘Clampfit’ v.9.2 (Axon Instruments). Long-term recordings were performed in conjunction with an A/D, D/A-converter PowerLab/400 (ADInstruments, Australia; sampling rate: 100 Hz) and analysed by the accessory software Chart v.5.0. A solenoid valve-driven fast solution change device similar to the ‘Y-tube’ system was used to rapidly apply drugs onto cells as performed previously (Shi et al. 2004). A liquid junction potential of ∼6 mV was corrected a posteriori.
Digital fluorescence imaging
Ba2+ fluorescence was measured using a dual-excitation wavelength spectrofluorometer (AQUACOSMOS, Hamamatsu Photonics, Japan). Transfected HEK cells which had been plated on a coverslip (3 mm × 10 mm) 24 h before experiments were loaded with the acetoxymethyl ester form of Fura 2 (Fura 2-AM; 1 μm) plus 0.01% pluronic acid F127 in the dark for 25–30 min at room temperature. After rinsing with physiological saline solution, the cells were alternately illuminated by UV lights (340 and 380 nm) and the emitted fluorescence filtered at 510 ± 10 nm was collected through a CCD camera (HISCA, Hamamatsu Photonics) with 200 times magnification (IX70, Olympus) for 100–200 ms at an interval of 5 s. Auto- and background fluorescences were determined by Mn quenching at the end of each experiment and subtracted from raw data. The ratio (R) of corrected fluorescence intensities at 340 and 380 nm excitations (F340/F380) was regarded as an indicator of intracellular Ba2+ concentration, and its rate of increase (△dRBa/△dt) as the magnitude of Ba2+ influx.
Immunocytochemistry
HEK cells overexpressing wild-type, mutant (T487E, T487A) or Myc-tagged TRPC6 proteins were reseeded on round coverslips and fixed with 4% paraformaldehyde for 20 min. After twice rinsing with PBS, the cells were either permeabilized with 0.2% Triton-containing PBS or simply soaked in PBS alone (for Fig. 7Ab lower row of images) for 15 min at room temperature, followed by thorough and careful rinsing. The cells were then subjected to the following single- or double-labelling immunostaining protocols of incubating successively with: 10% normal goat serum (Wako, Japan) for 1 h; rabbit anti-TRPC6 antibody (1:500 dilution; Alomone, Israel) with or without mouse anti-Myc antibody (1:1000 dilution; Invitrogen, USA) overnight at 4°C; Alexa 488-labelled goat anti-rabbit antiserum (1:300 dilution; Invitrogen) with or without Alexa 568-labelled goat-anti-mouse antiserum (1:300 dilution; Invitrogen) for 1 h. The cells were finally loaded with 4′,6-diamino-2-phenylindole (DAPI; dissolved in methanol at 1 μg ml−1 at 1:1000 dilution; Invitrogen) to counterstain all the nuclei. PBS rinsing was performed between each two steps above. The coverslips were then sealed with PermaFluor Aqueous (Immunon, Shandon, Pittsburgh, PA, USA) to prevent evaporation and kept in the dark to avoid photo-bleaching. Immunofluorescence was observed under a confocal laser scanning microscope (LSM 710-Zen2008, Carl Zeiss) equipped with argon/krypton laser sources. The spatial distribution of immunofluorescence was assessed by the ‘plot profile’ function of a free NIH software ‘ImageJ’ v.1.33u.
Figure 7. Immunocytochemical staining and electrophysiological properties of Myc-tagged TRPC6.
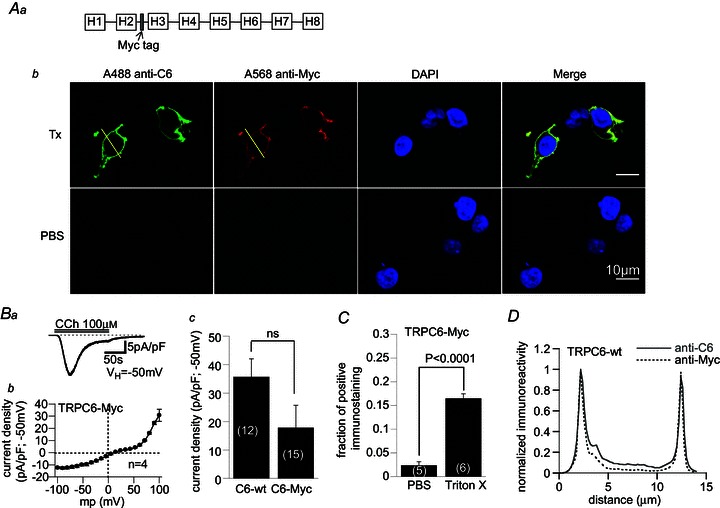
Aa, Myc sequence was inserted with two glycines at both sides in the H2–H3 intersegmental region of TRPC6 (see also Fig. 3A). Ab, typical micrographs showing immunofluorescent double-staining images with TRPC6 N-terminus (anti-C6)- and Myc (anti-Myc)-targeted antibodies before and after cell membrane permeabilization treatment with Triton X-100 (Tx). DAPI was used to counter-stain cell nuclei. B, Myc-tagged TRPC6 generated normal ICCh with characteristic I–V relationship; a, typical Myc-tagged TRPC6 current trace following perfusion of CCh (100 μm); b, I–V relationship of ICCh for Myc-tagged TRPC6 averaged from 4 experiments; c, comparison of ICCh density between wild-type and Myc-tagged TRPC6. C, fraction of cells positive for immunostaining with anti-Myc antibody with or without treatment by Triton X-100. D, immunofluorescence intensity plot for TRPC6- and Myc-staining along the lines drawn in Ab. For better comparison, the immunofluorescence is normalized with respect to its maximum value.
Immunoblotting
The plasma membrane (PM) protein fraction was extracted using a commercial protein extraction kit according to the manufacturer's instructions (BioVision, San Francisco, USA). Briefly, HEK cells expressing either wild-type or mutant TRPC6 proteins were homogenized and centrifuged at 4°C twice at different speed and time (700 g, 10 min; 10,000 g, 30 min) to yield the total membrane protein fraction. This faction was then used to separate a PM fraction in a two-phase solution tube by repeated centrifugation. The obtained PM fraction was lysed in the sample buffer. The total protein concentration of the sample was determined using the BCA protein assay kit (Pierce). Just before electrophoresis, 5% (v/v) 2-mercaptoethanol and 1% (w/v) bromophenol blue were added to the sample, and proteins were separated by 10% (w/v) SDS-PAGE and electrophoretically transferred to a PVDF membrane. The membrane was blocked with 5% (w/v) skimmed milk dissolved in Tween (0.1%)-PBS, and then incubated with 1:200 diluted anti-Na+/K+-ATPase (Abcam, Tokyo, Japan), or anti-TRPC6 (Alomone, Jerusalem, Israel) antibodies overnight. Protein expression was visualized by incubating the membrane with the secondary antibody linked to horseradish peroxidase.
Solutions and chemicals
The pipette solution contained (in mm): 120 CsOH, 120 aspartate, 20 CsCl, 2 MgCl2, 5 EGTA/1.5 CaCl2, 10 Hepes, 2 ATP, 0.01 GTP, 10 glucose (adjusted to pH 7.2 with Tris base). The external solution contained (in mm): 140 NaCl, 5 KCl, 1 CaCl2, 1.2 MgCl2, 10 Hepes, 10 glucose (pH 7.4, adjusted with Tris base).
