

Abstract
The abundant existence of proteins and regions that possess specific functions without being uniquely folded into unique 3D structures has become accepted by a significant number of protein scientists. Sequences of these intrinsically disordered proteins (IDPs) and IDP regions (IDPRs) are characterized by a number of specific features, such as low overall hydrophobicity and high net charge which makes these proteins predictable. IDPs/IDPRs possess large hydrodynamic volumes, low contents of ordered secondary structure, and are characterized by high structural heterogeneity. They are very flexible, but some may undergo disorder to order transitions in the presence of natural ligands. The degree of these structural rearrangements varies over a very wide range. IDPs/IDPRs are tightly controlled under the normal conditions and have numerous specific functions that complement functions of ordered proteins and domains. When lacking proper control, they have multiple roles in pathogenesis of various human diseases. Gaining structural and functional information about these proteins is a challenge, since they do not typically “freeze” while their “pictures are taken.” However, despite or perhaps because of the experimental challenges, these fuzzy objects with fuzzy structures and fuzzy functions are among the most interesting targets for modern protein research. This review briefly summarizes some of the recent advances in this exciting field and considers some of the basic lessons learned from the analysis of physics, chemistry, and biology of IDPs.
Keywords: intrinsically disordered protein, intrinsically disordered protein region, protein structure, protein function, protein evolution, protein–protein interaction, partially folded protein
Introduction
A bit more than ten years ago, Protein Science published a review entitled “Natively unfolded proteins: a point where biology waits for physics” (Protein Sci 2002 11(4):739–756).1 The major goal of that article was to bring an intriguing protein family of natively unfolded proteins (which are recognized now to constitute a subset of a very broad class of intrinsically disordered proteins, IDPs) out of shadow, to emphasize their lack of ordered structure under physiological conditions (at least ordered structure that could be detected by traditional low resolution techniques), to systemize their major structural properties, and to highlight their biological significance. The introduction of such biologically active but essentially unstructured proteins was used to challenge the hitherto dominant structure-centric viewpoint (structure–function paradigm), according to which a specific function of a protein is determined by its unique and rigid three-dimensional (3D) structure. The title of the review (“a point where biology waits for physics”) was inspired by the observations that many of such “structure-less” proteins analyzed by that time acted as “binders” that did undergo at least partial folding after interaction with their binding partners. These observations provoked an idea that these biologically important proteins with little or no ordered structure have to wait to become more folded (and functional) as a result of binding to their specific partners. In other words, for these proteins, “biology,” that is, the ability to have biological functions, seemed to wait for “physics” which is manifested in their ability to undergo binding-induced folding (at least partial), which is necessary to bring the functional state of these proteins to life.1
At the beginning, the idea that structure-less proteins can be biologically active was taken as a complete heresy by many researchers, since it was absolutely alien to then dominated structure–function paradigm which represented a foundation of the long-standing belief that the specific functionality of a given protein is determined by its unique 3-D structure. This structure–function paradigm that describes reasonably well the catalytic behavior of enzymes was based on the “lock-and-key” hypothesis formulated in 1894 by Emil Fischer.2 This viewpoint was solidified by the successful solution of X-ray crystallographic structures of many proteins (as of February 26, 2013 there were 81,922 protein structures in the Protein Data Bank,3 with 72,761 of these structures being determined by X-ray crystallography). These many crystal structures reinforced a static view of functional protein, where a rigid active site of an enzyme can be viewed as a sturdy lock that provides an exact fit to only one key, a specific and unique substrate.4 Despite numerous limitations, this lock-and-key model was an extremely fruitful concept that was responsible for the creation of modern protein science.1 Figure 1(A) shows some of the most obvious scientific consequences of the application of structure–function paradigm which is deservedly placed at the center of the “Big Bang” model that gives rise to the protein science universe.1
Figure 1.
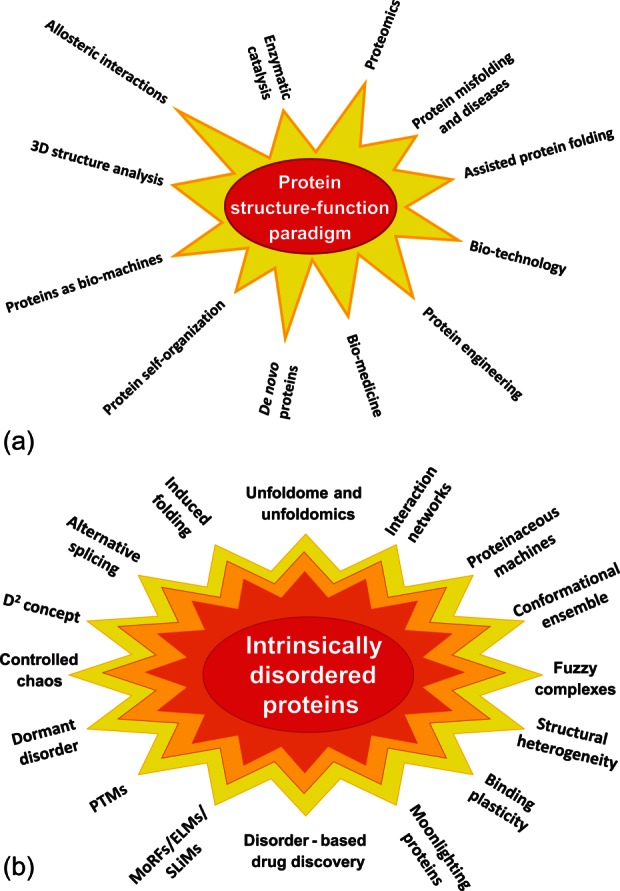
A: Protein structure–function paradigm is the “Big Bang” created universe of the modern protein science. Some major directions based on the consideration of protein function as lock-and-key mechanism are shown. Modified from Ref.1. B: Paradigm shift caused by the introduction of the protein intrinsic disorder concept opened a wide array of new directions in protein science. In essence, introduction of this concept can be considered as a scientific revolution that, according to Kuhn,5 “occurs when scientists encounter anomalies that cannot be explained by the universally accepted paradigm within which scientific progress has thereto been made” (http://en.wikipedia.org/wiki/Paradigm_shift).
Obviously, the consideration of a protein as a rigid crystal-like entity is an oversimplification, since even the most stable and well-folded proteins are dynamic systems that possess different degrees of conformational flexibility. This is because of the simple fact that so-called conformational forces, that is, forces stabilizing the secondary structure of a protein and its tertiary fold, are weak and can be broken even at ambient temperatures due to thermal fluctuations.4 The breaking of these weak interactions releases the groups that were involved in these interactions and gives them the possibility to be involved in the formation of new weak interactions of comparable energy.4 Since these structural rearrangements are of relatively small scale and since they occur typically in a time scale that is faster than the time required for structure determination by X-ray crystallography and many other physical techniques, the 3-D structures of proteins determined by these techniques represent averaged pictures.6 Furthermore, one should keep in mind that not all proteins structures which are deposited to PDB are structured throughout their entire lengths. Instead, many PDB proteins have portions of their sequences missing from the determined structures (so-called regions of missing electron density)7,8 due to the failure of the unobserved atom, side chain, residue, or region to scatter X-rays coherently caused by their flexible or disordered nature. Such flexible/disordered regions are rather common in the PDB, since only about 30% of protein structures deposited in the PDB do not have such regions of missing electron density.9
In addition to ordered proteins possessing disordered regions of varying length, the literature contains numerous examples of biologically active proteins with flexible structures.4 Therefore, there is another class of functional proteins and protein regions that contain smaller or larger highly dynamic fragments, and some proteins are even characterized by a complete or almost complete lack of ordered structure under physiological conditions (at least in vitro) which appears to be a critical aspect of these proteins' function in vivo.4,10–15 These proteins and protein regions (which are known now as IDPs and IDP regions (IDPRs)) have no single, well-defined equilibrium structure and exist as heterogeneous ensembles of conformers such that no single set of coordinates or backbone Ramachandran angles is sufficient to describe their conformational properties.
These proteins were independently discovered one-by-one over a long period of time and therefore they were considered as rare exceptions to the general rule. Although the phenomenon of biological functionality without stable structure was repeatedly observed, for a long time it was unnoticed by a wide audience because the authors frequently invented new terms to describe their protein of interest.16 In fact, an incomplete list of terms coined in the literature to describe these proteins includes floppy, pliable, rheomorphic,17 flexible,18 mobile,19 partially folded,20 natively denatured,21 natively unfolded,12,22 natively disordered,15 intrinsically unstructured,11,14 intrinsically denatured,21 intrinsically unfolded,22 intrinsically disordered,13 vulnerable,23 chameleon,24 malleable,25 4D,26 protein clouds,27 dancing proteins,28 proteins waiting for partners,29 and several other names often representing different combinations of “natively/naturally/inherently/intrinsically” with “unfolded/unstructured/disordered/denatured” among several others. Therefore, the majority of the names used in the early literature express that the “unfolded, unstructured, disordered, and denatured” state is a “native, natural, inherent, and intrinsic” property of these proteins.16
Although protein intrinsic disorder is considered now as an established concept and PubMed contains hundreds and hundreds of papers talking about different aspects of IDPs/IDPRs, the route to recognizing these proteins as a novel functional entity was complex and lengthy. As it is often the case for new scientific concepts, the idea of structure-less functionality went through the stages of passive ignorance and active denial to scrupulous examination and enthusiastic acceptance. For example, it took me more than a year to publish my first paper dedicated to the systematic analysis of such proteins, and the manuscript was successively rejected by 14 journals before it was finally accepted by Proteins.12 However, time showed that the concept of protein intrinsic disorder was a useful invention and could be considered as a universal lock-pick that helps in solving many of the seemingly unsolvable problems in protein science. One could say that this idea gave a new boost to the development of the protein science, generating a wide array of principally novel research directions [see Fig. 1(B)].
The goals of this review are: (i) to outline some recent advances in the field of IDPs/IDPRs; (ii) to illustrate the usefulness of intrinsic disorder for protein function; (iii) to show that intrinsic disorder can affect different levels of protein structural organization; (iv) to indicate intimate involvement of intrinsic disorder in pathogenesis of various maladies; (v) to emphasize the exceptional structural heterogeneity of IDPs/IDPRs and to show that IDPs are definitely much more structurally complex than random coil-like polypeptides; (vi) to accentuate that although this structural heterogeneity is very important for protein functionality, it represents a crucial hurdle for structural characterization of IDPs; (vii) to stress that new experimental and computational approaches and new theories and models are crucially needed for future progression of this field and protein science in general. These and other points highlight the current state of the field, where further advances in understanding of the “biology” of IDPs still waits for “physics,” with “physics” now being new theories, instrumentation, and analytical approaches.
Disorder Runs in their Blood: Disorderedness is Encoded in the Amino Acid Sequences of IDPs/IDPRs and can be Reliably Predicted
Identification of IDPs as unique entities belonging to a new protein tribe is directly related to the recognition that their amino acid sequences are dramatically different from those of ordered proteins.10,12,13,30–32 For example, it has been pointed out that the low content of hydrophobic residues combined with the high load of charged residues that often gives rise to high net charge of a polypeptide chain represents a characteristic feature of some IDPs (so called extended IDPs or natively unfolded proteins with coil-like or close to coil-like structures, see below).12 Therefore, compact proteins and extended IDPs can be distinguished based only on their net charges and hydropathies using a simple charge-hydropathy (CH) plot, where the IDPs are specifically localized within a specific region of CH phase space and are reliably separated from compact ordered proteins.12 More detailed comparison of amino acid sequences revealed that in comparison with ordered proteins and domains, the IDPs/IDPRs are significantly depleted in order-promoting amino acids (Trp, Tyr, Phe, Ile, Leu, Val, Cys, and Asn),10,33 being instead enriched in disorder-promoting residues, such as Ala, Arg, Gly, Gln, Ser, Glu, Lys, and Pro.13,31,32,34,35
Difference between ordered and disordered proteins goes far beyond these differences in their amino acid compositions. In fact, based on the comparison of the 265 amino acid physico-chemical property-based scales (such as hydropathy, net charge, flexibility index, helix propensities, strand propensities, aromaticity, etc.)34 and more than 6000 composition-based attributes (e.g., all possible combinations having one to four amino acids in the group)36 it has been concluded that ordered and disordered proteins and regions can be discriminated using many of these attributes.13 Based on the analysis of 517 amino acid scales, a novel amino acid scale, Top-IDP (Trp, Phe, Tyr, Ile Met, Leu, Val, Asn, Cys, Thr, Ala, Gly, Arg, Asp, His, Gln, Lys, Ser, Glu, and Pro), was built to provide ranking for the tendencies of the amino acid residue to promote order or disorder.30 The fact that the sequences of ordered and disordered proteins and regions are noticeably different suggested that IDPs clearly constitute a separate entity inside the protein kingdom, that these proteins can be reliably predicted using various computational tools,37–42 and structurally, that IDPs should be very different from ordered globular proteins since peculiarities of amino acid sequence determine protein structure.
Natural Abundance of IDPs: Touching the Tip of the Iceberg
Initial systematic analyses revealed that intrinsic disorder in proteins is a rather common phenomenon. In fact, as of 2002, the list of experimentally validated natively unfolded proteins with chain length greater than 50 amino acid residues contained more than 100 entries.1 It was also pointed out that this list would probably be doubled if shorter polypeptides 30–50 residues long were included,1 and that these 100 experimentally validated natively unfolded have at least 250 homologues, which are also expected to be natively unfolded.1,12 It happened that these “large” numbers (which actually were large enough to make a crucial point that biologically active structure-less proteins represent the new rule and not mere rare exceptions) constitute just a small tip of an iceberg. In fact, using computational tools developed for sequence-based intrinsic disorder prediction the wide spread of IDPs and hybrid proteins containing IDPRs was convincingly shown.43–46 For example, more than 15,000 out of 91,000 proteins in the then-current Swiss Protein database were identified as having long IDPRs.47 The published in 2000 analysis of 31 whole genomes that span the 3 kingdoms of life revealed that many proteins contained segments predicted to have ≥ 40 consecutive disordered residues and that the eukaryotes exhibited more disorder by these measures than either the prokaryotes or the archaea.43 Other studies on the abundance of intrinsic disorder in various evolutionary distant species supported these findings and consistently showed that the eukaryotic proteomes had higher fraction of intrinsic disorder than prokaryotic proteomes.44,48–52
This conclusion is in line with the results of a comprehensive bioinformatics investigation of the disorder distribution in almost 3500 proteomes from viruses and three kingdoms of life, results of which are shown in Figure 2 as the correlation between the intrinsic disorder content and proteome size for 3484 species from viruses, archaea, bacteria, and eukaryotes.46 Surprisingly, Figure 2 shows that there is a well-defined gap between the prokaryotes and eukaryotes in the plot of fraction of disordered residues on proteome size, where almost all eukaryotes have 32% or more disordered residues, whereas the majority of the prokaryotic species have 27% or fewer disordered residues.46 Therefore, it looks like the fraction of 30% disordered residues serves as a boundary between the prokaryotes and eukaryotes and reflects the existence of a complex step-wise correlation between the increase in the organism complexity and the increase in the amount of intrinsic disorder. A gap in the plot of fraction of disordered residues on proteome size parallels a morphological gap between prokaryotic and eukaryotic cells which contain many complex innovations that seemingly arose all at once. In other words, this sharp jump in the disorder content in proteomes associated with the transition from prokaryotic to eukaryotic cells suggests that the increase in the morphological complexity of the cell paralleled the increased usage of intrinsic disorder.46 The variability of disorder content in unicellular eukaryotes and rather weak correlation between disorder status and organism complexity (measured as the number of different cell types) is likely related to the wide variability of their habitats, with especially high levels of disorder being found in parasitic host-changing protozoa, the environment of which changes dramatically during their life-span.53 The further support for this hypothesis came from the fact that the intrinsic disorder content in multicellular eukaryotes (which are characterized by more stable and less variable environment of individual cells) was noticeably less variable than that in the unicellular eukaryotes.46
Figure 2.
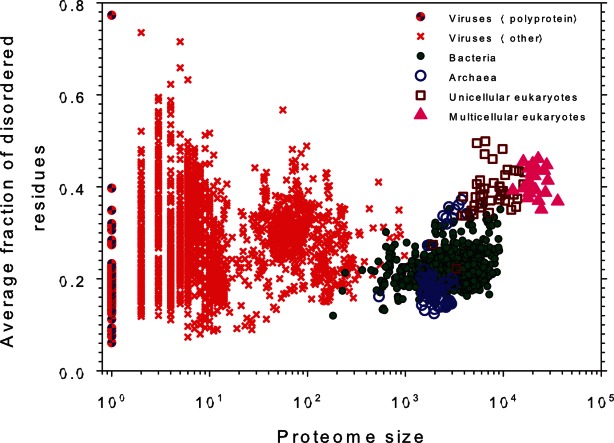
Correlation between the intrinsic disorder content and proteome size for 3484 species from viruses, archaea, bacteria, and eukaryotes. Each symbol indicates a species. There are totally six groups of species: viruses expressing one polyprotein precursor (small red circles filled with blue), other viruses (small red circles), bacteria (small green circles), archaea (blue circles), unicellular eukaryotes (brown squares), and multicellular eukaryotes (pink triangles). Each viral polyprotein was analyzed as a single polypeptide chain, without parsing it into the individual proteins before predictions. The proteome size is the number of proteins in the proteome of that species and is shown in log base. The average fraction of disordered residues is calculated by averaging the fraction of disordered residues of each sequence over the all sequences of that species. Disorder prediction is evaluated by PONDR-VSL2B. Modified from Ref.46.
Complex Simplicity of IDPs
It was pointed out that IDPs possess noticeable amino acid biases, and many IDPs/IDPRs are characterized by sequence redundancy and low sequence complexity, containing long stretches of various repeats and being completely devoid of some (often many) types of amino acid residues. These observations seem to indicate that the sequence space of IDPs/IDPRs should be simpler than that of ordered proteins. However, the reality is more complex than conventional wisdom might suggest, and the sequence space attainable by simple IDPs/IDPRs is more diversified than that of the structurally more sophisticated ordered proteins. In fact, a 100 residue-long protein in which any of the normally occurring 20 amino acids can be found has a sequence space of 20100 (∼10130) sequences.54 Obviously, not all random amino acid sequences can fold into unique structures. In other words, a sequence space of a foldable protein (or “foldable” sequence space) is noticeably smaller than the entire sequence space available for a random polypeptide chain. For decades, the actual size of “foldable” sequence space continues to be unsolved mystery despite a large body of theoretical, biochemical, and computational work that aims to unravel the relationship between a protein's primary sequence and its resulting 3D structure.55 However, the actual number of different amino acid residues in a given foldable sequence can be dramatically reduced,54 since all twenty residues are not necessary for protein folding and the actual physicochemical identity of most of the amino acids in a protein is irrelevant.56–63 In other words, folding alphabet can be noticeably reduced,55,64 and amino acids can be clustered based on some shared features such as homolog substitution frequency,65 local structural environments,66 or peculiarities of the tertiary structural environments.67 This simplified folding code further reduces the available “foldable” sequence space.68
Simply by virtue of their existence, IDPs/IDPRs add a new level of complexity to the sequence–structure relationship, dividing the population of protein sequences into two categories, sequences that yield natively ordered, and sequences that code natively disordered proteins.55 IDPs/IDPRs cannot fold spontaneously and some of them require specific partners to gain more ordered structure. Therefore, they do not possess an entire folding code that defines the ability of foldable proteins to fold spontaneously into a unique biologically active structure. The missing portion of the IDP folding code (or at least part of it) is supplemented by binding partner(s). This defines a principal difference between structured proteins and IDPs/IDPRs: foldable proteins fold first and then bind to their partners whereas IDPs/IDPRs remain disordered until they interact with their partners.68,69 Furthermore, many IDPs/IDPRs do not require folding to be functional,1,4,13,14,70–73 and some of them form fuzzy complexes, in which they preserve significant amount of disorder.74,75 All this suggests that the sequence space of IDPs (at least those which either do not fold at all or do not completely fold at binding) is noticeably greater than the “foldable” sequence space due to the removal of restrictions posed by the need to gain ordered structure spontaneously.68 This represents one of the conundrums of intrinsic disorder, where the apparent sequence redundancy and simplicity are combined with the lack of structural restrains leading to the increase in the dimensions and complexity of the available sequence space.
Also, the existence of a noticeable sequence–structure heterogeneity of IDPs should be emphasized.68 Since the unique 3D-structure of an ordered single-domain protein is defined by the interplay between all (or almost all) of its residues, one could expect that the structure-coding potential is homogeneously distributed within its amino acid sequence. On the other hand, a sequence of an IDP/IDPR contains multiple, relatively short functional elements and therefore represents a very complex structural and functional mosaic.68 This important feature defines the known ability of an IDP/IDPR to interact, regulate, and be controlled by multiple structurally unrelated partners.76 Such functional “anatomy” of IDPs/IDPRs is determined by the extremely high level of their sequence heterogeneity, which is further increased due to the ability of a single IDPR to bind to multiple partners gaining very different structures in the bound state.77
Structural Heterogeneity of IDPs: Continuous Spectrum of Protein Structures
One of the crucial consequences of an extended sequence space and non-homogeneous distribution of foldability (or the structure-coding potential) within amino acid sequences of IDPs and IDPRs is their astonishing structural heterogeneity. In fact, a typical IDP/IDPR contains a multitude of elements coding for potentially foldable, partially foldable, differently foldable, or not foldable at all protein segments.68 As a result, different parts of a molecule are ordered (or disordered) to a different degree. This distribution is constantly changing in time where a given segment of a protein molecule has different structures at different time points. As a result, at any given moment, an IDP has a structure which is different from a structure viewed at another moment.68
Another level of structural heterogeneity is determined by the fact that many proteins are hybrids of ordered and disordered domains and regions, and this mosaic structural organization is crucial for their functions.16 Also, even when they do not possess ordered domains, IDPs are known to have various levels and depth of disorder.78 Over a few past years, an understanding of the available conformational space of IDPs/IDPRs underwent significant evolution. In fact, for a long time, IDPs were considered mostly “unstructured” or “natively unfolded” polypeptide chains. This was mostly due to the fact that the majority of IDPs analyzed at early stages of the field contained very little ordered structure, that is, they were really mostly unstructured or unfolded. Finding and characterization of such “structure-less” proteins was important to build up a strong case to counter-point the dominant view represented by the classical sequence-to-structure-to-function paradigm, especially since such fully unstructured, yet functional proteins clearly represented the other extreme of the protein structure–function spectrum.16 The top half of the Figure 3 illustrates this situation by opposing rock-like ordered proteins and cooked spaghetti-like IDPs.
Figure 3.
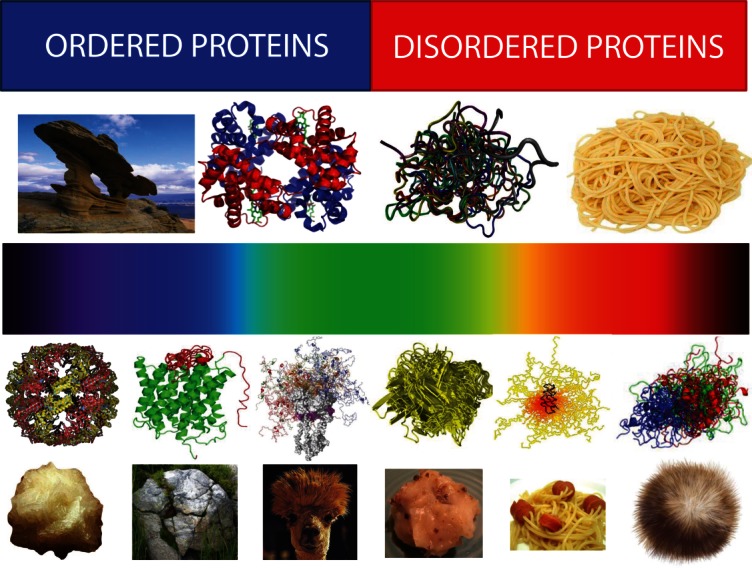
Structural heterogeneity of IDPs/IDPRs. Top half: Bi-colored view of functional proteins which are considered to be either ordered (folded, blue) or completely structure-less (disordered, red). Ordered proteins are taken as rigid rocks, whereas IDPs are considered as completely structure-less entities, kind of cooked noodles. Bottom half: A continuous emission spectrum representing the fact that functional proteins can extend from fully ordered to completely structure-less proteins, with everything in between. Intrinsic disorder can have multiple faces, can affect different levels of protein structural organization, and whole proteins, or various protein regions can be disordered to a different degree. Some illustrative examples includes ordered proteins that are completely devoid of disordered regions (rock-like type), ordered proteins with limited number of disordered regions (grass-on-the rock type), ordered proteins with significant amount of disordered regions (lhama/camel hair type), molten globule-like collapsed IDPs (greasy ball type), pre-molten globule-like extended IDPs (spaghetti-and-sausage type), and unstructured extended IDPs (hairball type).
However, already in some early studies, it was indicated that IDPs/IDRs could be crudely grouped into two major structural classes, proteins with compact and extended disorder.1,4,12,13,73 Based on these observations, the protein functionality was ascribed to at least three major protein conformational states, ordered, molten globular, and coil-like,13,79 indicating that functional IDPs can be less or more compact and possess smaller or larger amount of flexible secondary/tertiary structure.1,4,12,13,73,79 Roughly at the same time, it was emphasized that the extended IDPs (known as natively unfolded proteins) do not represent a uniform entity but contain two broad structural classes, native coils and native pre-molten globules.1
Currently available data suggest that intrinsic disorder possesses multiple flavors, can have multiple faces, and can affect different levels of protein structural organization, where whole proteins, or various protein regions can be disordered to a different degree.68 This new view of structural space of functional proteins can be visualized to form a continuous spectrum of differently disordered conformations extending from fully ordered to completely structure-less proteins, with everything in between (Fig. 3, bottom half). Here, functional proteins can be well-folded and be completely devoid of disordered regions (rock-like scenario). Other functional proteins may contain limited number of disordered regions (a grass-on-the rock scenario), or have significant amount of disordered regions (a llama/camel hair scenario), or be molten globule-like (a greasy ball scenario), or behave as pre-molten globules (a spaghetti-and-meatballs/sausage scenario), or be mostly unstructured (a hairball scenario).
Notably, in this representation, there is no boundary between ordered proteins and IDPs, and, the structure-disorder space of a protein is considered as a continuum. It is important to remember that even the most ordered proteins do not resemble “solid rocks” and have some degree of flexibility. In fact, a protein molecule is an inherently flexible entity and the presence of this flexibility (even for the most ordered proteins) is crucial for its biological activity.80 Also, another important point to remember is that due to their heteropolymeric nature, proteins are never random coils and always have some residual structure.68
Polymeric Roots of the Unusual Biophysics of IDPs
Protein biophysicists/biochemists working on different aspects of ordered proteins (e.g., analyzing their structural properties, functions, folding, etc.) would find biophysical properties of functional IDPs/IDPRs to be rather unusual since these highly dynamic proteins do not follow the well-accepted wisdom that a protein has to be well-folded to be biologically functional. However, the unusualness is a subjective feature, and from the viewpoint of polymer physics the extended IDPs/IDPRs possess the expected behavior of flexible and charged polymers, whereas the behavior of an ordered protein is rather unexpected (i.e., due to the existence of the native ensemble that for well-folded, ordered proteins can be approximated as a harmonic well around a unique, well-defined equilibrium structure). Therefore, one definitely should keep in mind that the “unusual” biophysics of extended IDPs/IDPRs has its roots in the usual polymer physics of highly charged and flexible polypeptides.
Each protein is believed to be a unique entity that has quite unique primary sequence which governs its 3D structure (or lack thereof) and ensures specific biological function(s). Therefore, understanding the effect of sequence variance on the biological performance presents a challenging task. However, natural polypeptides have originated as random copolymers of amino acids, which were adjusted or “selected” over evolution based on their functional capacities.56,81 Despite their differences in primary amino acid sequences, protein molecules in a number of conformational states behave as polymer homologues, suggesting that the volume interactions can be considered as a major driving force responsible for the formation of equilibrium structures or structural ensembles.82 For example, ordered globular proteins and molten globules (both as folding intermediates of globular proteins or as examples of collapsed IDPs) exhibit key properties of polymer globules, where the fluctuations of the molecular density are expected to be much less than the molecular density itself. Extended IDPs (both intrinsic coils and intrinsic pre-molten globules) and ordered proteins in the pre-molten globule intermediate state possess properties of squeezed coils, since water is a poor solvent for a polypeptide. In fact, even high concentrations of strong denaturants (e.g., urea and GdmCl) are very likely to be bad solvents for protein chains, resulting in the preservation of extensive residual structure even under these harsh denaturing conditions.82
Based on these and related observations, and taking into account the fact that many IDPs/IDPRs are characterized by significant amino acid composition biases, the overall polymeric behavior of these proteins and regions can be mimicked reasonably well by the behavior of low-complexity polypeptides (e.g., homopolypeptide and block copolypeptides). Following these ideas, it was shown that water is a poor solvent for polypeptide backbone alone and for the IDPs containing long tracts of polar amino acid residues since polar homo-polypeptides without hydrophobic groups (e.g., polyglutamine or glycine-serine block copolypeptides) were shown to prefer collapsed ensembles in aqueous media.83–88 Furthermore, even polyglycine was shown to have a tendency to form heterogeneous ensembles of collapsed structures in water.88
A systematic analysis of the conformational behavior of protamines, arginine-rich IDPs involved in the condensation of chromatin during spermatogenesis, and protamine-like peptides revealed that there is a charge-driven coil-to-globule transition in these highly charged polypeptides, where the net charge per residue serves as the discriminating order parameter.89 Overall, the increase in the hydrodynamic dimensions of a polypeptide chain with increase in its net charge per residue can be attributed to the increase in the intramolecular electrostatic repulsions between similarly charged side-chains and the favorable solvation of these moieties.89 Based on these premises, at least three different classes of globule-forming polar/charged IDPs were proposed. The first class is comprised by polar tracts which collapse due to water being a poor solvent for a backbone and non-charged side chains. The second class is represented by weak polyelectrolytes and weak polyampholytes, which have low per residue net charge and low fractions of positively and/or negatively charged residues. These IDPs/IDPRs form collapsed structures since the driving force responsible for the collapse is not overcome by the intramolecular electrostatic repulsion between the charged side-chains and by their favorable free energies of solvation. Furthermore, if such IDPs/IDPRs possess polyampholytic nature, their globular state could be additionally stabilized by electrostatic interactions between the oppositely charged side-chains. Finally, IDPs/IDPRs from the third class are strong polyampholytes characterized by high fractions of positively and/or negatively charged residues but have low per residue net charge. Such intrinsically disordered protein can form collapsed structures stabilized mostly by multiple electrostatic interactions between solvated side-chains of opposite sign.89
The extended IDPs/IDPRs were used as a model system for the analysis of the effect of electrostatic interactions on conformational properties of unfolded proteins, and for testing the quantitative descriptions and predictions of polymer theory related to the influence of charged amino acids on chain dimensions.90 For example, based on the analysis of the conformational equilibrium of coarse-grained polypeptides as a function of sequence hydrophobicity, charge, and length it has been concluded that the variations in sequence hydrophobicity and charge define a coil-to-globule transition comparable to that seeing in the empirical CH-plot,12,91 suggesting that a minimal, polymer physics-based model can capture the elements of global protein conformation.92
IDPs/IDPRs with very high net charges are expected to be more extended and behave more similar to random coils (i.e., similar to conformations adopted by proteins in the denaturant GdmCl). The analysis of the GdmCl-induced expansion of the unfolded states suggested that protein charge density plays a crucial role in defining the hydrodynamic behavior of the unfolded polypeptide chain.90 Here, highly charged proteins can exhibit a prominent expansion at low ionic strength that correlates with their net charges.90 It has been also hypothesized that the pronounced effect of charges on the dimensions of unfolded proteins might have important implications for their cellular functions.90
Similarly, a comprehensive analysis of the hydrodynamic dimensions of FG-nucleoporins containing large IDPRs with multiple phenylalanine-glycine repeats (FG-domains) revealed that under the physiologic conditions in vitro these domains adopt distinct categories of disordered structures, such as molten globule, pre-molten globule, relaxed-coil, extended-coil (as in urea), or very extended-coil (as in GdmCl).93 The category of intrinsically disordered structure in a given FG-domain was related to its amino acid composition, namely to the content of charged residues, where more charged FG-domains possessed larger hydrodynamic dimensions.94 Furthermore, FG-nucleporins with higher charge density were shown to be more dynamic than the collapsed-coil FG-domains, being also prone to repel other FG-domains. On the other hand, the collapsed-coil FG-domains were prone to oligomerize. These observations suggested that different types of FG-domains with different aggregation propensities provide molecular basis for two different gating mechanisms operating at the nuclear pore complex at distinct locations; one acting as a hydrogel, and the other as an entropic brush.94 Therefore, the abundance and peculiarities of the charged residues distribution within the protein sequences might determine physical and biological properties of extended IDPs and IDPRs.
Also, simple polymer physics-based reasoning can give reasonably well-justified explanation of the conformational behavior of extended IDPs. In general, the conformational behavior of IDPs is characterized by the low cooperativity (or the complete lack thereof) of the denaturant-induced unfolding, lack of the measurable excess heat absorption peak(s) characteristic for the melting of ordered proteins, “turned out” response to heat and changes in pH, and the ability to gain structure in the presence of various binding partners.95 The analysis of the temperature effects on structural properties of several extended IDPs revealed that native coils and native pre-molten globules partially fold as the temperature is increased.1,73,95–98 These heating-induced structural changes in extended IDPs were attributed to the increased strength of the hydrophobic interaction at higher temperatures, leading to a stronger hydrophobic attraction, which is the major driving force for folding. Similarly, extended IDPs/IDPRs are characterized by the “turned out” response to changes in pH,96,99–102 where a decrease (or increase) in pH induces their partial folding due to the minimization of their high net charges viewed at neutral pH, thereby decreasing charge/charge intramolecular repulsion and permitting hydrophobic-driven collapse to the partially folded conformation.95
Every Disordered Protein is Disordered in its Own Way
Data accumulated so far indicate that intrinsic disorder exists at multiple structural levels and might differently affect different regions/domains of IDPs. This defines noted structural complexity and heterogeneity of IDPs/IDPRs which are further enhanced by the way different proteins/protein regions respond to their environments. Furthermore, since intrinsic disorder is crucial for many biological functions and therefore must prevail in different environments, the amino acid sequences and compositions of IDPs and IDPRs are specifically shaped by the peculiarities of their global and local environments. All this makes the protein intrinsic disorder phenomenon to be so broad that one can even assume that every disordered protein (or at least every family of disordered proteins) is disordered in its own way. This hypothesis has far-reaching consequences since it implies that a general disorder predictor has limited accuracy and cannot predict with equally high accuracy disorder status of all protein sequences due to their heterogeneity. It also implies that some environmental factors definitely should be taken into account when assessing intrinsic disorder in proteins. Several examples are presented below to support the overall validity of these statements.