Carbachol (CCh) was purchased from Sigma-Aldrich (USA), calmidazolium (chloride salt) from Calbiochem (USA), Arg8-vasopressin (AVP) from Calbiochem-Merck (USA), myristoylated autocamtide-2-related inhibitory peptide (AIP) from Enzo (Tokyo, Japan), and ATP, GTP and EGTA from Dojindo (Kumamoto, Japan). Stock solutions for CCh and AVP were made by dissolving the powders in DMSO at >1000-fold higher concentrations than for use so that the final concentration of DMSO did not exceed 0.1%; this concentration did not affect the magnitude or kinetics of TRPC6 currents significantly, which had been ensured by our previous experiment (Shi et al. 2004).
Statistics
All data are expressed as means ± SEM. For a single comparison, paired and unpaired Student's t tests were used to evaluate statistical significance with a criterion of P < 0.05. For multiple comparisons, statistical significance was evaluated by Tukey's or Dunnett's multiple comparison tests. The numbers in parentheses in each figure denote the number of experiments. In fluorometric experiments (Fig. 4), the numbers in the columns denote those of independently performed experiments, in each of which data from 10–20 cells were averaged (see symbols and bars in Fig. 3B–D).
Results
Specific CaMKII inhibition suppresses both functional availability and voltage-dependent gating of TRPC6 channels
We previously showed that activation of TRPC6 channels by receptor stimulation was facilitated by increased Ca2+ concentration ([Ca2+]i) and blocked by overexpression of a Ca2+-insensitive form of mutant calmodulin or intracellular application of relatively specific CaMKII inhibitors (Shi et al. 2004). This was interpreted to indicate that CaMKII-mediated phosphorylation is necessary to accelerate or prime the channel for opening. We first confirmed this point by testing the effect of a specific CaMKII inhibitor autocamtide-2-related inhibitory peptide (AIP). AIP is a non-phosphorylatable analogue of a highly selective substrate of CaMKII autocamtide-2, thus being known to specifically inhibit CaMKII (Ishida et al. 1995).
Inclusion of AIP (10 μm) in the patch pipette progressively attenuated the activation of inward current by 100 μm CCh (ICCh) in a TRPC6-expressing HEK293 cell (10 min vs. 1 min in Fig. 1A). Statistical comparison between two sets of TRPC6-expressing cells with and without AIP treatment indicates that 10 min intracellular perfusion of AIP significantly reduces the density of ICCh (Fig. 1B). Current–voltage (I–V) relationships for ICCh obtained at 1 and 10 min after AIP treatment in the same cell show that suppression of ICCh by CaMKII inhibition occurs over a wide range of membrane potentials (Fig. 1C). The chord conductance (g)–voltage (V) relationship for ICCh calculated from these I–Vs indicates that the degree of TRPC6 channel activation increases by depolarization except for a range near the reversal potential where outward ionic flow is hindered via an as yet unknown mechanism (open circles in Fig. 1D). CaMKII inhibition by AIP treatment (for 10 min) markedly suppressed this g–V relationship to a more pronounced extent at hyperpolarizing potentials (filled circles in Fig. 1D). Such voltage-dependent inhibitory effects of AIP can also be seen as a decreased ratio of g values at −100 to 100 mV (g−100/g100). Comparison between two sets of TRPC6-expressing cells with and without AIP treatment indicates that decrease in the g−100/g100 ratio is statistically significant for AIP treatment (Fig. 1E), suggesting that CaMKII inhibition alters the voltage dependency of TRPC6 channels.
Figure 1. Selective inhibitor of CaMKII AIP causes strong suppression of receptor-activated currents due to TRPC6 heterologously expressed in HEK293 cells.

Holding potential (VH): −50 mV. A, typical traces of ICCh induced by carbachol (CCh), which stimulated the muscarinic acetylcholine receptor inherently expressed in a HEK293 cell. Including AIP (10 μm) in the pipette solution caused gradually developing inhibition of ICCh. Upper and lower traces: ICCh was induced 1 and 10 min after the onset of whole-cell conditions, respectively. The series resistance was maintained below 15 MΩ during the whole recording process. B, summary of ICCh density with or without AIP treatment. Evaluated from separate sets of experiments performed in different cells. C, typical current–voltage (I–V) relationships of ICCh obtained from the same cell at 1 and 10 min after the onset of intracellular perfusion with AIP (10 μm; San Francisco, USA). D, relationships between chord conductance (g) and membrane potential (V) shows the voltage-dependent action of AIP. The values of g are calculated from the data in C. E, comparison of g ratios at −100 mV relative to 100 mV (g−100/g100) with and without AIP treatment. Data were obtained from separate sets of experiments performed in different cells. P values in the figure are the result of unpaired Student's t tests.
These observations together suggest that CaMKII positively regulates functional availability and affects the voltage-dependent gating of TRPC6 channels.
The distal part of CIRB contains a critical site for CaMKII-mediated regulation of TRPC6 channels
In the next step, in order to narrow down a region(s) responsible for CaMKII-mediated regulation of TRPC6 channels, we employed chimeric channel constructs (TRPC6/C7) in which C- and N-termini and the transmembrane (TM) region are variably exchanged (Fig. 2A). These chimeras show which part of the channel confers the CaMKII sensitivity, because mouse TRPC7 is insensitive to intracellular ATP depletion or overexpression of Ca2+-insensitive mutant CaM, thus being an unlikely subject for CaMKII-mediated regulation (Shi et al. 2004).
Among six TRPC6/C7 chimeras tested, only in those containing the entire TRPC6 C-terminal sequence (Ch776, Ch676, Ch766), was ICCh effectively inhibited by pretreatment with a CaM antagonist calmidazolium (CMZ; 3 μm) (Fig. 2B). This points to the functional significance of the C-terminal region in CaMKII-mediated regulation. To further confirm this, we next tested the effect of the specific CaMKII inhibitor AIP on the Ch776 chimera whose C-terminus contains solely the TRPC6 sequence. As expected, AIP treatment markedly inhibited ICCh in HEK cells expressing this chimeric channel (Fig. 2C).
The results of chimeric TRPC6/C7 channels are consistent with the idea that the TRPC6 C-terminal sequence is crucial for CaMKII-mediated regulation, but puzzling because all consensus CaMKII phosphorylation motifs (‘RXXS/T’; Soderling, 1996) are located in either N-terminal or TM regions but absent in the C-terminus of TRPC6 (Fig. 3A). However, it deserves to be noted that the C-termini of TRPC subfamily members have been proposed to possess a common competitive binding domain for CaM and IP3R (named ‘CIRB’; in the case of TRPC6, this corresponds to His843–Asp872; Tang et al. 2001; Fig. 2D); deletion of a region encompassing the whole of this domain (Glu840–Glu874) has been reported to abolish the Ca2+ response to phospholipase C (PLC)-coupled receptor stimulation by endothelin-1 (Horinouchi et al. 2011). Indeed, we could confirm the latter observation by patch clamp experiments. A TRPC6 mutant lacking a region containing this domain (ΔLys824–Glu877) was unable to generate discernible currents in response to CCh stimulation (second column in Fig. 2E). We further found that a distal part of the CIRB domain after Gln855 is more essential than its proximal part by using two other deletion mutants ΔGln855–Glu877 and ΔLys824–Tyr854 (two rightmost columns in Fig. 2E). Thus, even though the C-terminus of the TRPC6 channel is devoid of CaMKII phosphorylation sites, it is possible that the distal CIRB domain serves as an indispensable site for Ca2+/CaM-dependent and CaMKII-mediated regulation of TRPC6 channel activity, e.g. as a docking site for CaMKII. However, we did not pursue these complex findings further in the present study (but see the Discussion).