Transmembrane proteins
The first example is given by transmembrane (TM) proteins, in which disorder is widely observed (e.g., 40% of human integral plasma proteins were predicted to contain long IDPRs).103–107 Furthermore, disorder is unevenly distributed between the cytoplasmic and the external surfaces of these proteins, with cytoplasmic domains being up to threefold more disordered than extracellular domains.105 Although these analyses gave interesting hints on the abundance of disorder in TM proteins, the obvious weakness of such evaluations is in the fact that they were performed using the disorder predictors developed from structured and disordered regions found in water-soluble proteins.108 However, the major physico-chemical properties of water-soluble and integral membrane proteins are very different due to the differences in their environments. For example, similar to typical water soluble proteins, the TM regions of membrane proteins are often highly structured, containing α-helices109 or β-structure,110 which are especially likely to occur due to the low dielectric constant values within the membrane bilayers.111,112 On the other hand, the exterior regions of TM proteins are much more apolar than the exteriors of water-soluble proteins.113–115 Therefore, the peculiarities of the membrane environment, with its highly nonpolar nature originating either from lipids or from protein interiors, are especially unfavorable for intrinsic disorder, since propensity for intrinsic disorder is typically encoded in a high content of polar and charged residues. Therefore, the IDPRs found in integral membrane proteins would be expected to be generally localized within the regions external to the membrane bilayer.108 Also, the distinctive environment of the membrane bilayer imposes constraints on the amino acid composition of integral membrane proteins, even on the regions external to the membrane bilayer.116,117 Comprehensive bioinformatics analysis revealed that integral membrane proteins commonly possess IDPRs defined as regions of missing electron density in their crystal structures.108 Comparison of the IDPRs found in the α-helical and the β-barrel bundle integral membrane proteins with the IDPRs viewed in typical water-soluble proteins revealed the existence of statistically distinct amino acid compositional biases characteristic for these three protein classes. Therefore, the use of specific amino acid signatures of IDPRs found in TM helical bundles and β-barrels can potentially lead to significantly more accurate disorder predictions for these two classes of integral membrane proteins.108
Proteins from archaea
Another illustrative example of the specific disorder-related and environment-dependent sequence features is given by archaeal proteins.46,51 Based on the levels of predicted disordered residues, archaeal proteins can be grouped into three classes, with ranges of the disordered residue content of 12–21%, 21%–32%, and 32%–38% (see Fig. 2). The archaeal proteomes with the highest disorder contents are halophiles and methanophiles.46,51 Similar to TM proteins, the estimation of intrinsic disorder in the extremophilic proteins of the microorganisms surviving under hypersaline conditions using predictors developed for the “normal” non-halophilic proteins existing under the normal physiological conditions of 100–150 mM NaCl may not be accurate.46 In fact, one of the strategies used by the halophilic archaea, which are salt-loving extremophilic organisms that grow optimally at high salt concentrations, to maintain proper osmotic pressure in their cytoplasm is a so-called “salt-in” strategy that involves accumulation of molar concentrations of potassium and chloride in their cytosoles.118 This strategy requires extensive adaptation of the intracellular proteins to the presence of near-saturating salt concentrations. The proteomes of such “salt-in” organisms are highly acidic,46,51 and their proteins are characterized by remarkable instability at conditions of low salt concentrations and by maintaining soluble and active conformations in hypersaline conditions that are generally detrimental to the non-halophilic proteins.118–127
Viral proteins
Finally, peculiarities of disorder distributions in viral proteins can be used to further support the importance of considering environmental factors.46,51 Here, the comprehensive analysis of intrinsic disorder in various completed proteomes revealed that the viral proteomes have the largest variation of disorder content, which ranges from 7.3% disordered residues in the human coronavirus NL63 to 77.3% disordered residues in the Avian carcinoma virus proteome (see Fig. 2).46 The high predicted intrinsic disorder content in viruses has multiple functional implications, where some IDPRs are used in the functioning of viral proteins and help viruses to highjack various pathways of the host cells, others likely have evolved to help viruses accommodate to their hostile habitats, and still others evolved to help viruses in managing their economic usage of genetic material via alternative splicing, overlapping genes, and anti-sense transcription.128 These findings are in agreement with another study revealing that in comparison with archaea and bacteria, viral and bacteriophagic proteins were significantly more enriched in polar residues and depleted in hydrophobic residues and were close to eukaryotic proteins in terms of their amino acid compositions and the reduced content of the order-promoting residues.129
Functional Manifoldness of IDPs
Functional protein clouds: Major functional advantages of being intrinsically disordered
The high natural abundance of IDPds/IDPRs and their specific structural features indicate that these proteins and regions might carry out important biological functions. This hypothesis has been confirmed by several comprehensive studies,1,11–14,71–73,78,130–134 which revealed that these structure-less members of the protein kingdom are abundantly involved in numerous biological processes, where they are frequently found to play different roles in regulation of the functions of their binding partners and in promotion of the assembly of supra-molecular complexes.1,4,11–15,31,70–73,76–78,79,131,132,134–149 The conformational plasticity of IDPs/IDPRs provides them with a wide spectrum of exceptional functional advantages over the functional modes of ordered proteins and domains.4,10,11,13,32,71,72,77,78,131,132,134,141,142,150,151 Some of these advantages are:
Increased speed of interaction due to greater capture radius and the ability to spatially search through interaction space;
Increased interaction (surface) area per residue;
Strengthened encounter complex allows for less stringent spatial orientation requirements;
Efficient regulation via rapid degradation;
The ability to be involved in one-to-many binding, where a single disordered region binds to several structurally diverse partners;
The ability to be involved in many-to-one binding, where many distinct (structured) proteins may bind a single disordered region;
The ability to overcome steric restrictions, enabling larger interaction surfaces in protein–protein and protein–ligand complexes than those obtained with rigid partners;
The ability to fold upon binding (completely or partially);
The ability of some IDPs/IDPRs to form very stable intertwined complexes;
The ability of some IDPs/IDPRs to stay substantially disordered in bound state;
Binding fuzziness, where different binding mechanisms (e.g., via stabilizing the binding-competent secondary structure elements within the contacting region, or by establishing the long-range electrostatic interactions, or being involved in transient physical contacts with the partner, or even without any apparent ordering) can be employed to accommodate peculiarities of interaction with various partners;
Binding plasticity, where an IDPR folds to specific bound conformations (which can be very different) according to the template provided by binding partners;
High accessibility of sites targeted for post-translational modifications (PTMs);
Efficient structural and functional regulation via PTMs such as phosphorylation, acetylation, lipidation, ubiquitination, sumoylation, and so forth, allowing for a simple means of modulation of their biological functions;
Efficient functional control via regulatory proteolytic attack sites of which are frequently associated with IDPRs;
Ease of regulation/redirection and production of otherwise diverse forms by alternative splicing (given the existence of multiple functions in a single disordered protein, and given that each functional element is typically relatively short, alternative splicing could readily generate a set of protein isoforms with a highly diverse set of regulatory elements152);
The possibility of overlapping binding sites due to extended linear conformation;
Decoupled binding affinity and specificity, where, due to the induced folding, IDP/IDPR can be involved in the formation of specific but weak complexes. In other words, IDP/IDPR might possess high specificity for given partners combined with high kon and koff rates that enable rapid association with the partner without an excessive binding strength. This combination of high specificity with low affinity defines the broad utilization of intrinsic disorder in regulatory interactions where turning a signal off is as important as turning it on;
Diverse evolutionary rates with some ID proteins being highly conserved and other ID proteins possessing high evolutionary rates. The latter ones can evolve into sophisticated and complex interaction centers (scaffolds) that can be easily tailored to the needs of divergent organisms;
Flexibility that allows masking (or not) of interaction sites or that allows interaction between bound partners;
The ability to be involved in the cascade interactions, where IDP binding to the first partner induces partial folding generating a new binding site suitable for interaction with the second partner, and so forth.
Functions of IDPs are complementary to the catalytic activities of ordered proteins.1,11–13,31,72,73,78,130,132,134,138–140,142,143 Many disorder-related functions (e.g., signaling, control, regulation, and recognition) are incompatible with well-defined, stable 3-D structures.1,11–14,31,73,78,79,132,134,138–140,142,144,153 Functions of many IDPs/IDPRs rely on interactions with specific binding partners, and many IDPs/IDPRs tend to undergo disorder-to-order transitions as a result of binding to their specific targets.12
Functionally, IDPs/IDPRs were grouped in at least six broad classes based on the mode of action.14,136 These broad classes included protein and RNA chaperones, entropic chains, effectors, scavengers, assemblers and display sites,14,136 and 28 separate functions, including molecular recognition via binding to other proteins, or to nucleic acids, were assigned for IDPRs in early studies.71,72 Later, a rich spectrum of biological functions associated with IDPs/IDPRs was found based on a comprehensive computational study of a correlation between the functional annotations in the Swiss-Prot database and predicted intrinsic disorder.138–140 The approach was based on the hypothesis that if a function described by a given keyword relies on intrinsic disorder, then the keyword-associated protein would be expected to have a greater level of predicted disorder compared to the protein randomly chosen from the Swiss-Prot. This analysis revealed that ∼44% and ∼34% of Swiss-Prot functional keywords were associated with ordered and disordered proteins, respectively, whereas 22% functional keywords yielded ambiguity in the likely function–structure associations.138–140 Interestingly, most of the structured protein-associated key words were shown to be related to enzymatic activities, whereas the majority of the disordered protein-associated keywords were related to signaling and regulation. These results agree well with the notion that enzymatic catalysis requires ordered structure and that effectiveness of signaling is dependent on binding reversibility, a property directly associated with the thermodynamics of disorder-to-order transition induced by binding.138–140
Many IDPs and IDPRs undergo a disorder-to-order transition upon functioning.11,13,15,71,72,78,79,130–132,134,154–157 When disordered regions bind to signaling partners, the free energy required to bring about the disorder to order transition takes away from the interfacial, contact free energy, with the net result that a highly specific interaction can be combined with a low net free energy of association.13,155 High specificity coupled with low affinity is a useful pair of properties for a reversible signaling interaction. Furthermore, a disordered protein can readily bind to multiple partners by changing shape to associate with different targets.13,158,159
All this clearly suggests that there is a new two-pathway protein structure–function paradigm, with sequence-to-structure-to-function for enzymes and membrane transport proteins, and sequence-to-disordered ensemble-to-function for proteins and protein regions involved in signaling, regulation, and control.1,13,71,73,79 One of the first generalization of this concept was given by The Protein Trinity Hypothesis, which suggested that native proteins can be in one of three states, the solid-like ordered state, the liquid-like collapsed-disordered state, or the gas-like extended-disordered state.79 Function is then viewed to arise from any one of the three states or from transitions between them. This model was subsequently expanded to include a fourth state (pre-molten globule) and transitions between all four states.1 In reality, based on the outlined above idea of the continuous spectrum of protein structures, functional proteins contain various amounts of intrinsic disorder and this continuous structural spectrum of protein defines their limitless functional variability.
Lock-picking with Ariadne's thread: Illustrative examples of disorder-dependent protein functions
Among intriguing protein functions relying on intrinsic disorder are moonlighting activities,137 actions of hub proteins,78,93,134,160–164 and scaffolding functions.141,165 Since all these functions illustrate the notions that the intrinsic disorder concept represents a universal skeleton key (or lock-pick) that helps unlocking seemingly unresolvable mysteries of protein science and therefore can be considered as a new Ariadne's thread that helps navigate the unusual twists of the sophisticated relationships between protein sequence, structure, and function, they are considered in some detail below.
Moonlighting proteins
Moonlighting is the ability of a protein to fulfill more than one function. Often, these functions are unrelated or at least are not obviously related to each other.137,166–168 The capability of a protein to be involved in moonlighting or multi-tasking activities represent one of the solutions used by the Nature to increase the organism's complexity without the expansion of the genome size, where by acting differently at distinct points of metabolic networks proteins increase network complexity without increasing the actual size of the network.137,166–168 Among various molecular mechanisms used by the moonlighting proteins to switch between functions are changes in cellular localization, changes in ligand binding, expression in different cell types, and variations of the oligomerization state.137 In addition to these mechanisms that can be explained within the frames of the traditional structure–function paradigm, consideration of the intrinsic disorder phenomenon opens new possibilities.137 In fact, one of the peculiar functional advantages of IDPs/IDPRs is their binding promiscuity and ability to be involved in one-to-many signaling, whereby an IDP/IDPR binds structurally different partners in a template-induced folding process.11,77,132,169 Therefore, IDPs/IDPRs can use the same region or overlapping interaction regions/surfaces to exert distinct effects and employ the disorder-based mechanisms to switch function that relies on their capability to form different conformations upon binding.137 Such structural malleability of IDPs/IDPRs defines their ability to participate in unprecedented moonlighting events, where these disordered moonlighting proteins or regions produce the opposing effects (inhibition and activation) on different partners or even the same partner molecule.137
Hub proteins
Signaling interactions inside the cell can be described as specific and complex networks that can be considered as “scale-free” or “small-world” networks, which have hubs, with many connections, and ends, that have the only connection to just one neighbor.170,171 Such scale-free networks combine the local clustering of connections characteristic of regular networks with occasional long-range connections between clusters, as can be expected to occur in random networks. In other words, the distance between nodes in these scale-free networks follows a power-law distribution.172 Based on their spatiotemporal peculiarities protein hubs were grouped into two broad categories, “date hubs” that binds their numerous partners sequentially, and “party hubs” simultaneously interacting with their partners.173 Since many IDPs are known to be involved in interaction with large number of distinct partners, they clearly can be considered as hubs in the scale-free protein–protein interaction networks.78,134 Based on the systematic analysis of several know hub proteins134 followed by a series of robust bioinformatics studies,93,160–164 it was concluded that hubs commonly use disordered regions to bind to multiple partners and that there are at least two primary mechanisms by which disorder is utilized in protein–protein interaction networks where one disordered region binds to many partners or many disordered region bind to one partner.134
Scaffold proteins
Scaffold proteins constitute an important subclass of hubs that typically have a modest number of interacting partners and that are commonly found at the central parts of functional complexes, where they interact with most of their partners at the same time and therefore act as party hubs.160 Besides being responsible for bringing together specific proteins within a signaling pathway and providing selective spatial orientation and temporal coordination to facilitate and promote interactions among interacting proteins, some scaffolds can influence the specificity and kinetics of signaling interactions via simultaneous binding to multiple participants in a particular pathway and facilitation and/or modifying the specificity of pathway interactions,174 other scaffold can change conformations of individual proteins and thus modulate their activities,174 still other scaffold proteins may modulate the activation of alternative pathways by promoting interactions between various signaling proteins.141 Analysis of several well-characterized signaling scaffold proteins reveled that their large IDPRs are crucial for the successful scaffold function.141 A more global bioinformatics analysis revealed that a typical design of a scaffold protein includes a set of short globular domains (∼80 amino acids on average) connected by long linker regions (∼150 residues on average) with crucial binding functions.165 This gave further support to the notion that signaling scaffold proteins utilize the various features of highly flexible ID regions to obtain more functionality from less structure.141
Disorder and transcription regulation
Conformational plasticity and adaptability associated with intrinsic disorder are crucial for various protein functions. Among the proteins whose functional life is strongly disorder-dependent are transcription factors (TFs)175,176 and other proteins involved in transcriptional regulation, such as the mediator complex,24,177 core and linker histones,178 and ribosomal proteins.179 For example, from 83 to 94% of TFs might possess long IDPRs, with the degree of disorder in eukaryotic TFs being significantly higher than in prokaryotic TFs.175,176 Also, TFs were shown to be depleted in order-promoting residues and enriched in disorder-promoting residues, and were characterized by high levels of α-molecular recognition feature (MoRF).175 Furthermore, disorder is unevenly distributed within the TFs, with the degree of disorder in their activation regions being much higher than that in DNA-binding domains. However, the AT-hooks (which are DNA-binding motifs present in many proteins which binds to the (ATAA) and (TATT) repeats of DNA) and basic regions of TF DNA-binding domains are highly disordered suggesting that eukaryotes with their well-developed gene transcription machinery require transcription factor flexibility to be more efficient.175 A number of interesting and important roles were also ascribed to intrinsic disorder in TFs related to the regulation of heat shock response (so called heat shock factors, HSFs)180 and in the reprogramming TFs (the Yamanaka factors, namely Sox2, Oct3/4 (Pou5f1), Klf4, and c-Myc, and the Thomson factors, namely Sox2, Oct3, Lin28, and Nanog) overexpression of which is known to generate induced pluripotent stem (iPS) cells from terminally differentiated somatic cells.181
Disorder in the regulation of cellular pathways
Of special interests are the vital roles of intrinsic disorder in regulation and orchestration of various cellular pathways. One of the illustrative examples of this regulatory role of intrinsic disorder is the canonical Wnt-pathway that involves five proteins, Axin, CKI-α, GSK-3β, APC (adenomatous polyposis coli, also known as deleted in polyposis 2.5 protein), and β-catenin (all shown to contain long IDPRs). This pathway is known to play a number of crucial roles in the development of organism, and the malfunctions of which might lead to various diseases including cancer.182 The comprehensive analysis of published data revealed that IDPRs found in Wnt-pathway proteins orchestrate protein–protein interactions, and facilitate PTMs and signaling.182 Furthermore, the scaffold protein Axin and another large protein, APC, are heavily enriched in disorder and act as flexible concentrators in gathering together all other proteins involved in the Wnt-pathway.182 Intriguingly, the multifarious roles of highly disordered APC in regulation of β-catenin function were established by showing that disordered APC helps the collection of β-catenin from cytoplasm, facilitates the β-catenin delivery to the binding sites on Axin, and controls the final detachment of β-catenin from Axin.182
Another important illustration of the involvement of intrinsic disorder in regulation of crucial pathway is given by the process of the programmed cell death (PCD), which is one of the most intricate cellular processes where the cell uses specialized cellular machinery and intracellular programs to kill itself and which enables metazoans to control cell numbers and eliminate cells that threaten the animal's survival.183 PCD includes several specific modules, such as apoptosis, autophagy, and programmed necrosis (necroptosis). These modules are not only tightly regulated but also intimately interconnected and are jointly controlled via a complex set of protein–protein interactions. Recently, several large sets of PCD-related proteins across 28 species were analyzed using a wide array of modern bioinformatics tools to understand the role of the intrinsic disorder in controlling and regulating the PCD.183 This analysis revealed that proteins involved in regulation and execution of PCD possess substantial amount of intrinsic disorder and IDPRs were implemented in a number of crucial functions, such as protein–protein interactions, interactions with other partners including nucleic acids and other ligands, were shown to be enriched in post-translational modification sites, and were characterized by specific evolutionary patterns.183
Molten globular Enzymes
Unique catalytic function of a protein is believed to be dictated by its unique 3D structure. This axiom constitutes a cornerstone of the lock-and-key paradigm and it seemed to be able to sustain the furious attack on protein structure–function relationship initiated by the discovery of IDPs and hybrid proteins with ordered domains and IDPRs. In fact, from the vast majority of experimental and computational studies a general conclusion was drawn over and over again, where the functional repertoire of IDPs complemented the functional arsenal of ordered proteins, with ordered proteins being mostly responsible for catalysis and transport and with IDPs doing the majority of other jobs in the cell.
On the other hand, all proteins (even the most ordered and tightly folded ones) are intrinsically flexible molecules that undergo conformational changes over a wide range of timescales and amplitudes.184 In fact, the combination of active site reactivity with the dynamic character of proteins allows enzymes to be promiscuous and remarkably efficient at the same time.185 Furthermore, in general, dynamic fluctuations are crucial for enzyme catalysis, since they can influence substrate binding and product release, and may even adjust the effective barriers of the catalyzed reactions.186–190 Often, dynamic changes in the enzyme during the catalytic reaction can be described using the induced-fit model, where a conversion of one tight conformational ensemble (free enzyme) to another distinct ensemble (bound enzyme) takes place through a series of local substrate-mediated structural rearrangements.191 Despite this crucial role of local flexibility in the enzymatic catalysis, enzymes are still relatively stable molecules whose dynamic character is restricted to a small set of tightly folded conformations and whose unique (albeit locally flexible) structures are needed for efficient catalysis. From this viewpoint, the presence of intrinsic disorder is expected to be poorly compatible with enzymatic catalysis, which requires a well-organized environment in the active site of the enzyme in order to facilitate the formation of the transition state of the chemical reaction to be catalyzed.192
In a sharp contrast to this common wisdom supported by a wide array of specific examples, several enzymes were shown to be much more dynamic than the catalytic machines are expected to be, clearly possessing, in their precatalytic states, many characteristic properties of molten globules and retaining unusually high flexibility in structurally defined enzyme–ligand complexes. One of the best characterized examples of such molten globular enzymes is the engineered monomeric form of chorismate mutase from Methanococcus jannaschii (MjCM).184,193–195 Here, a functional monomer (mMjCM) was created by inserting the hinge-loop sequence into the long, dimer-spanning N-terminal helix.193 In its unbound form, mMjCM was shown to exists as a native molten globule that was described as a dynamic ensemble of α-helical conformers rapidly interconverting on the millisecond timescale.193 Interaction with natural ligand induced global conformational changes in the molten globular mMjCM promoting formation of a defined enzyme–ligand complex, which, however, preserved unusually high flexibility.184 Catalytic mechanism of the molten globular mMjCM was described as follows: “Though probably stochastic in nature, internal motions in the complex may generate a collective dynamic matrix that samples catalytically active conformation(s) often enough to achieve rapid turnover in the presence of the true transition state.”184 Therefore, some enzymes can represent a highly dynamic heterogeneous conformational ensemble which is still compatible with efficient catalysis.
In agreement with this hypothesis, a molten globular character was described for circularly permuted dihydrofolate reductase (DHFR),196,197 and urease G from Bacillus pasteurii (BpUreG).198–200 Of these three enzymatic molten globules UreG is the only natural molten globular enzyme known to date, since both circularly permuted DHFR and monomeric MjCM were obtained as a result of some genetic manipulations. Although the number of known native molten globules with enzymatic activity is small, their existence provides an interesting hint on early protein evolution. In fact, simple logics suggests that well-ordered enzymes appear as a result of long evolutionary process, whose very likely starting point was a partially folded polypeptide with some general properties of the molten globule.
Multifarious Interactivity of IDPs
IDPs/IDPRs can form highly stable complexes, or be involved in signaling interactions where they undergo constant “bound–unbound” transitions, thus acting as dynamic and sensitive “on–off” switches. The ability of these proteins to return to the highly flexible conformations after the completion of a particular function, and their predisposition to gain different conformations depending on the environmental peculiarities, are unique physiological properties of IDPs which allow them to exert different functions in different cellular contests according to a specific conformational state.4
Static complexes
Due to their lack of rigid structure, combined with the high level of intrinsic dynamics and almost unrestricted flexibility at various structure levels in the non-bound state, as well as due to the unique capability to adjust to structure of the binding partner, IDPs are characterized by a very diverse range of binding modes, creating a multitude of unusual complexes, many of which are not attainable by ordered proteins.201 Some of these complexes are relatively static, resemble complexes of ordered proteins, and, therefore are suitable for the structure determination by X-ray crystallography. Among these static complexes are: MoRFs, wrappers, chameleons, penetrators, huggers, intertwined strings, long cylindrical containers, connectors, armature, tweezers and forceps, grabbers, tentacles, pullers, and stackers or β-arcs.201 These binding modes are shown in Supporting Information Figure 1S and briefly described in the Supporting Information Materials.
Disordered or fuzzy complexes of IDPs
In addition to the static complexes, where bound partners have fixed structures, some IDPs/IDPRs do not fold even in their bound state, forming so-called disordered, dynamic, or fuzzy complexes with ordered proteins,97,202–206 other disordered proteins,207–209 or biological membranes.210,211 In complexes of some of these IDPs with their binding partners, the disordered regions flanking the interaction interface but not the interface itself remain disordered. Such mode of interaction was recently described as “the flanking fuzziness” in contrast to “the random fuzziness” when the disordered protein remains entirely disordered in the bound state.75,212 It is also expected that the similar binding mode can be utilized by disordered protein while interacting with nucleic acids and other biological macromolecules.201
Physically, binding is considered as joining objects together and suggests spatial and temporal fixation of bound partners. The formation of protein complexes with specific binding partners is expected to bring some fixation (at least at the binding site). Therefore, disordered complexes where interaction of a disordered protein with the binding partners is not accompanied by a disorder-to-order transition within the interaction interface clearly cannot be described by the classical binding paradigm. This contradiction can be resolved assuming that the ordered binding partner and/or disordered protein contain multiple low affinity binding sites. The existence of several similar binding sites combined with a highly flexible and dynamic structure of disordered protein creates a unique situation where any binding site of disordered protein can interact with any binding site of its partner with almost equal probability, in a staccato manner. The low affinity of each individual contact implies that each of them is not stable and can be readily broken. Therefore, such disordered or fuzzy complex can be envisioned as a highly dynamic ensemble in which a disordered protein does not present a single binding site to its partner but resemble a “binding cloud,” in which multiple identical binding sites are dynamically distributed in a diffuse manner. In other words, in this staccato-type interaction mode, an disordered protein rapidly changes multiple binding sites while probing binding site(s) of its partner.201 An additional factor which can help holding a dynamic complex together could be a weak long-range attraction between protein molecules.213 This long-range attraction is universal for all protein solutions and has a range several times that of the diameter of the protein molecule, much greater than the range of the screened electrostatic repulsion.213
Upside-Down Functionality: Functional Unfolding or Order–Disorder Transition
The most common outcome of these function-related structural changes is the overall increase in the amount of ordered structure. However, functions of some ordered proteins require local or even global unfolding of a unique protein structure.68 Among specific features of these structural alterations are their induced nature and transient character combined with a wide range of molecular mechanisms by which they can be promoted.68 These functional unfolding-activating factors include light; mechanical force; changes in pH, temperature, or redox potential; interaction with membrane, ligands, nucleic acids, and proteins; various PTMs; release of autoinhibition due to the unfolding of autoinhibitory domains induced by their interaction with nucleic acids, proteins, membranes, PTMs, and so forth.68 Among rather unusual factors used by nature to activate proteins via functional unfolding are light and mechanical force. For example, exposure to blue light results in the activation of the photoactive yellow protein (PYP), which is an ordered, water-soluble ∼14 kDa protein that contains a thioester linked p-coumaric acid cofactor and serves as a photosensor in Ectothiorhodospira halophila.214,215 PYP is a bacterial blue light sensor that undergoes conformational changes upon signal transduction. The absorption of a photon triggers substantial protein unfolding and leads to the formation of the transient signaling state that interacts with the partner molecules. This allows the swimming bacterium to operate the directional switch that protects it from harmful illumination. Comprehensive analysis combining double electron electron resonance spectroscopy (DEER), high resolution NMR, and time-resolved pump–probe X-ray solution scattering (TR-SAXS/WAXS) revealed that the transiently activated and short-lived signaling state of the PYP possessed a large degree of disorder and existed as an ensemble of multiple conformers that exchange on a millisecond time scale.216,217 This unusual behavior is illustrated in Figure 4 that shows structures of inactive folded PYP and its light-activated functional form, which is highly disordered.68 Some proteins undergo local unfolding induced by the mechanical force and therefore can serve as force sensors.68 Among these natural force sensors are mechanosensitive ion channels that recognize and respond to the membrane tension, which is the mechanical forces applied along the plane of the cell membrane, rather than to the hydrostatic pressure perpendicular to the membrane plane.220 These ion channels are activated via partial unfolding of some of their functional parts induced by membrane tension.221
Figure 4.
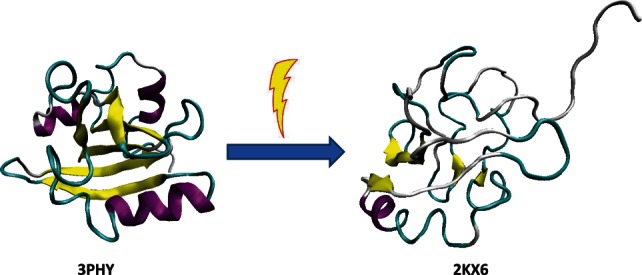
Comparison of the ground state (left structure, PDB ID: 3PHY) and the transient light activated signaling state of the PYP (right structure, PDB ID: 2KX6). Ground state structure was determined by multidimensional NMR spectroscopy.218 This structure is in agreement with an earlier published 1.4 Å crystal structure,219 and modeled structure based on combined DEER, TR-SAXS/WAXS, and NMR data.217 It consists of an open, twisted, 6-stranded, antiparallel β-sheet, which is flanked by four α-helices on both sides.217–219 On the contrary, the light-activated form is highly disordered. This structure satisfies DEER, SAXS/WAXS, and NMR data simultaneously.217
For a long time, the fact that IDPs/IDPRs undergo disorder-to-order transitions either during their functions or in order to be functional was used as one of the strongest arguments against the idea of protein intrinsic disorder. It was stated that most IDPs (those which are not the artifacts of current methods of protein production) are in fact proteins waiting for a partner (PWPs) that serve as parts of a multi-component complex and that do not fold correctly in the absence of other components.29 Therefore, when folded after binding to their partners, these proteins are not too different from typical ordered proteins. However, one need to keep in mind that a portion of “folding code” that defines the ability of ordered proteins to spontaneously gain a unique biologically active structure is missing for IDPs/IDPRs since they cannot fold spontaneously. This missing portion of the “folding code” (or a part of it) can be supplemented by binding partner(s). As a result, ordered and disordered proteins can be discriminated on a simple basis of temporal correlation between their folding and binding: ordered proteins fold first and then bind to their partners while the IDPs/IDPRs remain disordered until they bind their partners and often preserve substantial disorder in the bound state.69 Furthermore, numerous cases of functional unfolding (or transient disorder, or upside-down functionality) represent further support to the concept of functional disorder by clearly showing that many proteins possess dormant disorder that needs to be awakened in order to make these proteins functional.
Specific Surveillance for Special Control or Controlled Chaos
It is clear now that the IDPs and IDPRs are real, abundant, diversified, and vital. The highly dynamic nature of IDPs and IDPRs is a visual illustration of the chaos. However, the evolutionary persistence of these highly dynamic proteins (see below), their unique functionality, and involvement in all the major cellular processes evidence that this chaos is tightly controlled.147 To answer the question as to how these proteins are governed and regulated inside the cell, Gsponer et al. conducted a detailed study focused on the intricate mechanisms of IDP regulation.222 To this end, all the Saccharomyces cerevisiae proteins were grouped into three classes using one of the available disorder predictors, DisoPred244: (i) 1971 highly ordered proteins containing 0–10% of the predicted disorder; (ii) 2711 moderately disordered proteins with 10–30% predicted disordered residues; and (iii) 2020 highly disordered proteins containing 30–100% of the predicted disorder. Then, the correlations between intrinsic disorder and the various regulation steps of protein synthesis and degradation were evaluated.
This analysis revealed that the transcriptional rates of mRNAs encoding IDPs and ordered proteins were comparable. However the IDP-encoding transcripts were generally less abundant than transcripts encoding ordered proteins due to the increased decay rates of the transcripts of genes encoding IDPs.222 Furthermore, IDPs were shown to be less abundant than ordered proteins due to the lower rate of protein synthesis and shorter protein half-lives. As the abundance and half-life in a cell of certain proteins can be further modulated via their PTMs such as phosphorylation,223 the experimentally determined yeast kinase–substrate network was also analyzed. IDPs were shown to be substrates of twice as many kinases as were ordered proteins. Furthermore, the vast majority of kinases whose substrates were IDPs were either regulated in a cell-cycle dependent manner, or activated upon exposure to particular stimuli or stress.222 Therefore, PTMs may not only serve as important mechanism for the fine-tuning of the IDP functions but possibly they are necessary to tune the IDP availability under the different cellular conditions.222
In addition to S. cerevisiae, similar regulation trends were also found in Schizosaccharomyces pombe and Homo sapiens.222 Based on these observations it has been concluded that both unicellular and multicellular organisms appear to use similar mechanisms for regulation of the intrinsically disordered protein availability. Overall, this study clearly demonstrated that in eukaryotes, there is an evolutionarily conserved tight control of synthesis and clearance of most IDPs. This tight control is directly related to the major roles of IDPs in signaling, where it is crucial to be available in appropriate amounts and not to be present longer than needed.222 It has been also pointed out that although the abundance of many IDPs is under strict control, some IDPs could be present in cells in large amounts or/and for long periods of time due to either specific PTMs or via interactions with other factors, which could promote changes in cellular localization of IDPs or protect them from the degradation machinery.13,70,138,223,224 Overall, this study clearly showed that the chaos seemingly introduced into the protein world by the discovery of IDPs is under the tight control.147
In an independent study, a global scale relationship between the predicted fraction of protein disorder and protein expression in E. coli was analyzed.225 This study showed that the fraction of protein disorder was positively correlated with both measured RNA expression levels of E. coli genes in three different growth media and with predicted abundance levels of E. coli proteins.225 When a subset of 216 E. coli proteins that are known to be essential for the survival and growth of this bacterium were analyzed, the correlation between protein disorder and expression level became even more evident. In fact, essential proteins had on average a much higher fraction of disorder (0.36), had a higher number of proteins classified as completely disordered (19% vs. 2% for E. coli proteome), and were expressed at a higher level in all three media than an average E. coli gene.225 The manual literature mining for a group of E. coli proteins that had high levels of predicted intrinsic disorder revealed that the disorder predictions matched well with the experimentally elucidated regions of protein flexibility and disorder.225 A direct link between protein disorder and protein level in E. coli cells could be because the IDPs may carry out the essential control and regulation functions that are needed to respond to the various environmental conditions. Another possibility is that IDPs might undergo more rapid degradation compared to structured proteins, which cells can counter by increasing mRNA levels of the corresponding genes. In this case, higher synthesis and degradation rates could make the levels of these proteins very sensitive to the environment, with slight changes in either production or degradation leading to significant shifts in protein levels.225
Even more support for the tight control of IDPs inside the cell came from the analysis of cellular regulation of so-called “vulnerable” proteins.23 The integrity of the soluble protein functional structures is maintained in part by a precise network of hydrogen bonds linking the backbone amide and carbonyl groups. In a well-ordered protein, hydrogen bonds are shielded from water attack, preventing backbone hydration and the total or partial unfolding of the soluble structure under physiological conditions.226,227 Since soluble protein structures may be more or less vulnerable to water attack depending on their packing quality, a structural attribute, protein vulnerability, was introduced as the ratio of solvent-exposed backbone hydrogen bonds (which represent local weaknesses of the structure) to the overall number of hydrogen bonds.23 It has been also pointed out that structural vulnerability can be related to protein intrinsic disorder as the inability of a particular protein fold to protect intramolecular hydrogen bonds from water attack may result in backbone hydration leading to local or global unfolding. Since binding of a partner can help to exclude water molecules from the microenvironment of the preformed bonds, a vulnerable soluble structure gains extra protection of its backbone hydrogen bonds through the complex formation.226 To understand the role of structure vulnerability in transcriptome organization, the relationship between the structural vulnerability of a protein and the extent of co-expression of genes encoding its binding partners was analyzed. This study revealed that structural vulnerability can be considered as a determinant of transcriptome organization across tissues and temporal phases.23 Finally, by interrelating vulnerability, disorder propensity and co-expression patterns, the role of protein intrinsic disorder in transcriptome organization was confirmed, since the correlation between the extent of intrinsic disorder of the most disordered domain in an interacting pair and the expression correlation of the two genes encoding the respective interacting domains was evident.23
IDPs in Diseases
Because of the fact that IDPs are highly abundant and play crucial roles in numerous biological processes, it was not too surprising to find that some of them are involved in human diseases. For example, a number of human diseases originate from the deposition of stable, ordered, filamentous protein aggregates, commonly referred to as amyloid fibrils. In each of these pathological states, a specific protein or protein fragment changes from its natural soluble form into insoluble fibrils, which accumulate in a variety of organs and tissues.228–234 Several unrelated proteins including many IDPs are known to be involved in these protein deposition diseases.234,235 An illustrative examples of human neurodegenerative diseases associated with IDPs includes Alzheimer's disease (deposition of amyloid-β, tau-protein, α-synuclein fragment NAC)236–239; various taupathies (accumulation of tau-protein in the form of neurofibrillary tangles)238; Down's syndrome (nonfilamentous amyloid-β deposits)240; Parkinson's disease and other synucleinopathies (deposition of α-synuclein)241; prion diseases (deposition of PrPSC)242; and a family of polyQ diseases, a group of neurodegenerative disorders caused by expansion of GAC trinucleotide repeats coding for PolyQ in the gene products.243 Furthermore, most mutations in rigid globular proteins associated with accelerated fibrillation and protein deposition diseases have been shown to destabilize the native structure, increasing the steady-state concentration of partially folded (disordered) conformers.228–234
The maladies given above have been called conformational diseases, as they are characterized by the conformational changes, misfolding, and aggregation of an underlying protein. However, there is another side to this coin: protein functionality. In fact, many of the proteins associated with the conformational disorders are also involved in recognition, regulation, and cell signaling. For example, functions ascribed to α-synuclein, a protein involved in several neurodegenerative disorders, include binding fatty acids and metal ions; regulation of certain enzymes, transporters, and neurotransmitter vesicles; and regulation of neuronal survival (reviewed in Ref.241). Overall, there are about 50 proteins and ligands that interact and/or co-localize with this protein. Furthermore, α-synuclein has amazing structural plasticity and adopts a series of different monomeric, oligomeric, and insoluble conformations (reviewed in Ref.24). The choice between these conformations is determined by the peculiarities of the protein environment, suggesting that α-synuclein has an exceptional ability to fold in a template-dependent manner. Therefore, the development of the conformational diseases may originate not only from misfolding but also from the misidentification, misregulation, and missignaling of the related proteins.
Analysis of so-called polyglutamine diseases gives support to this hypothesis.244 Polyglutamine diseases are a specific group of hereditary neurodegeneration caused by expansion of CAG triplet repeats in an exon of disease genes which leads to the production of a disease protein containing an expanded polyglutamine, polyQ, stretch. Nine neurodegenerative disorders, including Kennedy's disease, Huntington's diseases, spinocerebellar atrophy-1, −2, −3, −6, 7, 17, and dentatorubral pallidoluysian atrophy are known to belong to this class of diseases.245–248 In most polyQ diseases, expansion to over 40 repeats leads to the onset.248 It has been emphasized that such molecular processes as unfolded protein response, protein transport, synaptic transmission, and transcription are implicated in the pathology of polyQ diseases.244 Importantly, more than 20 transcription-related factors have been reported to interact with pathological polyQ proteins. Furthermore, these interactions were shown to repress the transcription, leading finally to the neuronal dysfunction and death (reviewed in Ref.244). These results suggest that polyQ diseases represent kind of transcriptional disorder,244 supporting our misidentification hypothesis for at least some of the conformational disorders.
Disorder is very common in cancer-associated proteins too. In a 2002 study, it was found that 79% of cancer-associated and 66% of cell-signaling proteins contain predicted regions of disorder of 30 residues or longer.130 In contrast, only 13% of a set of proteins with well-defined ordered structures contained such long regions of predicted disorder.130 In experimental studies, the presence of disorder has been directly observed in several cancer-associated proteins, including p53,249 p57kip2,250 Bcl-XL and Bcl-2,251 c-Fos,252 a thyroid cancer associated protein TC-1,253 EWS-FLI1 fusion protein that includes a potent transcriptional activator, the EWS domain, alongside the highly conserved DNA-binding domain FLI1,254,255 among many other examples. The best characterized example of the important cancer-related IDP is the tumor suppressor protein p53, which occupies the center of a large signaling network. p53 regulates expression of genes involved in numerous cellular processes, including cell cycle progression, apoptosis induction, DNA repair, as well as others involved in responding to cellular stress.256 When p53 function is lost, either directly through mutation or indirectly through several other mechanisms, the cell often undergoes cancerous transformation.257,258 Cancers showing mutations in p53 are found in colon, lung, esophagus, breast, liver, brain, reticuloendothelial tissues, and hemopoietic tissues.257 p53 is regulated by several different mechanisms including inhibition of its activity by interaction with E3 ubiquitin ligase Mdm2, which binds to a short stretch of p53 located within the transactivation domain. Mdm2-bound p53 cannot activate or inhibit other genes. Mdm2 ubiquitinates p53 and thus targets it for destruction. Mdm2 also contains a nuclear export signal that causes p53 to be transported out of the nucleus.259,260
Druggability of IDPs
The possibility of interrupting the action of disease-associated proteins (including through modulation of protein–protein interactions) presents an extremely attractive objective for the development of new drugs. Since many proteins associated with various human diseases are either completely disordered or contain long disordered regions,261,262 and since some of these disease-related IDPs/IDPRs are involved in recognition, regulation, and signaling, these proteins/regions clearly represent novel potential drug targets.27 Due to failure to recognize the important role of disorder in protein function, current and evolving methods of drug discovery suffer from an overly rigid view of protein function. In fact, the rational design of enzyme inhibitors depends on the classical view where 3D-structure is an obligatory prerequisite for function. While generally applicable to many enzymatic domains, this view has persisted to influence thinking concerning all protein functions despite numerous examples to the contrary. This is most apparent in the observation that the vast majority of currently available drugs target the active site of enzymes, presumably since these are the only proteins for which the “unique structure–unique function” paradigm is generally applicable.