Thr487 is the most critical amino acid residue for CaMKII-mediated TRPC6 channel regulation
To pin down the phosphorylation site(s) responsible for TRPC6 channel regulation by CaMKII, we next performed site-directed scanning of all consensus motifs for CaMKII-mediated phosphorylation (Fig. 3A). To this end, we constructed eight mutants carrying an alanine (Ala)-substitution of the Ser/Thr residue in each motif (T9A, S14A, S28A, T69A, T193A, S321A, T401A, T487A), and evaluated their receptor responses by measuring the rate of Ba2+ fluorescence ratio increase (dRBa/dt) with Fura-2 fluorometry (Fig. 3B and C). The rationale behind using Ba2+ instead of Ca2+ was as follows. When Ca2+ was present in the bath, Ca2+ fluorescence changes during receptor stimulation were largely variable in magnitude and pattern, making statistical analysis difficult (data not shown). We reasoned that this would be due to active (and repeated) uptake and release of Ca2+ by internal organelles and vigorous extrusion to the outside, the extent of which may vary from cell to cell. In contrast, Ba2+ is almost free from these influences, and thus the value of dRBa/dt can be a more reliable measure of the magnitude of transmembrane divalent cation influx through mutant TRP channels. We also minimized the possible compromised activation of CaMKII by Ba2+ influx with the protocol shown in Fig. 3B; cells were first stimulated with CCh in the short absence of external Ca2+ which allowed intracellular Ca2+ release and activation of CaMKII and mutant TRP channels; then Ba2+ was added in the bath to measure the rate of initial Ba2+ fluorescence increase which should reflect the preceding phosphorylation state of mutant channels.
Figure 3C demonstrates typical time courses of CCh (100 μm)-induced Ba2+ influx (i.e. dRBa/dt) for wild-type and T487A-mutant TRPC6 channels. Amongst the eight mutants tested, the only T487A showed a markedly reduced Ba2+ influx in response to CCh as compared to wild-type TRPC6 (left nine columns in Fig. 4).
We previously reported that the Ch776 mutant exhibits a similar dependence on Ca2+/CaM to wild-type TRPC6 channels (Shi et al. 2004), and in the present study we demonstrated that this chimeric channel is subject to CaMKII-mediated regulation (Fig. 2C). This implies that the activation process of the Ch776 channel may involve a similar structural basis to wild-type TRPC6. The Ch776 channel has five consensus CaMKII phosphorylation motifs, all of which are located in N-terminal or TM regions (Supplemental Fig. 1B; Supplemental material is available online only). Among the Ser/Thr residues in these motifs, Thr433 is homologous to Thr487 of the wild-type TRPC6 channel, since both are predicted to be located between the putative 2nd and 3rd hydrophobic segments (i.e. H2–H3 intersegment) (Supplemental Fig. 1B vs. Fig. 3A). Strikingly, the magnitude of the CCh-induced Ba2+ influx represented by dRBa/dt was markedly reduced only in the Thr433 mutant (i.e. T433A), whereas no significant changes were observed in the other Ala-substituted mutants (T15A, S211A, S267A, S347A) (Fig. 3D and middle six columns in Fig. 4). In an interesting contrast, when the entire sequence was of TRPC7 (i.e. wild-type TRPC7), the functional impact of Ala substitution for Thr433 was totally lost (two rightmost columns in Fig. 4; Supplemental Fig. 1A). These results together suggest that both the Thr residue on the H2–H3 intersegment and the TRPC6 C-terminal sequence are critical for CaMKII-mediated regulation of wild-type TRPC6 and Ch776 chimeric channels.
We next tried to ensure the abrogating effect of the T487A mutation more directly by patch clamp experiments. As shown in Fig. 5A and B, only marginal ICCh was induced in HEK293 cells expressing the T487A mutant. Similar results were also observed with Thr487 mutations to two other non-polar amino acids, glutamine (T487Q) or asparagine (T487N) (two rightmost columns in Fig. 5B). On the other hand, phosphomimetic substitution of Thr487 with glutamate (T487E) or aspartate (T487D) resulted in less suppression of ICCh. In both T487A and T487E mutants, treatment with AIP (10 μm) did not further suppress ICCh (Fig. 5C), suggesting that these mutants are insensitive to CaMKII-mediated regulation. In addition, the phosphorylation site-dead mutation of Ch776 (T776–433A) which exhibited a markedly decreased ICCh was also unresponsive to AIP treatment (Supplemental Fig. 2B). Taken together, these results strongly support the view that Thr487 is a critical CaMKII-mediated phosphorylation site of the TRPC6 channel, and that essentially the same structural basis would exist for the Ch776 chimeric channel.
Figure 5. Mutation of Thr487 to neutral or permanently negatively charged amino acids abolishes ICCh.
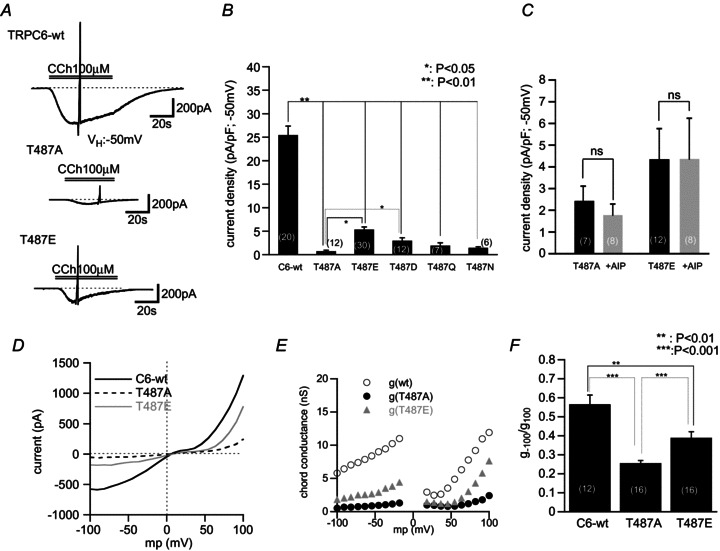
A, representative traces of ICCh for wt-, T487A- and T487E-expressing HEK293 cells. T487A mutation showed a very reduced development of ICCh, while T487E mutation resulted in an attenuated but more robust ICCh. Whole-cell recording at –50 mV. B, summary of ICCh density of various Thr487 mutants. C, application of AIP caused no discernible effects on ICCh in T487A and T487E. D, typical I–V curves of wild-type and mutant TRPC6 channels evaluated by slow ramp voltage application. E, g–voltage relationship calculated from D. Mutation of Thr487 to either alanine or glutamate showed more pronounced inhibition at more negatively clamped membrane potentials. F, comparison of g ratios (g−100/g100) of the three channels at −100 and +100 mV.