IDPs often bind their partners with relatively short regions that become ordered upon binding.263–265 Targeting disorder-based interactions should enable the development of more effective drug discovery techniques. There are at least two potential approaches for the inhibition of the disorder-based interactions, where small molecule either bind to the binding site of the ordered partner to outcompete the IDPs/IDPRs or interacts directly with the IDP/IDPR. The principles of small molecule binding to IDPRs have not been well studied, but sequence specific, small molecule binding to short peptides has been observed.266 An interesting twist here is that small molecules can inhibit disorder-based protein–protein interactions via induction of the dysfunctional ordered structures in targeted IDPR, that is, via the drug-induced misfolding.
In agreement with these concepts, small molecules “Nutlins” have been discovered that inhibited the p53-Mdm2 interaction by mimicking the inducible α-helix in p53 (residues 13–29) that binds to Mdm2.259,260 Although X-ray crystallographic studies of the p53-Mdm2 complex revealed that the Mdm2 binding region of p53 forms an α-helical structure that binds into a deep groove on the surface of Mdm2,267 NMR studies showed that the unbound N-terminal region of p53 lacks fixed structure, although it does possess an amphipathic helix part of the time.249 A close examination of the interface between the proteins reveals that Phe19, Trp23, and Leu26 of p53 are the major contributors to the interaction, with the side chains of these three amino acids pointing down into a crevice on the Mdm2 surface.259,260 The structure of Nutlin-2 was shown to mimic the crucial residues of p53, with two bromophenyl groups fitting into Mdm2 in the same pockets as Trp23 and Leu26, and an ethyl-ether side chain filling the spot normally taken by Phe19.268–270 Nutlins and related small molecules increased the level of p53 in cancer cell lines. This drastically decreased the viability of these cells, causing most of them to undergo apoptosis. When one of the nutlins was given orally to mice, a 90% inhibition of tumor growth compared to the control was induced.260,268–270
This successful nutlin story marks the potential beginning of a new era, the signaling-modulation era, in targeting drugs to protein–protein interactions. Importantly, this druggable p53-Mdm2 interaction involves a disorder-to-order transition. Principles of such transitions are generally understood and therefore can use to find similar drug targets, which are inducible α-helices.271 In addition to nutlins inhibiting p53-Mdm2 interaction, several other small molecules also act by blocking protein–protein interactions.272,273 Some of these interactions involve one structured partner and one disordered partner, with disordered segments becoming α-helix upon binding.271 Therefore, the p53–Mdm2 complex is not a unique exception and many other disorder-based protein–protein interactions are blocked by a small molecule. All this suggest that there is a cornucopia of new drug targets that would operate by blocking disorder-based protein–protein interactions.
For these p53–Mdm2-type examples, the drug molecules mimic a critical region of the disordered partner (which folds upon binding) and compete with this region for its binding site on the structured partner. These druggable interaction sites operate by the coupled binding and folding mechanism. They are small enough and compact enough to be easily mimicked by small molecules.25 Methods for predicting such binding sites in disordered regions have been developed274 and the bioinformatics tools to identify which disordered binding regions can be easily mimicked by small molecules have been elaborated.271
A complementary approach for small molecules to inhibit the disorder-based protein–protein interactions relies on the direct binding of drugs to the IDPs/IDPRs, which is illustrated by the c-Myc-Max story.275 In order to bind DNA, regulate expression of target genes, and function in most biological contexts, c-Myc transcription factor must dimerize with its obligate heterodimerization partner, Max, which lacks a transactivation segment. Both c-Myc and Max are intrinsically disordered in their monomeric forms. Upon heterodimerization, they undergo coupled binding and folding of their basic-helix-loop-helix-leucine zipper domains (bHLHZips). Since the deregulation of c-Myc is related to many types of cancer, the disruption of the c-Myc–Max dimeric complex is one of the approaches for c-Myc inhibition. Several small molecules were found to inhibit the c-Myc–Max dimer formation.275 These molecules were shown to bind to one of the three discrete sites within the 85-residue bHLHZip domain of c-Myc, which are composed of short contiguous stretches of amino acids that can selectively and independently bind small molecules.275 Inhibitor binding induces only local conformational changes, preserves the overall disorder of c-Myc, and inhibits interaction with Max.275 Furthermore, binding of inhibitors to c-Myc was shown to occur simultaneously and independently on the three independent sites. Based on these observations it has been concluded that a rational and generic approach to the inhibition of protein–protein interactions involving IDPs may therefore be possible through the targeting of intrinsically disordered sequence.275
Recently, a functional misfolding concept was introduced to describe a mechanism preventing IDPs from unwanted interactions with non-native partners.276 IDPs/IDPRs are characterized by high conformational dynamics and flexibility, the presence of sticky preformed binding elements, and the ability to morph into differently-shaped bound configurations. However, detailed analyses of the conformational behavior and fine structure of several IDPs revealed that the preformed binding elements might be involved in a set of non-native intramolecular interactions. Based on these observations it was proposed that an intrinsically disordered polypeptide chain in its unbound state can be misfolded to sequester the preformed elements inside the non-interactive or less-interactive cage, therefore preventing these elements from the unnecessary and unwanted interactions with non-native binding partners.276 It is important to remember, however, that the mentioned functional misfolding is related to the ensemble behavior of transiently populated elements of structure. In other words, it describes the behavior of a globally disordered polypeptide chain containing highly dynamic elements of residual structure, so-called interaction-prone preformed fragments, some of which could potentially be related to protein function.276
This ability of IDRPs/IDPRs to functionally misfold can be used for finding small molecules which would potentially stabilize different members of the functionally misfolded ensemble, and therefore prevent the targeted protein from establishing biological interactions.277 This approach is very different from the discussed above direct targeting of short IDPRs since it is based on a small molecule binding to a highly dynamic surface created via the transient interaction of preformed interaction-prone fragments. In essence, this approach can be considered as an extension of the well-established structure-based rational drug design elaborated for ordered proteins. In fact, if the structure of a member(s) of the functionally misfolded ensemble can be guessed, then this structure can be used to find small molecules that are potentially able to interact with this structure, utilizing tools originally developed for the rational structure-based drug design for ordered proteins.277
Ideally, a drug that targets a given protein–protein interaction should be tissue specific. Although some proteins are unique for a given tissue, many more proteins have very wide distribution, being present in several tissues and organs. How can one develop tissue-specific drugs targeting such abundant proteins? Often, tissue specificity for many of the abundant proteins is achieved via the alternative splicing of the corresponding pre-mRNAs, which generates two or more protein isoforms from a single gene. Estimates indicate that between 35 and 60% of human genes yield protein isoforms by means of alternatively spliced mRNA.278 The added protein diversity from alternative splicing is thought to be important for tissue-specific signaling and regulatory networks in the multicellular organisms. The regions of alternative splicing in proteins are enriched in intrinsic disorder, and it was proposed that associating alternative splicing with protein disorder enables the time- and tissue-specific modulation of protein function.152 Since disorder is frequently utilized in protein binding regions, having alternative splicing of pre-mRNA coupled to IDPRs can define tissue-specific signaling and regulatory diversity.152 These findings open a unique opportunity to develop tissue-specific drugs modulating the function of a given ID protein/region (with a unique profile of disorder distribution) in a target tissue and not affecting the functionality of this same protein (with different disorder distribution profile) in other tissues.
Evolution of Intrinsically Disordered Proteins
Wavy pattern of global evolution of intrinsic disorder
IDPs/IDPRs are more common in eukaryotes than in less complex organisms.43,44,48–52 This suggests that disorder, with its ability to be implemented in various signaling, recognition, and regulation pathways and networks, is important for the maintenance of life in eukaryotic and especially muticellular eukaryotic organisms.4,45,78,134 Also, the finding that alternatively spliced regions of mRNA code for IDPRs much more often than for structured regions suggests that there is a linkage between alternative splicing and signaling by IDPRs that constitutes a plausible mechanism that could underlie and support cell differentiation, which ultimately gave rise to the multicellular eukaryotic organisms.152
Therefore, one can assume that intrinsic disorder represents a relatively recent evolutionary invention. However, this hypothesis obviously would be wrong if earlier stages of evolution would be taken into account. In fact, the chances that the first polypeptides that appeared in the primordial soup of the primitive Earth possessed well-developed and unique 3D structures are minimal. The Earth formed about 4.5 billion years ago. Scientists dated the first fossils to 3.85 billion years ago. There are still debates and different theories about what happened in those years between the time the earth was cool enough to spawn life and the time the first fossils were formed. At the beginning of the 20th century, Oparin279 and Haldane280 proposed that some organic molecules could have been spontaneously produced from the gases of the primitive Earth atmosphere, assuming that this primitive atmosphere was reducing (as opposed to oxygen-rich), and there was an appropriate supply of energy, such as lightning or ultraviolet light. Thirty year later, this hypothesis (that constitutes a cornerstone of the theory of molecular evolution) received strong support from the elegant experiments of Stanley L. Miller and Harold C. Urey who were able to synthesize various organic compounds including some amino acids from non-organic compounds which were believed to represent the major components of the early Earth's atmosphere (water vapor, hydrogen, methane, and ammonia) by putting them into a closed system and running a continuous electric current through the system, to simulate lightning storms believed to be common on the early Earth.281,282 However, the Miller–Urey experiment yielded only about half of the modern amino acids281,282 suggesting that the first proteins on Earth may have contained only a few amino acids.
These findings go in parallel with the biosynthetic theory of the genetic code evolution suggesting that the genetic code evolved from a simpler form that encoded fewer amino acids,283 probably paralleled by the invention of biosynthetic pathways for new and chemically more complex amino acids.284 Furthermore, some additional support of the validity of this hypothesis can be found in the standard genetic code (that consists of 4 × 4 × 4 = 64 triplets of nucleotides, codons), which is redundant (64 codons encodes for 20 amino acids). In fact, with only two exceptions, codons encoding one amino acid may differ in any of their three positions. However, only the third positions of some codons may be fourfold degenerate, that is, any nucleotide at this position specifies the same amino acid and all nucleotide substitutions at this site are synonymous. Using these observations as a reflection of the evolutionary development, it was proposed that there was a period during code evolution where the third position was not needed at all and a doublet code preceded the triplet code, giving rise to 4 × 4 = 16 codons encoding for 16 or fewer amino acids, if a termination codon is taken into account.285
Based on these and many other premises, one can discriminate evolutionary old and new amino acids. In 2000, Eduard N. Trifonov combined 40 different single-factor criteria into a consensus scale and proposed the following temporal order of addition for the amino acids: G/A, V/D, P, S, E/L, T, R, N, K, Q, I, C, H, F, M, Y, W.286 Even superficial analysis of this sequence reveals that many of the early amino acids (such as G, D, E, P, and S) are disorder-promoting, as they are very abundant in modern IDPs. On the other hand, the major order-promoting residues (C, W, Y, and F) were added to the genetic code late. This observation is further illustrated by Figure 5(A) which represents modern genetic code, contains information on the early and late codons (shown by light red and light blue colors, respectively), and on corresponding disorder- and order-promoting residues (shown by red and blue colors, respectively). Codons with intermediate age and disorder-neutral residues are shown by light pink and pink colors, respectively. Figure 5(A) illustrates that there is relatively good agreement between the “age” of the residue and its disorder-promoting capacity, with early residues being mostly disorder-promoting, and with the majority of late residues being mostly order-promoting. This conclusion follows from the abundance of the matching colors (light red-red, light blue-blue, and light pink-pink). There are only two noticeable exceptions from these rule, valine and leucine, which are early but order-promoting residues. This strongly suggests that the primordial polypeptides were intrinsically disordered.
Figure 5.
Peculiarities of disorder evolution. A: Modern genetic code with information on the early and late codons (shown by light red and light blue colors, respectively) and disorder- and order-promoting residues (shown by red and blue colors, respectively). Codons with intermediate ages (i.e., those located between early and late codons) are shown by light pink color, whereas disorder-neutral residues are shown by pink color. B: Wavy pattern of the global disorder evolution. X-axis represents evolutionary time and Y-axis shows disorder content in proteins at given evolutionary time point. Here, primordial proteins are expected to be mostly disordered (left-hand side of the plot), proteins in LUA likely are mostly structured (center of the plot), whereas many protein in eukaryotes are either totally disordered or hybrids containing both ordered and disordered regions (right-hand side of the plot).
It is very unlikely that these disordered primordial polypeptides possessed catalytic activity.287 This hypothesis is in line with the RNA world theory suggesting that during the evolution of enzymatic activity, catalysis was transferred from RNA first to ribonucleoprotein (RNP) and only then to protein.288 Therefore, the first proteins in the “breakthrough organism” (the first to have encoded protein synthesis) would be nonspecific chaperone-like proteins rather than catalysts.136,287 Such RNA chaperone activities of early proteins conferred to their carriers a significant selective advantage in the RNA world, where RNA, which is especially prone to misfolding,289,290 was used for both information storage and catalysis.291 Since the variability of physicochemical properties of amino acids greatly exceeds that of nucleotides and since protein structures are noticeably more stable than RNA structures, the transition from RNAs (ribozymes) to proteins as carriers of enzymatic activity was a logical evolutionary step. However, efficient catalysis relies on the proper spatial arrangement of catalytic residues which requires a stable structure.292 Therefore, grafting of the enzymatic activity to proteins generated strong evolutionary pressure toward the well-folded structures.
In other words, the global evolution of intrinsic disorder is characterized by a wavy pattern [see Fig. 5(B)], where highly disordered primordial proteins with primarily RNA-chaperone activities were gradually substituted by the well-folded, highly ordered enzymes that evolved to catalyze the production of all the complex “goodies” crucial for the independent existence of the first cellular organisms. Due to its specific features crucial for the regulation of complex processes, protein intrinsic disorder was reinvented at the subsequent evolutionary steps leading to the development of more complex organisms from the last universal ancestor (i.e., the most recent organism from which all organisms now living on Earth descend293,294), and culminating in the appearance of the highly elaborated eukaryotic cells [see Fig. 5(B)].
Diversified evolution of IDPs in modern organisms
There is no simple answer to the question on the comparative evolutionary rates of ordered and IDPs and regions in modern organisms. In fact, it looks like everything is possible, and intrinsically disordered sequences may evolve faster, slower or similar to ordered sequences. For example, disordered and ordered domains of the same protein (e.g., papillomavirus E7 oncoprotein) were shown to possess similar degrees of conservation and co-evolution.295 Many other IDPs/IDPRs were shown to be characterized by high evolutionary rates151,296,297 determined by the lack of specific structural restrictions. In fact, the analysis of calcineurins,10 topoisomerase,298 ribosomal protein S4,299 β-subunits of the potassium channel Kvβ1.1,300 and many other proteins showed that disordered regions in these proteins contained more amino acid substitutions, insertions, and deletions than the ordered regions of the same proteins.151,301 Furthermore, based on the observation that a significantly higher degree of positive Darwinian selection was observed in IDPRs of proteins compared to regions of α-helix, β-sheet or tertiary structures, it was hypothesized that IDPRs may be required for the genetic variation with adaptive potential and that these regions may be of “central significance for the evolvability of the organism or cell in which they occur.”302
On the other hand, some IDPs and IDPRs are highly conserved. Human α-synuclein (a canonical IDP of 140 residues140,303) differs from its mouse counterpart by merely six residues (4%), and there are just 21 residue differences (12%, which include residue differences at 18 positions and 3 insertions/deletions) between the human and canary α-synucleins.304 In flagellin, the ordered central region has greater sequence diversity than its disordered termini.305 Functionally important conserved regions of predicted disorder were shown to be rather common in proteins from all kingdoms of life, including viruses.306,307 Furthermore, many functional domains of a significant size were shown to be intrinsically disordered.165
Overall, a systematic study of several families of proteins with at least one structurally characterized disordered region revealed that their IDPRs are characterized by highly heterogeneous evolutionary rates, with some disordered amino acid sequences evolving slowly, and others evolving more rapidly than ordered sequences.151 Also, even different parts of the same disordered region can possess noticeable variability in their divergence during the evolutionary process.308 Finally, in some disordered proteins, peculiarities of the amino acid composition, and not the amino acid sequence might be conserved.309,310
Some Future Directions
The last 15 years witnessed a real revolution in our understanding of the protein structure–function relationships. The fact that there is an entire class of polypeptides which do not have rigid structures but possess crucial biological function was heavily underappreciated and ignored for a very long time despite numerous examples scattered in literature. The work which started in my group as an attempt to understand what is so special about several natively unfolded proteins produced a real explosion of interest to structure-less proteins with biological functions. A new field was created and a lot of intriguing information was produced related to structures and functions of IDPs/IDPRs. There is no need to list once again all the discoveries and findings made in this field—they are subjects of many recent reviews and some of them are briefly covered in this article. Although the amount of data generated during the past decade and a half on specific features related to the structural properties of IDPs and IDPRs, their abundance, distribution, functional repertoire, regulation, involvement into the disease pathogenesis, and so forth is vast, it seems that this mass of data produced so far is just a small tip of a humongous iceberg. IDPs/IDPRs continue to bring discoveries almost on a daily basis and even more breakthroughs are expected in future. Modern protein science is at the turning point, but biology still waits for physics. New models explaining various functions of IDPs, their evolution, and involvement in diseases are in great demand, together with the general theory unifying current knowledge on protein structure and function, and with novel experimental and computational tools for focused studies of IDPs/IDPRs.
Acknowledgments
This work would be impossible without numerous collaborators whose enthusiasm and help drove the studies on intrinsically disordered proteins for many years. Over the years I collaborated with more than 550 colleagues around the globe, and I am grateful to all former and current colleagues for their priceless contributions, assistance and support.
Supplementary material
Additional Supporting Information may be found in the online version of this article.
References
- 1.Uversky VN. Natively unfolded proteins: a point where biology waits for physics. Protein Sci. 2002;11:739–756. doi: 10.1110/ps.4210102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fischer E. Einfluss der configuration auf die wirkung der enzyme. Ber Dt Chem Ges. 1894;27:2985–2993. [Google Scholar]
- 3.Bernstein FC, Koetzle TF, Williams GJ, Meyer EF, Jr, Brice MD, Rodgers JR, Kennard O, Shimanouchi T, Tasumi M. The Protein Data Bank: a computer-based archival file for macromolecular structures. J Mol Biol. 1977;112:535–542. doi: 10.1016/s0022-2836(77)80200-3. [DOI] [PubMed] [Google Scholar]
- 4.Uversky VN, Dunker AK. Understanding protein non-folding. Biochim Biophys Acta. 2010;1804:1231–1264. doi: 10.1016/j.bbapap.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kuhn TS. The structure of scientific revolutions. Chicago: University Of Chicago Press; 2012. [Google Scholar]
- 6.Petsko GA, Ringe D. Primers in biology. Protein structure and function. London: New Science Press Ltd., Sinauer Associates, Inc. Publishers, Blackwell Publishing; 2004. [Google Scholar]
- 7.Bloomer AC, Champness JN, Bricogne G, Staden R, Klug A. Protein disk of tobacco mosaic virus at 2.8 A resolution showing the interactions within and between subunits. Nature. 1978;276:362–368. doi: 10.1038/276362a0. [DOI] [PubMed] [Google Scholar]
- 8.Bode W, Schwager P, Huber R. The transition of bovine trypsinogen to a trypsin-like state upon strong ligand binding. The refined crystal structures of the bovine trypsinogen-pancreatic trypsin inhibitor complex and of its ternary complex with Ile-Val at 1.9 A resolution. J Mol Biol. 1978;118:99–112. doi: 10.1016/0022-2836(78)90246-2. [DOI] [PubMed] [Google Scholar]
- 9.Le Gall T, Romero PR, Cortese MS, Uversky VN, Dunker AK. Intrinsic disorder in the Protein Data Bank. J Biomol Struct Dyn. 2007;24:325–342. doi: 10.1080/07391102.2007.10507123. [DOI] [PubMed] [Google Scholar]
- 10.Dunker AK, Garner E, Guilliot S, Romero P, Albrecht K, Hart J, Obradovic Z, Kissinger C, Villafranca JE. Protein disorder and the evolution of molecular recognition: theory, predictions and observations. Pac Symp Biocomput. 1998:473–484. [PubMed] [Google Scholar]
- 11.Wright PE, Dyson HJ. Intrinsically unstructured proteins: re-assessing the protein structure–function paradigm. J Mol Biol. 1999;293:321–331. doi: 10.1006/jmbi.1999.3110. [DOI] [PubMed] [Google Scholar]
- 12.Uversky VN, Gillespie JR, Fink AL. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins. 2000;41:415–427. doi: 10.1002/1097-0134(20001115)41:3<415::aid-prot130>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 13.Dunker AK, Lawson JD, Brown CJ, Williams RM, Romero P, Oh JS, Oldfield CJ, Campen AM, Ratliff CM, Hipps KW, Ausio J, Nissen MS, Reeves R, Kang C, Kissinger CR, Bailey RW, Griswold MD, Chiu W, Garner EC, Obradovic Z. Intrinsically disordered protein. J Mol Graph Model. 2001;19:26–59. doi: 10.1016/s1093-3263(00)00138-8. [DOI] [PubMed] [Google Scholar]
- 14.Tompa P. Intrinsically unstructured proteins. Trends Biochem Sci. 2002;27:527–533. doi: 10.1016/s0968-0004(02)02169-2. [DOI] [PubMed] [Google Scholar]
- 15.Daughdrill GW, Pielak GJ, Uversky VN, Cortese MS, Dunker AK. Natively disordered proteins. In: Buchner J, Kiefhaber T, editors. Handbook of protein folding. Weinheim, Germany: Wiley-VCH, Verlag GmbH & Co. KGaA; 2005. pp. 271–353. [Google Scholar]
- 16.Dunker AK, Babu MM, Barbar E, Blackledge M, Bondos SE, Dosztányi Z, Dyson HJ, Forman-Kay J, Fuxreiter M, Gsponer J, Han K-H, Jones DT, Longhi S, Metallo SJ, Nishikawa K, Nussinov R, Obradovic Z, Pappu R, Rost B, Selenko P, Subramaniam V, Sussman JL, Tompa P, Uversky VN. 2013. What's in a name? Why these proteins are intrinsically disordered. Intrinsically Disordered Proteins 1:e24157. [DOI] [PMC free article] [PubMed]
- 17.Holt C, Sawyer L. Caseins as rheomorphic proteins: interpretation of primary and secondary structures of the as1-, b-, and k-caseins. J Chem Soc Faraday Trans. 1993;89:2683–2692. [Google Scholar]
- 18.Pullen RA, Jenkins JA, Tickle IJ, Wood SP, Blundell TL. The relation of polypeptide hormone structure and flexibility to receptor binding: the relevance of X-ray studies on insulins, glucagon and human placental lactogen. Mol Cell Biochem. 1975;8:5–20. doi: 10.1007/BF01731645. [DOI] [PubMed] [Google Scholar]
- 19.Cary PD, Moss T, Bradbury EM. High-resolution proton-magnetic-resonance studies of chromatin core particles. Eur J Biochem. 1978;89:475–482. doi: 10.1111/j.1432-1033.1978.tb12551.x. [DOI] [PubMed] [Google Scholar]
- 20.Linderstrom-Lang K, Schellman JA. In: Protein structure and enzyme activity. Boyer PD, Lardy H, Myrback K, editors. The enzymes. New York: Academic Press; 1959. pp. 443–510. [Google Scholar]
- 21.Schweers O, Schonbrunn-Hanebeck E, Marx A, Mandelkow E. Structural studies of tau protein and Alzheimer paired helical filaments show no evidence for beta-structure. J Biol Chem. 1994;269:24290–24297. [PubMed] [Google Scholar]
- 22.Weinreb PH, Zhen W, Poon AW, Conway KA, Lansbury PT., Jr NACP, a protein implicated in Alzheimer's disease and learning, is natively unfolded. Biochemistry. 1996;35:13709–13715. doi: 10.1021/bi961799n. [DOI] [PubMed] [Google Scholar]
- 23.Chen J, Liang H, Fernandez A. Protein structure protection commits gene expression patterns. Genome Biol. 2008;9:R107. doi: 10.1186/gb-2008-9-7-r107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Uversky VN. A protein-chameleon: conformational plasticity of alpha-synuclein, a disordered protein involved in neurodegenerative disorders. J Biomol Struct Dyn. 2003;21:211–234. doi: 10.1080/07391102.2003.10506918. [DOI] [PubMed] [Google Scholar]
- 25.Fuxreiter M, Tompa P, Simon I, Uversky VN, Hansen JC, Asturias FJ. Malleable machines take shape in eukaryotic transcriptional regulation. Nat Chem Biol. 2008;4:728–737. doi: 10.1038/nchembio.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tsvetkov P, Asher G, Paz A, Reuven N, Sussman JL, Silman I, Shaul Y. Operational definition of intrinsically unstructured protein sequences based on susceptibility to the 20S proteasome. Proteins. 2008;70:1357–1366. doi: 10.1002/prot.21614. [DOI] [PubMed] [Google Scholar]
- 27.Dunker AK, Uversky VN. Drugs for ‘protein clouds’: targeting intrinsically disordered transcription factors. Curr Opin Pharmacol. 2010;10:782–788. doi: 10.1016/j.coph.2010.09.005. [DOI] [PubMed] [Google Scholar]
- 28.Livesay DR. Protein dynamics: dancing on an ever-changing free energy stage. Curr Opin Pharmacol. 2010;10:706–708. doi: 10.1016/j.coph.2010.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Janin J, Sternberg MJE. Protein flexibility, not disorder, is intrinsic to molecular recognition. F1000 Biol Rep. 2013;5:2. doi: 10.3410/B5-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Campen A, Williams RM, Brown CJ, Meng J, Uversky VN, Dunker AK. TOP-IDP-scale: a new amino acid scale measuring propensity for intrinsic disorder. Protein Pept Lett. 2008;15:956–963. doi: 10.2174/092986608785849164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Radivojac P, Iakoucheva LM, Oldfield CJ, Obradovic Z, Uversky VN, Dunker AK. Intrinsic disorder and functional proteomics. Biophys J. 2007;92:1439–1456. doi: 10.1529/biophysj.106.094045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Romero P, Obradovic Z, Li X, Garner EC, Brown CJ, Dunker AK. Sequence complexity of disordered protein. Proteins. 2001;42:38–48. doi: 10.1002/1097-0134(20010101)42:1<38::aid-prot50>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 33.Garner E, Cannon P, Romero P, Obradovic Z, Dunker AK. Predicting disordered regions from amino acid sequence: common themes despite differing structural characterization. Genome Inform Ser Workshop Genome Inform. 1998;9:201–213. [PubMed] [Google Scholar]
- 34.Williams RM, Obradovi Z, Mathura V, Braun W, Garner EC, Young J, Takayama S, Brown CJ, Dunker AK. The protein non-folding problem: amino acid determinants of intrinsic order and disorder. Pac Symp Biocomput. 2001:89–100. doi: 10.1142/9789814447362_0010. [DOI] [PubMed] [Google Scholar]
- 35.Vacic V, Uversky VN, Dunker AK, Lonardi S. Composition Profiler: a tool for discovery and visualization of amino acid composition differences. BMC Bioinformatics. 2007;8:211. doi: 10.1186/1471-2105-8-211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li X, Obradovic Z, Brown CJ, Garner EC, Dunker AK. Comparing predictors of disordered protein. Genome Inform Ser Workshop Genome Inform. 2000;11:172–184. [PubMed] [Google Scholar]
- 37.Ferron F, Longhi S, Canard B, Karlin D. A practical overview of protein disorder prediction methods. Proteins. 2006;65:1–14. doi: 10.1002/prot.21075. [DOI] [PubMed] [Google Scholar]
- 38.Bourhis JM, Canard B, Longhi S. Predicting protein disorder and induced folding: from theoretical principles to practical applications. Curr Protein Pept Sci. 2007;8:135–149. doi: 10.2174/138920307780363451. [DOI] [PubMed] [Google Scholar]
- 39.Dosztanyi Z, Sandor M, Tompa P, Simon I. Prediction of protein disorder at the domain level. Curr Protein Pept Sci. 2007;8:161–171. doi: 10.2174/138920307780363406. [DOI] [PubMed] [Google Scholar]
- 40.Dosztanyi Z, Tompa P. Prediction of protein disorder. Methods Mol Biol. 2008;426:103–115. doi: 10.1007/978-1-60327-058-8_6. [DOI] [PubMed] [Google Scholar]
- 41.He B, Wang K, Liu Y, Xue B, Uversky VN, Dunker AK. Predicting intrinsic disorder in proteins: an overview. Cell Res. 2009;19:929–949. doi: 10.1038/cr.2009.87. [DOI] [PubMed] [Google Scholar]
- 42.Jin F, Liu Z. Inherent relationships among different biophysical prediction methods for intrinsically disordered proteins. Biophys J. 2013;104:488–495. doi: 10.1016/j.bpj.2012.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dunker AK, Obradovic Z, Romero P, Garner EC, Brown CJ. Intrinsic protein disorder in complete genomes. Genome Inform Ser Workshop Genome Inform. 2000;11:161–171. [PubMed] [Google Scholar]
- 44.Ward JJ, Sodhi JS, McGuffin LJ, Buxton BF, Jones DT. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J Mol Biol. 2004;337:635–645. doi: 10.1016/j.jmb.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 45.Uversky VN. The mysterious unfoldome: structureless, underappreciated, yet vital part of any given proteome. J Biomed Biotechnol. 2010;2010:568068. doi: 10.1155/2010/568068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xue B, Dunker AK, Uversky VN. Orderly order in protein intrinsic disorder distribution: disorder in 3500 proteomes from viruses and the three domains of life. J Biomol Struct Dyn. 2012;30:137–149. doi: 10.1080/07391102.2012.675145. [DOI] [PubMed] [Google Scholar]
- 47.Romero P, Obradovic Z, Kissinger CR, Villafranca JE, Garner E, Guilliot S, Dunker AK. Thousands of proteins likely to have long disordered regions. Pac Symp Biocomput. 1998:437–448. [PubMed] [Google Scholar]
- 48.Feng ZP, Zhang X, Han P, Arora N, Anders RF, Norton RS. Abundance of intrinsically unstructured proteins in P. falciparum and other apicomplexan parasite proteomes. Mol Biochem Parasitol. 2006;150:256–267. doi: 10.1016/j.molbiopara.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 49.Tompa P, Dosztanyi Z, Simon I. Prevalent structural disorder in E. coli and S. cerevisiae proteomes. J Proteome Res. 2006;5:1996–2000. doi: 10.1021/pr0600881. [DOI] [PubMed] [Google Scholar]
- 50.Galea CA, High AA, Obenauer JC, Mishra A, Park CG, Punta M, Schlessinger A, Ma J, Rost B, Slaughter CA, Kriwacki RW. Large-scale analysis of thermostable, mammalian proteins provides insights into the intrinsically disordered proteome. J Proteome Res. 2009;8:211–226. doi: 10.1021/pr800308v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xue B, Williams RW, Oldfield CJ, Dunker AK, Uversky VN. Archaic chaos: intrinsically disordered proteins in Archaea. BMC Syst Biol 4 (Suppl. 2010;1):S1. doi: 10.1186/1752-0509-4-S1-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Burra PV, Kalmar L, Tompa P. Reduction in structural disorder and functional complexity in the thermal adaptation of prokaryotes. PLoS One. 2010;5:e12069. doi: 10.1371/journal.pone.0012069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mohan A, Sullivan WJ, Jr, Radivojac P, Dunker AK, Uversky VN. Intrinsic disorder in pathogenic and non-pathogenic microbes: discovering and analyzing the unfoldomes of early-branching eukaryotes. Mol Biosyst. 2008;4:328–340. doi: 10.1039/b719168e. [DOI] [PubMed] [Google Scholar]
- 54.Dryden DT, Thomson AR, White JH. How much of protein sequence space has been explored by life on Earth? J R Soc Interface. 2008;5:953–956. doi: 10.1098/rsif.2008.0085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wathen B, Jia Z. Folding by numbers: primary sequence statistics and their use in studying protein folding. Int J Mol Sci. 2009;10:1567–1589. doi: 10.3390/ijms10041567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lau KF, Dill KA. Theory for protein mutability and biogenesis. Proc Natl Acad Sci USA. 1990;87:638–642. doi: 10.1073/pnas.87.2.638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chan HS, Dill KA. Polymer principles in protein structure and stability. Annu Rev Biophys Biophys Chem. 1991;20:447–490. doi: 10.1146/annurev.bb.20.060191.002311. [DOI] [PubMed] [Google Scholar]
- 58.Cordes MH, Davidson AR, Sauer RT. Sequence space, folding and protein design. Curr Opin Struct Biol. 1996;6:3–10. doi: 10.1016/s0959-440x(96)80088-1. [DOI] [PubMed] [Google Scholar]
- 59.Riddle DS, Santiago JV, Bray-Hall ST, Doshi N, Grantcharova VP, Yi Q, Baker D. Functional rapidly folding proteins from simplified amino acid sequences. Nat Struct Biol. 1997;4:805–809. doi: 10.1038/nsb1097-805. [DOI] [PubMed] [Google Scholar]
- 60.Plaxco KW, Riddle DS, Grantcharova V, Baker D. Simplified proteins: minimalist solutions to the ‘protein folding problem’. Curr Opin Struct Biol. 1998;8:80–85. doi: 10.1016/s0959-440x(98)80013-4. [DOI] [PubMed] [Google Scholar]
- 61.Larson SM, England JL, Desjarlais JR, Pande VS. Thoroughly sampling sequence space: large-scale protein design of structural ensembles. Protein Sci. 2002;11:2804–2813. doi: 10.1110/ps.0203902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Guo HH, Choe J, Loeb LA. Protein tolerance to random amino acid change. Proc Natl Acad Sci USA. 2004;101:9205–9210. doi: 10.1073/pnas.0403255101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Doi N, Kakukawa K, Oishi Y, Yanagawa H. High solubility of random-sequence proteins consisting of five kinds of primitive amino acids. Protein Eng Des Sel. 2005;18:279–284. doi: 10.1093/protein/gzi034. [DOI] [PubMed] [Google Scholar]
- 64.White SH, Jacobs RE. Statistical distribution of hydrophobic residues along the length of protein chains. Implications for protein folding and evolution. Biophys J. 1990;57:911–921. doi: 10.1016/S0006-3495(90)82611-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Murphy LR, Wallqvist A, Levy RM. Simplified amino acid alphabets for protein fold recognition and implications for folding. Protein Eng. 2000;13:149–152. doi: 10.1093/protein/13.3.149. [DOI] [PubMed] [Google Scholar]
- 66.Etchebest C, Benros C, Bornot A, Camproux AC, de Brevern AG. A reduced amino acid alphabet for understanding and designing protein adaptation to mutation. Eur Biophys J. 2007;36:1059–1069. doi: 10.1007/s00249-007-0188-5. [DOI] [PubMed] [Google Scholar]
- 67.Wang J, Wang W. A computational approach to simplifying the protein folding alphabet. Nat Struct Biol. 1999;6:1033–1038. doi: 10.1038/14918. [DOI] [PubMed] [Google Scholar]
- 68.Uversky VN. Unusual biophysics of intrinsically disordered proteins. Biochim Biophys Acta. 2013;1834:932–951. doi: 10.1016/j.bbapap.2012.12.008. [DOI] [PubMed] [Google Scholar]
- 69.Uversky VN, Dunker AK. The case for intrinsically disordered proteins playing contributory roles in molecular recognition without a stable 3D structure. F1000 Biol Rep. 2013;5:1. doi: 10.3410/B5-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tompa P. The interplay between structure and function in intrinsically unstructured proteins. FEBS Lett. 2005;579:3346–3354. doi: 10.1016/j.febslet.2005.03.072. [DOI] [PubMed] [Google Scholar]
- 71.Dunker AK, Brown CJ, Obradovic Z. Identification and functions of usefully disordered proteins. Adv Protein Chem. 2002;62:25–49. doi: 10.1016/s0065-3233(02)62004-2. [DOI] [PubMed] [Google Scholar]
- 72.Dunker AK, Brown CJ, Lawson JD, Iakoucheva LM, Obradovic Z. Intrinsic disorder and protein function. Biochemistry. 2002;41:6573–6582. doi: 10.1021/bi012159+. [DOI] [PubMed] [Google Scholar]
- 73.Uversky VN. What does it mean to be natively unfolded? Eur J Biochem. 2002;269:2–12. doi: 10.1046/j.0014-2956.2001.02649.x. [DOI] [PubMed] [Google Scholar]
- 74.Fuxreiter M, Tompa P. Fuzzy complexes: a more stochastic view of protein function. Adv Exp Med Biol. 2012;725:1–14. doi: 10.1007/978-1-4614-0659-4_1. [DOI] [PubMed] [Google Scholar]