The negative impact of the Thr487 mutation on TRPC6 channel activation appears to occur through a similar mechanism to that with AIP treatment. In both T487A and T487E, albeit the latter to a lesser extent, the I–V relationship of ICCh becomes flattened (Fig. 5D), and its chord conductance g is greatly decreased over a wide range of membrane potentials (Fig. 5E). Further, the relative degree of voltage-dependent activation of ICCh at −100 mV (g−100/g100) is significantly smaller in Thr487 mutants than wild-type TRPC6 (Fig. 5F). All these changes are indistinguishable from those observed after CaMKII inhibition with AIP treatment (Fig. 1).
Thr487 mutations do not affect the cellular expression of the TRPC6 channel
Attenuated channel activation can result from impaired expression or trafficking of channel proteins. We tested this possibility by comparing the cellular distribution of immunoreactivity between wild-type and Thr487-mutant TRPC6 channels by confocal laser scanning microscopy. As demonstrated in Fig. 6A, the brightest immunofluorescence was seen near the cell membrane in both wild-type TRPC6 and Thr487-mutant (T487A, T487E)-expressing cells. Intensity plots of immunofluorescence across the cell (along the line i–ii) more clearly indicate that these channel proteins are most densely localized within ∼1 μm from the cell membrane and expressed at comparable levels (see the σ and A values, respectively; Fig. 6B and C). Consistent with these findings, protein expression levels in the plasma membrane fraction evaluated by immunoblot analysis were similar between wild-type and Thr487 mutants (T487A, T487E) (Fig. 6D). These results strongly suggest that functional changes (e.g. modified gating kinetics) rather than impaired expression/trafficking are responsible for markedly compromised activation of the mutant channels.
Figure 6. Immunocytochemical staining of wt-TRPC6 and Thr487 mutants expressed in HEK cells.
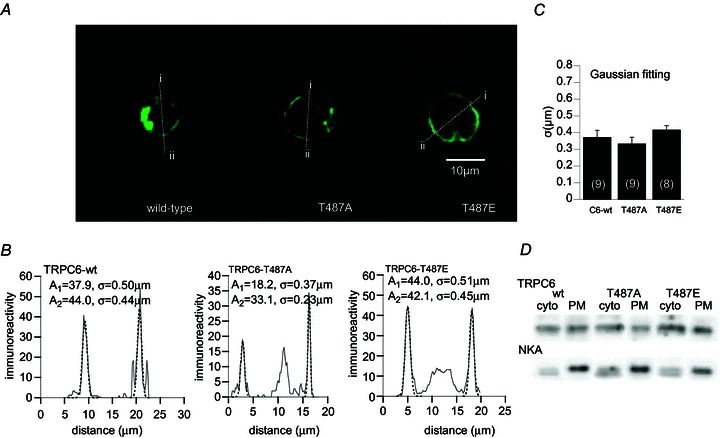
A, typical immunostained images of three heterologously expressed TRP channels (wild-type, T487A, T487E). Both mutants show a clear membrane-targeted expression profile similar to wt-TRPC6. B, distribution of immunofluorescence across the cell for the three TRP channels which was measured along the lines i–ii in A. Dotted curves are the results of the Gaussian fitting of each peak with the relationship: A× exp(–(x–m)2/(2σ2), where A, x, m and σ denote the peak intensity of immunofluorescence, distance (μm), position of the peak (μm), and standard deviation of the peak distribution (μm), respectively. C, comparison of immunofluorescence distribution pattern (σ values) among the three channels. D, immunoblots of wild-type (wt) and Thr487 mutant (T487A, T487E) TRPC6 proteins in the plasma membrane (PM) and cytosolic (cyto) fractions. Na+/K+-ATPase (NKA) was used as a PM marker. Representative of 3 experiments.
Thr487 probably locates on the first intracellular loop of the TRPC6 channel
A previous study proposed that the human TRPC6 channel possesses two extracellularly facing asparagine residues (Asn473, Asn561) susceptible to N-glycosylation which are essential for the tight regulation of the channel activity by PLC-coupled receptors (Dietrich et al. 2003). However, the predicted location of Asn473 by the Kite–Doolittle analysis is in the same H2–H3 intersegment as that of Thr488 (Thr487 in mouse) (Fig. 3A) which should intracellularly be located so as to be accessed by CaMKII. To solve this apparent contradiction, we evaluated the membrane sidedness of Thr487 by constructing a Myc-tagged TRPC6 channel together with its functionality. The Myc sequence ‘EQKLISEEDL’ was inserted between Ser475 and Thr476 in the putative H2–H3 intersegmental domain of the TRPC6 protein (Figs 3A and 7Aa), and its immunoreactivity was evaluated by anti-Myc antibody before and after permeabilization of the cell membrane.
HEK cells expressing Myc-tagged TRPC6 protein generated comparably large ICCh to wild-type TRPC6 (Fig. 7Ba and c). The I–V relationship of the Myc-tagged TRPC6 channel shows the hallmark of the wild-type TRPC6 channel, i.e. ‘S’-shaped inward suppression and outward rectification (Fig. 7Bb). Further, inhibition of CaMKII by intracellular perfusion of AIP (10 μm) strongly suppressed the generation of ICCh due to Myc-tagged TRPC6 (Supplemental Fig. 3). These results strongly suggest that the functional properties of this engineered TRPC6 channel remain unchanged by the insertion of the Myc tag.
The anti-Myc immunoreactivity was virtually not detected in non-permeabilized cells even 48 h after the transfection of trpc6 DNA (PBS; Fig. 7Ab and C). The observed very low fraction of positive staining under these conditions is probably due to cell damage caused by the procedures of cell dispersion, plating or fixation. After permeabilization with Triton X-100, however, about 15% of cells were clearly stained by anti-Myc antibody (Fig. 7Ab (second panels) and C (right column)). This percentage of positive staining was similar to that of successfully transfected cells under random selection without the expression marker CD8. The adequacy of permeabilization and robust expression of TRPC6 protein were confirmed by simultaneous double staining with anti-TRPC6 antibody targeting an intracellular N-terminal sequence. This antibody could also stain the cells only after treatment with Triton X-100 (first panels in Fig. 7Ab), and the coincidence of positive immunostaining with anti-Myc and anti-TRPC6 antibodies was 100% (in 165 out of 165 cells tested). In addition, the merged images and intensity plots of immunofluorescence indicate that anti-Myc and anti-TRPC6 immunoreactivities are both restricted near the cell membrane with almost perfectly overlapping distributions (Fig. 7Ab (rightmost panels) and D). In aggregate, these results strongly support the view that the region harbouring the Thr487 residue faces the cytosolic space and is thus accessible by CaMKII.
Physiological implications of Thr487 phosphorylation by CaMKII
Finally, to explore the physiological significance of Thr487 phosphorylation in a more native situation, we transfected the T487A mutant into an A7r5 myocyte, which comes from an established vascular smooth muscle cell line. As has been reported previously (Takahashi et al. 2008; Inoue et al. 2009), these cells responded to Arg8-vasopressin (AVP) by generating cationic currents with features reminiscent of the TRPC6 channel, such as potentiation by flufenamate (upper trace in the left panel; Fig. 8A). The density of AVP-induced cationic current was significantly decreased by 10 min intracellular perfusion of AIP (Supplemental Fig. 4) and markedly reduced after transfection of the T487A mutant as compared with the empty vector (Fig. 8B). These results support the idea that CaMKII-mediated Thr487 phosphorylation positively regulates TRPC6-like channel activity induced by AVP receptor stimulation.
Figure 8. Overexpression of TRPC6 T487A mutant in a smooth muscle cell line, A7r5, greatly inhibited endogenous AVP-induced TRPC6-like currents.
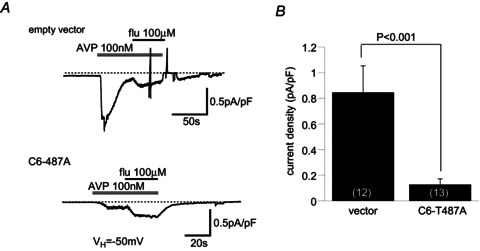
A, typical trace of whole-cell current induced by AVP (100 nm) in an empty vector-transfected A7r5 cell. Application of flufenamate (flu) caused clear potentiation of the current, which is the hallmark feature of a TRPC6 channel. B, overexpression of T487A into A7r5 cells significantly inhibited AVP-induced current, which still exhibited flu sensitivity. C, comparison of AVP-induced current density between empty vector and T487A mutant transfection.
Discussion
The aim of the present study was to define the molecular basis for CaMKII-mediated positive regulation of TRPC6 channel activity. For this purpose, we employed three approaches, chimera, deletion and site-directed analyses. The results of chimera experiments unexpectedly revealed that the C-terminus, which entirely lacks CaMKII phosphorylation motifs, is essential for channel activation. Further, experiments with deletion mutants of CIRB, reportedly a common binding domain for CaM and IP3R (Tang et al. 2001), showed that its distal region (Gln855–Glu877) is crucial for receptor-mediated activation of the TRPC6 channel. The functional importance of the distal CIRB region in Ca2+-dependent TRPC6 channel activation is in good agreement with the fact that the Ca2+ dependence of CaM binding to a peptide fragment corresponding to this region (Arg852–Lys874) in living cells (Mori et al. 2011) faithfully reproduces the [Ca2+]–activity relationship of receptor-activated TRPC6 channels (Shi et al. 2004). In contrast, site-directed mutagenesis analysis based on consensus CaMKII phosphorylation motifs indicated that a single threonine residue located on the TM region (Thr487) would be sufficient for CaMKII-mediated regulation. The threonine residue homologous to Thr487 is also present in the T776 chimeric channel (Thr433). We found that this channel is also susceptible to CaMKII inhibition by AIP, whereas wild-type TRPC7 is not, despite the fact that they share a common sequence except for the C-terminus. The most consistent interpretation to account for these results is that, even though spatially separated, the C-terminal domain and Thr487 should have a tight functional collaboration in regulating the gating of the TRPC6 channel via CaMKII activation (for more discussion, see below).
It remains entirely unclear how Ca2+/CaM binding is converted into subsequent activation of CaMKII thereby modulating TRPC6 channel activity. Recent investigation of L-type voltage-dependent Ca2+ channels suggests that neuronal CaMKII is constitutively tethered to a long II–III linker of the CaV1.2 α1-subunit via the β1 or β2 subunit. This structural arrangement allows enhanced phosphorylation of both subunits to facilitate the Ca2+ channel activity in response to Ca2+ influx during repetitive membrane excitations (Abiria & Colbran, 2010). In another mode of activity-dependent CaV1.2 facilitation, the autophosphorylated form of CaMKII docks to a C-terminal domain of the α1-subunit to enhance the channel activity independently of Ca2+/CaM binding (Hudmon et al. 2005). Similar positive feedback regulations through Ca2+ influx-dependent CaMKII activation have also been documented for other types of voltage-dependent calcium channels (Pitt, 2007). In neuronal NMDA receptor channels, Ca2+ influx through the channels promotes the recruitment of CaMKIIα to a C-terminal region of the NR2B subunit comprising the channel, thereby enhancing its activity through phosphorylation. This mechanism serves as an effective positive feedback to potentiate the NMDA receptor channels, preferentially at synapses experiencing high-frequency neuronal activity capable of inducing long-term potentiation (Bayer et al. 2001; Merrill et al. 2005).
The above-mentioned examples demonstrate the cell activity-dependent activation of CaMKII regulating Ca2+-permeable cation channels by their own Ca2+ influxes. This is strategically advantageous to enhance the ensuing functions specifically at the site of the Ca2+ influx. In the case of TRPC6 channels, the presence of external Ca2+ facilitates the activation of the channel by PLC-coupled receptor stimulation. Thus, a similar activity-dependent mechanism appears to operate therein. However, there is a different line of evidence that static [Ca2+]i levels also affect the subsequent receptor-mediated activation of TRPC6 channels via CaMKII (Shi et al. 2004). In this situation, a biphasic Ca2+ dependence of the channel activation with a maximum near the resting [Ca2+]i (100–200 nm) was observed upon subsequent receptor stimulation, which was prevented by specific inhibition of CaMKII with AIP (Shi et al. 2004; Fig. 1). An analogous role of CaMKII-mediated regulation has been found for capsaicin-induced activation of the TRPV1 channel, where a serine residue (Ser502) located on the first intracellular loop as well as a C-terminal threonine residue (Thr704) are already phosphorylated prior to receptor stimulation (Jung et al. 2004). Phosphorylation of these residues by CaMKII (Ser502 is a common phosphorylation site for CaMKII, and protein kinases A and C) sensitizes the channel for subsequent activation by capsaicin through enhancement of its binding to the channel. Conversely, after the binding of capsaicin, Ca2+ influx occurring through the activated channel per se activates the calcineurin activity via binding of Ca2+/CaM, which in turn dephosphorylates these residues resulting in decreased capsaicin binding and desensitization. Thus, dynamic equilibrium between CaMKII-mediated sensitization and calcineurin-mediated desensitization elaborately controls the TRPV1 channel activity in a Ca2+/CaM-dependent manner. However, it remains unknown whether the sensitizing action of CaMKII at resting [Ca2+]i requires its constitutive binding to the channel protein.
The molecular model for CaMKII–channel interaction has been proposed for the CaV1.2 channel. In this model, the C-terminal domains of both α1C- and β2a-subunits, which act as a molecular mimickry of the CaMKII autoinhibitory (AI) region, replace the AI regions of multimeric CaMKII facilitating the formation of the channel–kinase complex (Pitt, 2007). Similar AI-like sequences are also found by genome-wide screening in a variety of proteins involved in cellular signalling and dynamics including ion channels (Hund et al. 2010). The C-terminal region containing the distal CIRB (Ile857–Gln888) of TRPC6 shows some degree of sequence similarity to the AI-like region of the CaV1.2 α1-subunit (Thr1644–Gln1675) (Supplemental Fig. 6). Thus, it is conceivable that this region might serve as an important site for the interaction between CaMKII and the TRPC6 channel. Further studies based on detailed structural analyses are awaited to answer these questions properly.