- 75.Tompa P, Fuxreiter M. Fuzzy complexes: polymorphism and structural disorder in protein–protein interactions. Trends Biochem Sci. 2008;33:2–8. doi: 10.1016/j.tibs.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 76.Uversky VN. Intrinsic disorder-based protein interactions and their modulators. Curr Pharm Des. doi: 10.2174/1381612811319230005. (in press) [DOI] [PubMed] [Google Scholar]
- 77.Oldfield CJ, Meng J, Yang JY, Yang MQ, Uversky VN, Dunker AK. Flexible nets: disorder and induced fit in the associations of p53 and 14-3-3 with their partners. BMC Genomics. 2008;9(Suppl 1):S1. doi: 10.1186/1471-2164-9-S1-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Uversky VN, Oldfield CJ, Dunker AK. Showing your ID: intrinsic disorder as an ID for recognition, regulation and cell signaling. J Mol Recognit. 2005;18:343–384. doi: 10.1002/jmr.747. [DOI] [PubMed] [Google Scholar]
- 79.Dunker AK, Obradovic Z. The protein trinity—linking function and disorder. Nat Biotechnol. 2001;19:805–806. doi: 10.1038/nbt0901-805. [DOI] [PubMed] [Google Scholar]
- 80.Ma B, Kumar S, Tsai CJ, Nussinov R. Folding funnels and binding mechanisms. Protein Eng. 1999;12:713–720. doi: 10.1093/protein/12.9.713. [DOI] [PubMed] [Google Scholar]
- 81.Ptitsyn O. Molten globule and protein folding. Adv Protein Chem. 1995;47:83–229. doi: 10.1016/s0065-3233(08)60546-x. [DOI] [PubMed] [Google Scholar]
- 82.Tcherkasskaya O, Uversky VN. Polymeric aspects of protein folding: a brief overview. Protein Pept Lett. 2003;10:239–245. doi: 10.2174/0929866033478988. [DOI] [PubMed] [Google Scholar]
- 83.Vitalis A, Pappu RV. ABSINTH: a new continuum solvation model for simulations of polypeptides in aqueous solutions. J Comput Chem. 2009;30:673–699. doi: 10.1002/jcc.21005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Vitalis A, Wang X, Pappu RV. Quantitative characterization of intrinsic disorder in polyglutamine: insights from analysis based on polymer theories. Biophys J. 2007;93:1923–1937. doi: 10.1529/biophysj.107.110080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Vitalis A, Wang X, Pappu RV. Atomistic simulations of the effects of polyglutamine chain length and solvent quality on conformational equilibria and spontaneous homodimerization. J Mol Biol. 2008;384:279–297. doi: 10.1016/j.jmb.2008.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wang X, Vitalis A, Wyczalkowski MA, Pappu RV. Characterizing the conformational ensemble of monomeric polyglutamine. Proteins. 2006;63:297–311. doi: 10.1002/prot.20761. [DOI] [PubMed] [Google Scholar]
- 87.Williamson TE, Vitalis A, Crick SL, Pappu RV. Modulation of polyglutamine conformations and dimer formation by the N-terminus of huntingtin. J Mol Biol. 2010;396:1295–1306. doi: 10.1016/j.jmb.2009.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tran HT, Mao A, Pappu RV. Role of backbone-solvent interactions in determining conformational equilibria of intrinsically disordered proteins. J Am Chem Soc. 2008;130:7380–7392. doi: 10.1021/ja710446s. [DOI] [PubMed] [Google Scholar]
- 89.Mao AH, Crick SL, Vitalis A. Chicoine CL, Pappu RV (2010) Net charge per residue modulates conformational ensembles of intrinsically disordered proteins. Proc Natl Acad Sci USA. 107:8183–8188. doi: 10.1073/pnas.0911107107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Muller-Spath S, Soranno A, Hirschfeld V, Hofmann H, Ruegger S, Reymond L, Nettels D, Schuler B. From the cover: charge interactions can dominate the dimensions of intrinsically disordered proteins. Proc Natl Acad Sci USA. 2010;107:14609–14614. doi: 10.1073/pnas.1001743107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Oldfield CJ, Cheng Y, Cortese MS, Brown CJ, Uversky VN, Dunker AK. Comparing and combining predictors of mostly disordered proteins. Biochemistry. 2005;44:1989–2000. doi: 10.1021/bi047993o. [DOI] [PubMed] [Google Scholar]
- 92.Ashbaugh HS, Hatch HW. Natively unfolded protein stability as a coil-to-globule transition in charge/hydropathy space. J Am Chem Soc. 2008;130:9536–9542. doi: 10.1021/ja802124e. [DOI] [PubMed] [Google Scholar]
- 93.Patil A, Nakamura H. Disordered domains and high surface charge confer hubs with the ability to interact with multiple proteins in interaction networks. FEBS Lett. 2006;580:2041–2045. doi: 10.1016/j.febslet.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 94.Yamada J, Phillips JL, Patel S, Goldfien G, Calestagne-Morelli A, Huang H, Reza R, Acheson J, Krishnan VV, Newsam S, Gopinathan A, Lau EY, Colvin ME, Uversky VN, Rexach MF. A bimodal distribution of two distinct categories of intrinsically disordered structures with separate functions in FG nucleoporins. Mol Cell Proteomics. 2010;9:2205–2224. doi: 10.1074/mcp.M000035-MCP201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Uversky VN. Intrinsically disordered proteins and their environment: effects of strong denaturants, temperature, pH, counter ions, membranes, binding partners, osmolytes, and macromolecular crowding. Protein J. 2009;28:305–325. doi: 10.1007/s10930-009-9201-4. [DOI] [PubMed] [Google Scholar]
- 96.Uversky VN, Li J, Fink AL. Evidence for a partially folded intermediate in alpha-synuclein fibril formation. J Biol Chem. 2001;276:10737–10744. doi: 10.1074/jbc.M010907200. [DOI] [PubMed] [Google Scholar]
- 97.Permyakov SE, Millett IS, Doniach S, Permyakov EA, Uversky VN. Natively unfolded C-terminal domain of caldesmon remains substantially unstructured after the effective binding to calmodulin. Proteins. 2003;53:855–862. doi: 10.1002/prot.10481. [DOI] [PubMed] [Google Scholar]
- 98.Uversky VN, Permyakov SE, Zagranichny VE, Rodionov IL, Fink AL, Cherskaya AM, Wasserman LA, Permyakov EA. Effect of zinc and temperature on the conformation of the gamma subunit of retinal phosphodiesterase: a natively unfolded protein. J Proteome Res. 2002;1:149–159. doi: 10.1021/pr0155127. [DOI] [PubMed] [Google Scholar]
- 99.Uversky VN, Gillespie JR, Millett IS, Khodyakova AV, Vasiliev AM, Chernovskaya TV, Vasilenko RN, Kozlovskaya GD, Dolgikh DA, Fink AL, Doniach S, Abramov VM. Natively unfolded human prothymosin alpha adopts partially folded collapsed conformation at acidic pH. Biochemistry. 1999;38:15009–15016. doi: 10.1021/bi990752+. [DOI] [PubMed] [Google Scholar]
- 100.Konno T, Tanaka N, Kataoka M, Takano E, Maki M. A circular dichroism study of preferential hydration and alcohol effects on a denatured protein, pig calpastatin domain I. Biochim Biophys Acta. 1997;1342:73–82. doi: 10.1016/s0167-4838(97)00092-7. [DOI] [PubMed] [Google Scholar]
- 101.Lynn A, Chandra S, Malhotra P, Chauhan VS. Heme binding and polymerization by Plasmodium falciparum histidine rich protein. II. Influence of pH on activity and conformation. FEBS Lett. 1999;459:267–271. doi: 10.1016/s0014-5793(99)01260-0. [DOI] [PubMed] [Google Scholar]
- 102.Johansson J, Gudmundsson GH, Rottenberg ME, Berndt KD, Agerberth B. Conformation-dependent antibacterial activity of the naturally occurring human peptide LL-37. J Biol Chem. 1998;273:3718–3724. doi: 10.1074/jbc.273.6.3718. [DOI] [PubMed] [Google Scholar]
- 103.Magidovich E, Fleishman SJ, Yifrach O. Intrinsically disordered C-terminal segments of voltage-activated potassium channels: a possible fishing rod-like mechanism for channel binding to scaffold proteins. Bioinformatics. 2006;22:1546–1550. doi: 10.1093/bioinformatics/btl137. [DOI] [PubMed] [Google Scholar]
- 104.Roy S, Schnell S, Radivojac P. Unraveling the nature of the segmentation clock: intrinsic disorder of clock proteins and their interaction map. Comput Biol Chem. 2006;30:241–248. doi: 10.1016/j.compbiolchem.2006.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Minezaki Y, Homma K, Nishikawa K. Intrinsically disordered regions of human plasma membrane proteins preferentially occur in the cytoplasmic segment. J Mol Biol. 2007;368:902–913. doi: 10.1016/j.jmb.2007.02.033. [DOI] [PubMed] [Google Scholar]
- 106.De Biasio A, Guarnaccia C, Popovic M, Uversky VN, Pintar A, Pongor S. Prevalence of intrinsic disorder in the intracellular region of human single-pass type I proteins: the case of the notch ligand Delta-4. J Proteome Res. 2008;7:2496–2506. doi: 10.1021/pr800063u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Yang JY, Yang MQ, Dunker AK, Deng Y, Huang X. Investigation of transmembrane proteins using a computational approach. BMC Genomics. 2008;9(Suppl 1):S7. doi: 10.1186/1471-2164-9-S1-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Xue B, Li L, Meroueh SO, Uversky VN, Dunker AK. Analysis of structured and intrinsically disordered regions of transmembrane proteins. Mol Biosyst. 2009;5:1688–1702. doi: 10.1039/B905913J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lenard J, Singer SJ. Protein conformation in cell membrane preparations as studied by optical rotatory dispersion and circular dichroism. Proc Natl Acad Sci USA. 1966;56:1828–1835. doi: 10.1073/pnas.56.6.1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Rosenbusch JP. Characterization of the major envelope protein from Escherichia coli. Regular arrangement on the peptidoglycan and unusual dodecyl sulfate binding. J Biol Chem. 1974;249:8019–8029. [PubMed] [Google Scholar]
- 111.Dunker AK, Jones TC. Proposed knobs-into-holes packing for several membrane proteins. Membr Biochem. 1978;2:1–16. doi: 10.3109/09687687809063855. [DOI] [PubMed] [Google Scholar]
- 112.Paul C, Rosenbusch JP. Folding patterns of porin and bacteriorhodopsin. EMBO J. 1985;4:1593–1597. doi: 10.1002/j.1460-2075.1985.tb03822.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Rees DC, DeAntonio L, Eisenberg D. Hydrophobic organization of membrane proteins. Science. 1989;245:510–513. doi: 10.1126/science.2667138. [DOI] [PubMed] [Google Scholar]
- 114.Stevens TJ, Arkin IT. Are membrane proteins “inside-out” proteins? Proteins. 1999;36:135–143. doi: 10.1002/(sici)1097-0134(19990701)36:1<135::aid-prot11>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 115.Rees DC, Eisenberg D. Turning a reference inside-out: commentary on an article by Stevens and Arkin entitled: “Are membrane proteins ‘inside-out’ proteins?” (Proteins 1999;36:135–143) Proteins. 2000;38:121–122. [PubMed] [Google Scholar]
- 116.Von Heijne G. The distribution of positively charged residues in bacterial innter membrane proteins correlates with the transmembrane topology. EMBO J. 1986;5:3021–3027. doi: 10.1002/j.1460-2075.1986.tb04601.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Bernsel A, Viklund H, Falk J, Lindahl E, von Heijne G, Elofsson A. Prediction of membrane-protein topology from first principles. Proc Natl Acad Sci USA. 2008;105:7177–7181. doi: 10.1073/pnas.0711151105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Siglioccolo A, Paiardini A, Piscitelli M, Pascarella S. Structural adaptation of extreme halophilic proteins through decrease of conserved hydrophobic contact surface. BMC Struct Biol. 2011;11:50. doi: 10.1186/1472-6807-11-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zaccai G, Eisenberg H. Halophilic proteins and the influence of solvent on protein stabilization. Trends Biochem Sci. 1990;15:333–337. doi: 10.1016/0968-0004(90)90068-m. [DOI] [PubMed] [Google Scholar]
- 120.Madern D, Ebel C, Zaccai G. Halophilic adaptation of enzymes. Extremophiles. 2000;4:91–98. doi: 10.1007/s007920050142. [DOI] [PubMed] [Google Scholar]
- 121.Mevarech M, Frolow F, Gloss LM. Halophilic enzymes: proteins with a grain of salt. Biophys Chem. 2000;86:155–164. doi: 10.1016/s0301-4622(00)00126-5. [DOI] [PubMed] [Google Scholar]
- 122.Wright DB, Banks DD, Lohman JR, Hilsenbeck JL, Gloss LM. The effect of salts on the activity and stability of Escherichia coli and Haloferax volcanii dihydrofolate reductases. J Mol Biol. 2002;323:327–344. doi: 10.1016/s0022-2836(02)00916-6. [DOI] [PubMed] [Google Scholar]
- 123.Bergqvist S, Williams MA, O'Brien R, Ladbury JE. Halophilic adaptation of protein–DNA interactions. Biochem Soc Trans. 2003;31:677–680. doi: 10.1042/bst0310677. [DOI] [PubMed] [Google Scholar]
- 124.Fukuchi S, Yoshimune K, Wakayama M, Moriguchi M, Nishikawa K. Unique amino acid composition of proteins in halophilic bacteria. J Mol Biol. 2003;327:347–357. doi: 10.1016/s0022-2836(03)00150-5. [DOI] [PubMed] [Google Scholar]
- 125.Paul S, Bag SK, Das S, Harvill ET, Dutta C. Molecular signature of hypersaline adaptation: insights from genome and proteome composition of halophilic prokaryotes. Genome Biol. 2008;9:R70. doi: 10.1186/gb-2008-9-4-r70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Tadeo X, Lopez-Mendez B, Trigueros T, Lain A, Castano D, Millet O. Structural basis for the aminoacid composition of proteins from halophilic archea. PLoS Biol. 2009;7:e1000257. doi: 10.1371/journal.pbio.1000257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Elcock AH, McCammon JA. Electrostatic contributions to the stability of halophilic proteins. J Mol Biol. 1998;280:731–748. doi: 10.1006/jmbi.1998.1904. [DOI] [PubMed] [Google Scholar]
- 128.Xue B, Williams RW, Oldfield CJ, Goh GK, Dunker AK, Uversky VN. Viral disorder or disordered viruses: do viral proteins possess unique features? Protein Pept Lett. 2010;17:932–951. doi: 10.2174/092986610791498984. [DOI] [PubMed] [Google Scholar]
- 129.Berezovsky IN. The diversity of physical forces and mechanisms in intermolecular interactions. Phys Biol. 2011;8:035002. doi: 10.1088/1478-3975/8/3/035002. [DOI] [PubMed] [Google Scholar]
- 130.Iakoucheva LM, Brown CJ, Lawson JD, Obradovic Z, Dunker AK. Intrinsic disorder in cell-signaling and cancer-associated proteins. J Mol Biol. 2002;323:573–584. doi: 10.1016/s0022-2836(02)00969-5. [DOI] [PubMed] [Google Scholar]
- 131.Dyson HJ, Wright PE. Coupling of folding and binding for unstructured proteins. Curr Opin Struct Biol. 2002;12:54–60. doi: 10.1016/s0959-440x(02)00289-0. [DOI] [PubMed] [Google Scholar]
- 132.Dyson HJ, Wright PE. Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol. 2005;6:197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- 133.Fink AL. Natively unfolded proteins. Curr Opin Struct Biol. 2005;15:35–41. doi: 10.1016/j.sbi.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 134.Dunker AK, Cortese MS, Romero P, Iakoucheva LM, Uversky VN. Flexible nets. The roles of intrinsic disorder in protein interaction networks. FEBS J. 2005;272:5129–5148. doi: 10.1111/j.1742-4658.2005.04948.x. [DOI] [PubMed] [Google Scholar]
- 135.Uversky VN. Protein folding revisited. A polypeptide chain at the folding-misfolding-nonfolding cross-roads: which way to go? Cell Mol Life Sci. 2003;60:1852–1871. doi: 10.1007/s00018-003-3096-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Tompa P, Csermely P. The role of structural disorder in the function of RNA and protein chaperones. FASEB J. 2004;18:1169–1175. doi: 10.1096/fj.04-1584rev. [DOI] [PubMed] [Google Scholar]
- 137.Tompa P, Szasz C, Buday L. Structural disorder throws new light on moonlighting. Trends Biochem Sci. 2005;30:484–489. doi: 10.1016/j.tibs.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 138.Xie H, Vucetic S, Iakoucheva LM, Oldfield CJ, Dunker AK, Uversky VN, Obradovic Z. Functional anthology of intrinsic disorder. I. Biological processes and functions of proteins with long disordered regions. J Proteome Res. 2007;6:1882–1898. doi: 10.1021/pr060392u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Xie H, Vucetic S, Iakoucheva LM, Oldfield CJ, Dunker AK, Obradovic Z, Uversky VN. Functional anthology of intrinsic disorder. III. Ligands, post-translational modifications, and diseases associated with intrinsically disordered proteins. J Proteome Res. 2007;6:1917–1932. doi: 10.1021/pr060394e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Vucetic S, Xie H, Iakoucheva LM, Oldfield CJ, Dunker AK, Obradovic Z, Uversky VN. Functional anthology of intrinsic disorder. II. Cellular components, domains, technical terms, developmental processes, and coding sequence diversities correlated with long disordered regions. J Proteome Res. 2007;6:1899–1916. doi: 10.1021/pr060393m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Cortese MS, Uversky VN, Dunker AK. Intrinsic disorder in scaffold proteins: getting more from less. Prog Biophys Mol Biol. 2008;98:85–106. doi: 10.1016/j.pbiomolbio.2008.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Dunker AK, Silman I, Uversky VN, Sussman JL. Function and structure of inherently disordered proteins. Curr Opin Struct Biol. 2008;18:756–764. doi: 10.1016/j.sbi.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 143.Dunker AK, Uversky VN. Signal transduction via unstructured protein conduits. Nat Chem Biol. 2008;4:229–230. doi: 10.1038/nchembio0408-229. [DOI] [PubMed] [Google Scholar]
- 144.Dunker AK, Oldfield CJ, Meng J, Romero P, Yang JY, Chen JW, Vacic V, Obradovic Z, Uversky VN. The unfoldomics decade: an update on intrinsically disordered proteins. BMC Genomics. 2008;9(Suppl 2):S1. doi: 10.1186/1471-2164-9-S2-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Russell RB, Gibson TJ. A careful disorderliness in the proteome: sites for interaction and targets for future therapies. FEBS Lett. 2008;582:1271–1275. doi: 10.1016/j.febslet.2008.02.027. [DOI] [PubMed] [Google Scholar]