Site-directed substitution of Ser/Thr residues with non-phosphorylatable amino acids (alanine, glutamine or asparagine) suggested that a single Thr487 residue is critical for CaMKII-mediated regulation of TRPC6 channels. The rationale behind this conclusion is: firstly, Ala-mutation of Thr487 remarkably reduced the degree of TRPC6 channel activation by receptor stimulation, whereas this was partially rescued by phosphomimetic substitution of Thr487 with glutamate or aspartate; secondly, attenuated TRPC6 channel activation by loss-of-function mutations of Thr487 was not further inhibited by a specific CaMKII inhibitor AIP; finally, the observed changes in functional availability and voltage-dependent gating of TRPC6 channels were indistinguishable between AIP-mediated CaMKII inhibition and loss-of-function mutations of Thr487. However, it should cautiously be considered that the present mutagenesis scanning restricted to the ‘RXX(S/T)’ motif may have missed additional critical residues involved in CaMKII-mediated regulation of TRPC6 channels. In fact, a previous study examining the substrate specificity of several serine/threonine kinases with a degenerate peptide library suggests that potential substrates for CaMKII may be much broader than those predicted from ‘RXX(S/T)’; e.g. Lys (instead of Arg) could be another preferred amino acid residue at the 3-position of the CaMKII substrate sequence (Songyang et al. 1996). Thus, although the critical significance of Thr487 is undoubted, a more extensive survey will be further required to comprehensively understand the molecular basis of CaMKII-mediated regulation of TRPC6 channels.
The location of Thr487 is predicted to be on the same H2–H3 intersegment as that of Asn472 which, in human TRPC6, has been assigned to an extracellular N-glycosylation site (Dietrich et al. 2003). However, the present experiments using Myc-tagged TRPC6 channels rather support the view that Thr487 resides on the first intracellular loop of the TRPC6 channel. The same membrane topology appears to be true for the Ch776 chimeric channel, in which the Ala-mutation of a homologous threonine residue (Thr433) caused almost total loss of responsiveness to receptor stimulation. Predicted membrane topology of TRPC members by the Kite–Doolittle algorithm suggests that the H3 segment, which is just distal to Thr487 or Thr433 in TRPC6 and Ch776 channels, respectively, consists of a rather short stretch of hydrophobic amino acids with a low transmembrane score compared to H1 or H2 segments (Fig. 3A and Supplemental Fig. 1B; e.g. see also the initial study of Zhu et al. 1996). In good agreement with this prediction, systematic analysis of the TRPC1 channel's membrane topology assigns H1, H2, H4–H6 and H8 to the 1st to 6th TM segments (S1–S6), respectively, H3 to a part of the intracellular loop, and H7 to a pore loop (Dohke et al. 2004). Provided that this membrane topology applies for the TRPC6 channel, its first intracellular loop containing Thr487 should be as long as ∼65 amino acids (AAs), being far longer than the other intracellular loops comprising ∼20 AAs (Supplemental Fig. 5). On the other hand, the location of the CIRB domain on the TRPC6 C-terminus is 127 AAs apart from the C-terminal end of the last TM segment (S6), which is 19 AAs longer than that of TRPC7 (108 AAs) (Fig. 3A vs. Supplemental Fig. 1A). This may enable the TRPC6 CIRB domain to reach more distant targets than that of TRPC7, presumably explaining why TRPC7 is not, and Ch776 is, susceptible to CaMKII-mediated regulation. In other words, the above two separately located regions, i.e. the target site and interacting domain for CaMKII's action might be optimally placed in the TRPC6 channel, but it would not be so in the TRPC7 channel. In this regard, it is notable that a homologous site (Ser502) to Thr487 of TRPC6, which is located on the first intracellular loop of the TRPV1 channel, is also susceptible to CaMKII-mediated phosphorylation that sensitizes the channel for activation by capsaicin (Jung et al. 2004).
In summary, the present results indicate that the two spatially separated sites of the TRPC6 channel, i.e. the distal domain of CIRB on the C-terminus and Thr487 located on the putative first intracellular loop, are crucial for CaMKII-mediated positive regulation of TRPC6 channels. This regulation probably occurs in both homeostatic and activity-dependent manners at basal and through an induced rise in [Ca2+]i via receptor-activated Ca2+ influx, respectively. The former may sensitize (or prime) the channel for subsequent receptor stimulation, while the latter further accelerates the channel activation. In the cardiovascular system in which the TRPC6 channel is abundantly expressed, this mechanism may serve as an effective positive feedback regulation coupled with Ca2+ mobilization from the internal store as well as across the cell membrane in response to PLC-linked receptor stimulation by neurohormonal factors such as sympathetically released noradrenaline and vasoconstrictive hormones (e.g. angiotensin II, endothelin, vasopressin). Such self-stimulatory regulations of Ca2+ influx is a common strategy for Ca2+-permeable channels to fine-tune a variety of cardiovascular functions including facilitating effects on cardiac excitation–contraction coupling, vascular tone regulation, and endothelial nitric oxide production. Its dysregulation may lead to pathophysiological states such as arrhythmia, hypertension and atherosclerosis (Couchonnal & Anderson, 2008; Ching et al. 2011).
Acknowledgments
This work was supported mainly by a grant-in-aid to R.I. from Scientific Research (C) (No. 17590221), a postdoctoral fellowship to J.S. from the Japan Society for Promotion of Sciences (P09134) and from an overseas funding granted to J.S. (National Natural Science Foundation of China: No. 30871004). Part of the work was also supported by Grants-in-aid to R.I. for Scientific Research (C) (No. 21590246) and from Scientific Research on Innovative Areas (No. 22136008), and grants to R.I. from Seizon Kagaku Institute, Tokyo Biochemical Research Foundation, and Vehicle Racing Commemorative Foundation.
Glossary
- AIP
autocamtide-2-related inhibitory peptide
- AVP
Arg8-vasopressin
- CaM
Ca2+/calmodulin
- CaMKII
Ca2+/CaM-dependent kinase II
- CCh
carbachol
- CIRB
CaM/IP3R binding domain
- IP3R
inositol-1,4,5-trisphosphate receptor
- PLC
phospholipase C
- PM
plasma membrane
- TM
transmembrane
- TRPC6
transient receptor potential channel 6
Author contributions
All experiments were performed in the corresponding author's labs at the Department of Physiology, Graduate School of Medical Sciences, Kyushu University, and Department of Physiology, Graduate School of Medical Sciences, Fukuoka University. R.I. was responsible for conception and design of the experiments. J.S., N.G., S.T., J.I., Y.H. and R.I. collected, analysed and interpreted data. N.G., S.K., Y.M. and R.I. constructed chimeras, deletion and site-directed mutants. J.S. and R.I. drafted the paper, and S.K., Y.I. and Y.M. revised it critically for important intellectual content. All authors approved the final version of the manuscript.
Authors’ present addresses
J. Shi: Department of Anatomy and K.K. Leung Brain Research Center, the Fourth Military Medical University, Xi’an 710032, China.
N. Geshi: Department of Plant and Environmental Sciences, University of Copenhagen, DK-1871 Frederiksberg C, Denmark.
Y. Ito: Kumamoto Health Science University, Izumi-machi 325, Kita-ku, Kumamoto 861-5598, Japan.
R. Inoue: Department of Physiology, Graduate School of Medical Sciences, Fukuoka University, Nanakuma 7-45-1, Johnan-ku, Fukuoka 814-0180, Japan.
References
- Abiria SA, Colbran RJ. CaMKII associates with CaV1.2 L-type calcium channels via selected β subunits to enhance regulatory phosphorylation. J Neurochem. 2010;112:150–161. doi: 10.1111/j.1471-4159.2009.06436.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ai X, Curran JW, Shannon TR, Bers DM, Pogwizd SM. Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ Res. 2005;97:1314–1322. doi: 10.1161/01.RES.0000194329.41863.89. [DOI] [PubMed] [Google Scholar]
- Aiba T, Hesketh GG, Liu T, Carlisle R, Villa-Abrille MC, O’Rourke B, Akar FG, Tomaselli GF. Na+ channel regulation by Ca2+/calmodulin and Ca2+/calmodulin-dependent protein kinase II in guinea-pig ventricular myocytes. Cardiovasc Res. 2010;85:454–463. doi: 10.1093/cvr/cvp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banumathi E, O’Connor A, Gurunathan S, Simpson DA, McGeown JG, Curtis TM. VEGF-induced retinal angiogenic signaling is critically dependent on Ca2+ signaling by Ca2+/calmodulin-dependent protein kinase II. Invest Ophthalmol Vis Sci. 2011;52:3103–3111. doi: 10.1167/iovs.10-6574. [DOI] [PubMed] [Google Scholar]
- Bare DJ, Kettlun CS, Liang M, Bers DM, Mignery GA. Cardiac type 2 inositol 1,4,5-trisphosphate receptor: interaction and modulation by calcium/calmodulin-dependent protein kinase II. J Biol Chem. 2005;280:15912–15920. doi: 10.1074/jbc.M414212200. [DOI] [PubMed] [Google Scholar]
- Bayer KU, De Koninck P, Leonard AS, Hell JW, Schulman H. Interaction with the NMDA receptor locks CaMKII in an active conformation. Nature. 2001;411:801–805. doi: 10.1038/35081080. [DOI] [PubMed] [Google Scholar]
- Bers DM, Grandi E. Calcium/calmodulin-dependent kinase II regulation of cardiac ion channels. J Cardiovasc Pharmacol. 2009;54:180–187. doi: 10.1097/FJC.0b013e3181a25078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaich A, Welling A, Fischer S, Wegener JW, Köstner K, Hofmann F, Moosmang S. Facilitation of murine cardiac L-type CaV1.2 channel is modulated by calmodulin kinase II-dependent phosphorylation of S1512 and S1570. Proc Natl Acad Sci U S A. 2010;107:10285–10289. doi: 10.1073/pnas.0914287107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chelu MG, Sarma S, Sood S, Wang S, van Oort RJ, Skapura DG, Li N, Santonastasi M, Müller FU, Schmitz W, Schotten U, Anderson ME, Valderrábano M, Dobrev D, Wehrens XH. Calmodulin kinase II-mediated sarcoplasmic reticulum Ca2+ leak promotes atrial fibrillation in mice. J Clin Invest. 2009;119:1940–1951. doi: 10.1172/JCI37059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ching LC, Kou YR, Shyue SK, Su KH, Wei J, Cheng LC, Yu YB, Pan CC, Lee TS. Molecular mechanisms of activation of endothelial nitric oxide synthase mediated by transient receptor potential vanilloid type 1. Cardiovasc Res. 2011;91:492–501. doi: 10.1093/cvr/cvr104. [DOI] [PubMed] [Google Scholar]
- Couchonnal LF, Anderson ME. The role of calmodulin kinase II in myocardial physiology and disease. Physiology (Bethesda) 2008;23:151–159. doi: 10.1152/physiol.00043.2007. [DOI] [PubMed] [Google Scholar]
- Dietrich A, Mederos y Schnitzler M, Emmel J, Kalwa H, Hofmann T, Gudermann T. N-linked protein glycosylation is a major determinant for basal TRPC3 and TRPC6 channel activity. J Biol Chem. 2003;278:47842–47852. doi: 10.1074/jbc.M302983200. [DOI] [PubMed] [Google Scholar]
- Dohke Y, Oh YS, Ambudkar IS, Turner RJ. Biogenesis and topology of the transient receptor potential Ca2+ channel TRPC1. J Biol Chem. 2004;279:12242–12248. doi: 10.1074/jbc.M312456200. [DOI] [PubMed] [Google Scholar]
- Horinouchi T, Terada K, Higa T, Aoyagi H, Nishiya T, Suzuki H, Miwa S. Function and regulation of endothelin type A receptor-operated transient receptor potential canonical channels. J Pharmacol Sci. 2011;117:295–306. doi: 10.1254/jphs.11162fp. [DOI] [PubMed] [Google Scholar]
- Hudmon A, Schulman H. Neuronal Ca2+/calmodulin-dependent protein kinase II: the role of structure and autoregulation in cellular function. Annu Rev Biochem. 2002;71:473–510. doi: 10.1146/annurev.biochem.71.110601.135410. [DOI] [PubMed] [Google Scholar]
- Hudmon A, Schulman H, Kim J, Maltez JM, Tsien RW, Pitt GS. CaMKII tethers to L-type Ca2+ channels, establishing a local and dedicated integrator of Ca2+ signals for facilitation. J Cell Biol. 2005;171:537–547. doi: 10.1083/jcb.200505155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hund TJ, Koval OM, Li J, Wright PJ, Qian L, Snyder JS, Gudmundsson H, Kline CF, Davidson NP, Cardona N, Rasband MN, Anderson ME, Mohler PJ. A βIV-spectrin/CaMKII signaling complex is essential for membrane excitability in mice. J Clin Invest. 2010;120:3508–3519. doi: 10.1172/JCI43621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue R, Jensen LJ, Jian Z, Shi J, Hai L, Lurie AI, Henriksen FH, Salomonsson M, Morita H, Kawarabayashi Y, Mori M, Mori Y, Ito Y. Synergistic activation of vascular TRPC6 channel by receptor and mechanical stimulation via phospholipase C/diacylglycerol and phospholipase A2/ω-hydroxylase/20-HETE pathways. Circ Res. 2009;104:1399–1409. doi: 10.1161/CIRCRESAHA.108.193227. [DOI] [PubMed] [Google Scholar]
- Ishida A, Kameshita I, Okuno S, Kitani T, Fujisawa H. A novel highly specific and potent inhibitor of calmodulin-dependent protein kinase II. Biochem Biophys Res Comm. 1995;212:806–812. doi: 10.1006/bbrc.1995.2040. [DOI] [PubMed] [Google Scholar]
- Jung J, Shin JS, Lee SY, Hwang SW, Koo J, Cho H, Oh U. Phosphorylation of vanilloid receptor 1 by Ca2+/calmodulin-dependent kinase II regulates its vanilloid binding. J Biol Chem. 2004;279:7048–7054. doi: 10.1074/jbc.M311448200. [DOI] [PubMed] [Google Scholar]
- Kim MT, Park WJ, Kim S, Lee JW, Lee SY, Jeon JH, So I, Kim BJ, Kim SJ. Involvement of calmodulin kinase II in the action of sulphur mustard on the contraction of vascular smooth muscle. Basic Clin Pharmacol Toxicol. 2011;108:28–33. doi: 10.1111/j.1742-7843.2010.00623.x. [DOI] [PubMed] [Google Scholar]
- Li W, Li H, Sanders PN, Mohler PJ, Backs J, Olson EN, Anderson ME, Grumbach IM. The multifunctional Ca2+/calmodulin-dependent kinase II δ (CaMKIIδ) controls neointima formation after carotid ligation and vascular smooth muscle cell proliferation through cell cycle regulation by p21. J Biol Chem. 2011;286:7990–7999. doi: 10.1074/jbc.M110.163006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrill MA, Chen Y, Strack S, Hell JW. Activity-driven postsynaptic translocation of CaMKII. Trends Pharmacol Sci. 2005;26:645–653. doi: 10.1016/j.tips.2005.10.003. [DOI] [PubMed] [Google Scholar]
- Mori MX, Imai Y, Itsuki K, Inoue R. Quantitative measurement of Ca2+-dependent calmodulin-target binding by Fura-2 and CFP and YFP FRET imaging in living cells. Biochemistry. 2011;50:4685–4696. doi: 10.1021/bi200287x. [DOI] [PubMed] [Google Scholar]
- Nishioka K, Nishida M, Ariyoshi M, Jian Z, Saiki S, Hirano M, Nakaya M, Sato Y, Kita S, Iwamoto T, Hirano K, Inoue R, Kurose H. Cilostazol suppresses angiotensin II-induced vasoconstriction via protein kinase A-mediated phosphorylation of the transient receptor potential canonical 6 channel. Arterioscler Thromb Vasc Biol. 2011;31:2278–2286. doi: 10.1161/ATVBAHA.110.221010. [DOI] [PubMed] [Google Scholar]
- Pitt GS. Calmodulin and CaMKII as molecular switches for cardiac ion channels. Cardiovasc Res. 2007;73:641–647. doi: 10.1016/j.cardiores.2006.10.019. [DOI] [PubMed] [Google Scholar]
- Schulman H. Activity-dependent regulation of calcium/calmodulin-dependent protein kinase II localization. J Neurosci. 2004;24:8399–8403. doi: 10.1523/JNEUROSCI.3606-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott JA, Xie L, Li H, Li W, He JB, Sanders PN, Carter AB, Backs J, Anderson ME, Grumbach IM. The multifunctional Ca2+/calmodulin-dependent kinase II regulates vascular smooth muscle migration through matrix metalloproteinase 9. Am J Physiol Heart Circ Physiol. 2012;302:H1953–H1964. doi: 10.1152/ajpheart.00978.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen B, Kwan HY, Ma X, Wong CO, Du J, Huang Y, Yao X. cAMP activates TRPC6 channels via the phosphatidylinositol 3-kinase (PI3K)-protein kinase B (PKB)-mitogen-activated protein kinase kinase (MEK)-ERK1/2 signaling pathway. J Biol Chem. 2011;286:19439–19445. doi: 10.1074/jbc.M110.210294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Mori E, Mori Y, Mori M, Li J, Ito Y, Inoue R. Multiple regulation by calcium of murine homologues of transient receptor potential proteins TRPC6 and TRPC7 expressed in HEK293 cells. J Physiol. 2004;561:415–432. doi: 10.1113/jphysiol.2004.075051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer HA. Ca2+/calmodulin-dependent protein kinase II function in vascular remodelling. J Physiol. 2012;590:1349–1356. doi: 10.1113/jphysiol.2011.222232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soderling TR. Structure and regulation of calcium/calmodulin-dependent protein kinases II and IV. Biochim Biophys Acta. 1996;1297:131–138. doi: 10.1016/s0167-4838(96)00105-7. [DOI] [PubMed] [Google Scholar]
- Songyang Z, Lu KP, Kwon YT, Tsai LH, Filhol O, Cochet C, Brickey DA, Soderling TR, Bartleson C, Graves DJ, DeMaggio AJ, Hoekstra MF, Blenis J, Hunter T, Cantley LC. A structural basis for substrate specificities of protein Ser/Thr kinases: primary sequence preference of casein kinases I and II, NIMA, phosphorylase kinase, calmodulin-dependent kinase II, CDK5, and Erk1. Mol Cell Biol. 1996;16:6486–6493. doi: 10.1128/mcb.16.11.6486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi S, Lin H, Geshi N, Mori Y, Kawarabayashi Y, Takami N, Mori MX, Honda A, Inoue R. Nitric oxide-cGMP-protein kinase G pathway negatively regulates vascular transient receptor potential channel TRPC6. J Physiol. 2008;586:4209–4223. doi: 10.1113/jphysiol.2008.156083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J, Lin Y, Zhang Z, Tikunova S, Birnbaumer L, Zhu MX. Identification of common binding sites for calmodulin and inositol 1,4,5-trisphosphate receptors on the carboxyl termini of Trp channels. J Biol Chem. 2001;276:21303–21310. doi: 10.1074/jbc.M102316200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang WY, Hao LY, Minobe E, Saud ZA, Han DY, Kameyama M. CaMKII phosphorylates a threonine residue in the C-terminal tail of Cav1.2 Ca2+ channel and modulates the interaction of the channel with calmodulin. J Physiol Sci. 2009;59:283–290. doi: 10.1007/s12576-009-0033-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsby PJ, Wang H, Wolfe JT, Colbran RJ, Johnson ML, Barrett PQ. A mechanism for the direct regulation of T-type calcium channels by Ca2+/calmodulin-dependent kinase II. J Neurosci. 2003;23:10116–10121. doi: 10.1523/JNEUROSCI.23-31-10116.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J, Davies LA, Howard JD, Adney SK, Welsby PJ, Howell N, Carey RM, Colbran RJ, Barrett PQ. Molecular basis for the modulation of native T-type Ca2+ channels in vivo by Ca2+/calmodulin-dependent protein kinase II. J Clin Invest. 2006;116:2403–2412. doi: 10.1172/JCI27918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao X, Kwan HY, Huang Y. Regulation of TRP channels by phosphorylation. Neurosignals. 2005;14:273–280. doi: 10.1159/000093042. [DOI] [PubMed] [Google Scholar]
- Yousif MH, Akhtar S, Walther T, Benter IF. Role of Ca2+/calmodulin-dependent protein kinase II in development of vascular dysfunction in diabetic rats with hypertension. Cell Biochem Funct. 2008;26:256–263. doi: 10.1002/cbf.1446. [DOI] [PubMed] [Google Scholar]
- Zhang T, Brown JH. Role of Ca2+/calmodulin-dependent protein kinase II in cardiac hypertrophy and heart failure. Cardiovasc Res. 2004;63:476–486. doi: 10.1016/j.cardiores.2004.04.026. [DOI] [PubMed] [Google Scholar]
- Zhu X, Jiang M, Peyton M, Boulay G, Hurst R, Stefani E, Birnbaumer L. trp, a novel mammalian gene family essential for agonist-activated capacitative Ca2+ entry. Cell. 1996;85:661–671. doi: 10.1016/s0092-8674(00)81233-7. [DOI] [PubMed] [Google Scholar]