- 146.Tompa P, Fuxreiter M, Oldfield CJ, Simon I, Dunker AK, Uversky VN. Close encounters of the third kind: disordered domains and the interactions of proteins. Bioessays. 2009;31:328–335. doi: 10.1002/bies.200800151. [DOI] [PubMed] [Google Scholar]
- 147.Uversky VN, Dunker AK. Biochemistry. Controlled chaos. Science. 2008;322:1340–1341. doi: 10.1126/science.1167453. [DOI] [PubMed] [Google Scholar]
- 148.Wright PE, Dyson HJ. Linking folding and binding. Curr Opin Struct Biol. 2009;19:31–38. doi: 10.1016/j.sbi.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Uversky VN. Intrinsically disordered proteins from A to Z. Int J Biochem Cell Biol. 2012;43:1090–1103. doi: 10.1016/j.biocel.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 150.Dunker AK, Obradovic Z, Romero P, Kissinger C, Villafranca E. On the importance of being disordered. PDB Newsletter. 1997;81:3–5. [Google Scholar]
- 151.Brown CJ, Takayama S, Campen AM, Vise P, Marshall TW, Oldfield CJ, Williams CJ, Dunker AK. Evolutionary rate heterogeneity in proteins with long disordered regions. J Mol Evol. 2002;55:104–110. doi: 10.1007/s00239-001-2309-6. [DOI] [PubMed] [Google Scholar]
- 152.Romero PR, Zaidi S, Fang YY, Uversky VN, Radivojac P, Oldfield CJ, Cortese MS, Sickmeier M, LeGall T, Obradovic Z, Dunker AK. Alternative splicing in concert with protein intrinsic disorder enables increased functional diversity in multicellular organisms. Proc Natl Acad Sci USA. 2006;103:8390–8395. doi: 10.1073/pnas.0507916103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Dosztanyi Z, Csizmok V, Tompa P, Simon I. IUPred: web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics. 2005;21:3433–3434. doi: 10.1093/bioinformatics/bti541. [DOI] [PubMed] [Google Scholar]
- 154.Spolar RS, Record MT., Jr Coupling of local folding to site-specific binding of proteins to DNA. Science. 1994;263:777–784. doi: 10.1126/science.8303294. [DOI] [PubMed] [Google Scholar]
- 155.Schulz GE. Nucleotide binding proteins. In: Balaban M, editor. Molecular mechanism of biological recognition. New York: Elsevier/North-Holland Biomedical Press; 1979. pp. 79–94. [Google Scholar]
- 156.Pontius BW. Close encounters: why unstructured, polymeric domains can increase rates of specific macromolecular association. Trends Biochem Sci. 1993;18:181–186. doi: 10.1016/0968-0004(93)90111-y. [DOI] [PubMed] [Google Scholar]
- 157.Plaxco KW, Gross M. Cell biology. The importance of being unfolded. Nature. 1997;386:657. doi: 10.1038/386657a0. 659. [DOI] [PubMed] [Google Scholar]
- 158.Karush F. Heterogeneity of the binding sites of bovine serum albumin. J Am Chem Soc. 1950;72:2705–2713. [Google Scholar]
- 159.Kriwacki RW, Hengst L, Tennant L, Reed SI, Wright PE. Structural studies of p21Waf1/Cip1/Sdi1 in the free and Cdk2-bound state: conformational disorder mediates binding diversity. Proc Natl Acad Sci USA. 1996;93:11504–11509. doi: 10.1073/pnas.93.21.11504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Ekman D, Light S, Bjorklund AK, Elofsson A. What properties characterize the hub proteins of the protein–protein interaction network of Saccharomyces cerevisiae? Genome Biol. 2006;7:R45. doi: 10.1186/gb-2006-7-6-r45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Haynes C, Oldfield CJ, Ji F, Klitgord N, Cusick ME, Radivojac P, Uversky VN, Vidal M, Iakoucheva LM. Intrinsic disorder is a common feature of hub proteins from four eukaryotic interactomes. PLoS Comput Biol. 2006;2:e100. doi: 10.1371/journal.pcbi.0020100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Dosztanyi Z, Chen J, Dunker AK, Simon I, Tompa P. Disorder and sequence repeats in hub proteins and their implications for network evolution. J Proteome Res. 2006;5:2985–2995. doi: 10.1021/pr060171o. [DOI] [PubMed] [Google Scholar]
- 163.Singh GP, Ganapathi M, Dash D. Role of intrinsic disorder in transient interactions of hub proteins. Proteins. 2007;66:761–765. doi: 10.1002/prot.21281. [DOI] [PubMed] [Google Scholar]
- 164.Singh GP, Dash D. Intrinsic disorder in yeast transcriptional regulatory network. Proteins. 2007;68:602–605. doi: 10.1002/prot.21497. [DOI] [PubMed] [Google Scholar]
- 165.Balazs A, Csizmok V, Buday L, Rakacs M, Kiss R, Bokor M, Udupa R, Tompa K, Tompa P. High levels of structural disorder in scaffold proteins as exemplified by a novel neuronal protein, CASK-interactive protein1. FEBS J. 2009;276:3744–3756. doi: 10.1111/j.1742-4658.2009.07090.x. [DOI] [PubMed] [Google Scholar]
- 166.Jeffery CJ. Moonlighting proteins. Trends Biochem Sci. 1999;24:8–11. doi: 10.1016/s0968-0004(98)01335-8. [DOI] [PubMed] [Google Scholar]
- 167.Jeffery CJ. Multifunctional proteins: examples of gene sharing. Ann Med. 2003;35:28–35. doi: 10.1080/07853890310004101. [DOI] [PubMed] [Google Scholar]
- 168.Jeffery CJ. Molecular mechanisms for multitasking: recent crystal structures of moonlighting proteins. Curr Opin Struct Biol. 2004;14:663–668. doi: 10.1016/j.sbi.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 169.Hsu WL, Oldfield CJ, Xue B, Meng J, Huang F, Romero P, Uversky VN, Dunker AK. Exploring the binding diversity of intrinsically disordered proteins involved in one-to-many binding. Protein Sci. 2013;22:258–273. doi: 10.1002/pro.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Watts DJ, Strogatz SH. Collective dynamics of ‘small-world’ networks. Nature. 1998;393:440–442. doi: 10.1038/30918. [DOI] [PubMed] [Google Scholar]
- 171.Goh KI, Oh E, Jeong H, Kahng B, Kim D. Classification of scale-free networks. Proc Natl Acad Sci USA. 2002;99:12583–12588. doi: 10.1073/pnas.202301299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Barabasi AL, Albert R. Emergence of scaling in random networks. Science. 1999;286:509–512. doi: 10.1126/science.286.5439.509. [DOI] [PubMed] [Google Scholar]
- 173.Agarwal S, Deane CM, Porter MA, Jones NS. Revisiting date and party hubs: novel approaches to role assignment in protein interaction networks. PLoS Comput Biol. 2010;6:e1000817. doi: 10.1371/journal.pcbi.1000817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Liu W, Rui H, Wang J, Lin S, He Y, Chen M, Li Q, Ye Z, Zhang S, Chan SC, Chen YG, Han J, Lin SC. Axin is a scaffold protein in TGF-beta signaling that promotes degradation of Smad7 by Arkadia. EMBO J. 2006;25:1646–1658. doi: 10.1038/sj.emboj.7601057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Liu J, Perumal NB, Oldfield CJ, Su EW, Uversky VN, Dunker AK. Intrinsic disorder in transcription factors. Biochemistry. 2006;45:6873–6888. doi: 10.1021/bi0602718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Minezaki Y, Homma K, Kinjo AR, Nishikawa K. Human transcription factors contain a high fraction of intrinsically disordered regions essential for transcriptional regulation. J Mol Biol. 2006;359:1137–1149. doi: 10.1016/j.jmb.2006.04.016. [DOI] [PubMed] [Google Scholar]
- 177.Toth-Petroczy A, Oldfield CJ, Simon I, Takagi Y, Dunker AK, Uversky VN, Fuxreiter M. Malleable machines in transcription regulation: the mediator complex. PLoS Comput Biol. 2008;4:e1000243. doi: 10.1371/journal.pcbi.1000243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Peng Z, Mizianty MJ, Xue B, Kurgan L, Uversky VN. More than just tails: intrinsic disorder in histone proteins. Mol Biosyst. 2012;8:1886–1901. doi: 10.1039/c2mb25102g. [DOI] [PubMed] [Google Scholar]
- 179.Peng Z, Xue B, Mizianty MJ, Oldfield CJ, Kurgan L, Uversky VN. A creature with hundred of waggly tails: intrinsically disordered proteins in ribosome. Biochim Biophys Acta Mol Cell Res. doi: 10.1007/s00018-013-1446-6. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Westerheide SD, Raynes R, Powell C, Xue B, Uversky VN. HSF transcription factor family, heat shock response, and protein intrinsic disorder. Curr Protein Pept Sci. 2012;13:86–103. doi: 10.2174/138920312799277956. [DOI] [PubMed] [Google Scholar]
- 181.Xue B, Oldfield CJ, Van YY, Dunker AK, Uversky VN. Protein intrinsic disorder and induced pluripotent stem cells. Mol Biosyst. 2012;8:134–150. doi: 10.1039/c1mb05163f. [DOI] [PubMed] [Google Scholar]
- 182.Xue B, Dunker AK, Uversky VN. The roles of intrinsic disorder in orchestrating the Wnt-pathway. J Biomol Struct Dyn. 2012;29:843–861. doi: 10.1080/073911012010525024. [DOI] [PubMed] [Google Scholar]
- 183.Peng Z, Xue B, Kurgan L, Uversky VN. Resilience of death: Intrinsic disorder in proteins involved in the programmed cell death. Cell Death Differ. doi: 10.1038/cdd.2013.65. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Pervushin K, Vamvaca K, Vogeli B, Hilvert D. Structure and dynamics of a molten globular enzyme. Nat Struct Mol Biol. 2007;14:1202–1206. doi: 10.1038/nsmb1325. [DOI] [PubMed] [Google Scholar]
- 185.Gatti-Lafranconi P, Hollfelder F. Flexibility and reactivity in promiscuous enzymes. Chembiochem. 2013;14:285–292. doi: 10.1002/cbic.201200628. [DOI] [PubMed] [Google Scholar]
- 186.Karplus M. Dynamics of proteins. Adv Biophys. 1984;18:165–190. doi: 10.1016/0065-227x(84)90011-x. [DOI] [PubMed] [Google Scholar]
- 187.Hammes GG. Multiple conformational changes in enzyme catalysis. Biochemistry. 2002;41:8221–8228. doi: 10.1021/bi0260839. [DOI] [PubMed] [Google Scholar]
- 188.Boehr DD, Dyson HJ, Wright PE. An NMR perspective on enzyme dynamics. Chem Rev. 2006;106:3055–3079. doi: 10.1021/cr050312q. [DOI] [PubMed] [Google Scholar]
- 189.Hammes-Schiffer S, Benkovic SJ. Relating protein motion to catalysis. Annu Rev Biochem. 2006;75:519–541. doi: 10.1146/annurev.biochem.75.103004.142800. [DOI] [PubMed] [Google Scholar]
- 190.Olsson MH, Parson WW, Warshel A. Dynamical contributions to enzyme catalysis: critical tests of a popular hypothesis. Chem Rev. 2006;106:1737–1756. doi: 10.1021/cr040427e. [DOI] [PubMed] [Google Scholar]
- 191.Koshland DE, Jr, Neet KE. The catalytic and regulatory properties of enzymes. Annu Rev Biochem. 1968;37:359–410. doi: 10.1146/annurev.bi.37.070168.002043. [DOI] [PubMed] [Google Scholar]
- 192.Vendruscolo M. Enzymatic activity in disordered states of proteins. Curr Opin Chem Biol. 2010;14:671–675. doi: 10.1016/j.cbpa.2010.08.022. [DOI] [PubMed] [Google Scholar]
- 193.Vamvaca K, Vogeli B, Kast P, Pervushin K, Hilvert D. An enzymatic molten globule: efficient coupling of folding and catalysis. Proc Natl Acad Sci USA. 2004;101:12860–12864. doi: 10.1073/pnas.0404109101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Vamvaca K, Jelesarov I, Hilvert D. Kinetics and thermodynamics of ligand binding to a molten globular enzyme and its native counterpart. J Mol Biol. 2008;382:971–977. doi: 10.1016/j.jmb.2008.07.049. [DOI] [PubMed] [Google Scholar]
- 195.Woycechowsky KJ, Choutko A, Vamvaca K, Hilvert D. Relative tolerance of an enzymatic molten globule and its thermostable counterpart to point mutation. Biochemistry. 2008;47:13489–13496. doi: 10.1021/bi801108a. [DOI] [PubMed] [Google Scholar]
- 196.Uversky VN, Kutyshenko VP, Protasova N, Rogov VV, Vassilenko KS, Gudkov AT. Circularly permuted dihydrofolate reductase possesses all the properties of the molten globule state, but can resume functional tertiary structure by interaction with its ligands. Protein Sci. 1996;5:1844–1851. doi: 10.1002/pro.5560050910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Protasova N, Kireeva ML, Murzina NV, Murzin AG, Uversky VN, Gryaznova OI, Gudkov AT. Circularly permuted dihydrofolate reductase of E. coli has functional activity and a destabilized tertiary structure. Protein Eng. 1994;7:1373–1377. doi: 10.1093/protein/7.11.1373. [DOI] [PubMed] [Google Scholar]
- 198.Zambelli B, Stola M, Musiani F, De Vriendt K, Samyn B, Devreese B, Van Beeumen J, Turano P, Dikiy A, Bryant DA, Ciurli S. UreG, a chaperone in the urease assembly process, is an intrinsically unstructured GTPase that specifically binds Zn2+ J Biol Chem. 2005;280:4684–4695. doi: 10.1074/jbc.M408483200. [DOI] [PubMed] [Google Scholar]
- 199.Neyroz P, Zambelli B, Ciurli S. Intrinsically disordered structure of Bacillus pasteurii UreG as revealed by steady-state and time-resolved fluorescence spectroscopy. Biochemistry. 2006;45:8918–8930. doi: 10.1021/bi060227s. [DOI] [PubMed] [Google Scholar]
- 200.Zambelli B, Cremades N, Neyroz P, Turano P, Uversky VN, Ciurli S. Insights in the (un)structural organization of Bacillus pasteurii UreG, an intrinsically disordered GTPase enzyme. Mol Biosyst. 2012;8:220–228. doi: 10.1039/c1mb05227f. [DOI] [PubMed] [Google Scholar]
- 201.Uversky VN. Multitude of binding modes attainable by intrinsically disordered proteins: a portrait gallery of disorder-based complexes. Chem Soc Rev. 2011;40:1623–1634. doi: 10.1039/c0cs00057d. [DOI] [PubMed] [Google Scholar]
- 202.Borg M, Mittag T, Pawson T, Tyers M, Forman-Kay JD, Chan HS. Polyelectrostatic interactions of disordered ligands suggest a physical basis for ultrasensitivity. Proc Natl Acad Sci USA. 2007;104:9650–9655. doi: 10.1073/pnas.0702580104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Mittag T, Orlicky S, Choy WY, Tang X, Lin H, Sicheri F, Kay LE, Tyers M, Forman-Kay JD. Dynamic equilibrium engagement of a polyvalent ligand with a single-site receptor. Proc Natl Acad Sci USA. 2008;105:17772–17777. doi: 10.1073/pnas.0809222105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Mittag T, Kay LE, Forman-Kay JD. Protein dynamics and conformational disorder in molecular recognition. J Mol Recognit. 2010;23:105–116. doi: 10.1002/jmr.961. [DOI] [PubMed] [Google Scholar]
- 205.Mittag T, Marsh J, Grishaev A, Orlicky S, Lin H, Sicheri F, Tyers M, Forman-Kay JD. Structure/function implications in a dynamic complex of the intrinsically disordered Sic1 with the Cdc4 subunit of an SCF ubiquitin ligase. Structure. 2010;18:494–506. doi: 10.1016/j.str.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Sigalov AB, Kim WM, Saline M, Stern LJ. The intrinsically disordered cytoplasmic domain of the T cell receptor zeta chain binds to the nef protein of simian immunodeficiency virus without a disorder-to-order transition. Biochemistry. 2008;47:12942–12944. doi: 10.1021/bi801602p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Sigalov A, Aivazian D, Stern L. Homooligomerization of the cytoplasmic domain of the T cell receptor zeta chain and of other proteins containing the immunoreceptor tyrosine-based activation motif. Biochemistry. 2004;43:2049–2061. doi: 10.1021/bi035900h. [DOI] [PubMed] [Google Scholar]
- 208.Sigalov AB, Zhuravleva AV, Orekhov VY. Binding of intrinsically disordered proteins is not necessarily accompanied by a structural transition to a folded form. Biochimie. 2007;89:419–421. doi: 10.1016/j.biochi.2006.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Pometun MS, Chekmenev EY, Wittebort RJ. Quantitative observation of backbone disorder in native elastin. J Biol Chem. 2004;279:7982–7987. doi: 10.1074/jbc.M310948200. [DOI] [PubMed] [Google Scholar]
- 210.Sigalov AB, Hendricks GM. Membrane binding mode of intrinsically disordered cytoplasmic domains of T cell receptor signaling subunits depends on lipid composition. Biochem Biophys Res Commun. 2009;389:388–393. doi: 10.1016/j.bbrc.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Sigalov AB, Aivazian DA, Uversky VN, Stern LJ. Lipid-binding activity of intrinsically unstructured cytoplasmic domains of multichain immune recognition receptor signaling subunits. Biochemistry. 2006;45:15731–15739. doi: 10.1021/bi061108f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Hazy E, Tompa P. Limitations of induced folding in molecular recognition by intrinsically disordered proteins. Chemphyschem. 2009;10:1415–1419. doi: 10.1002/cphc.200900205. [DOI] [PubMed] [Google Scholar]
- 213.Liu Y, Fratini E, Baglioni P, Chen WR, Chen SH. Effective long-range attraction between protein molecules in solutions studied by small angle neutron scattering. Phys Rev Lett. 2005;95:118102. doi: 10.1103/PhysRevLett.95.118102. [DOI] [PubMed] [Google Scholar]
- 214.Meyer TE, Yakali E, Cusanovich MA, Tollin G. Properties of a water-soluble, yellow protein isolated from a halophilic phototrophic bacterium that has photochemical activity analogous to sensory rhodopsin. Biochemistry. 1987;26:418–423. doi: 10.1021/bi00376a012. [DOI] [PubMed] [Google Scholar]
- 215.Hoff WD, van Stokkum IH, van Ramesdonk HJ, van Brederode ME, Brouwer AM, Fitch JC, Meyer TE, van Grondelle R, Hellingwerf KJ. Measurement and global analysis of the absorbance changes in the photocycle of the photoactive yellow protein from Ectothiorhodospira halophila. Biophys J. 1994;67:1691–1705. doi: 10.1016/S0006-3495(94)80643-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Rubinstenn G, Vuister GW, Mulder FA, Dux PE, Boelens R, Hellingwerf KJ, Kaptein R. Structural and dynamic changes of photoactive yellow protein during its photocycle in solution. Nat Struct Biol. 1998;5:568–570. doi: 10.1038/823. [DOI] [PubMed] [Google Scholar]
- 217.Ramachandran PL, Lovett JE, Carl PJ, Cammarata M, Lee JH, Jung YO, Ihee H, Timmel CR, van Thor JJ. The short-lived signaling state of the photoactive yellow protein photoreceptor revealed by combined structural probes. J Am Chem Soc. 2011;133:9395–9404. doi: 10.1021/ja200617t. [DOI] [PubMed] [Google Scholar]
- 218.Dux P, Rubinstenn G, Vuister GW, Boelens R, Mulder FA, Hard K, Hoff WD, Kroon AR, Crielaard W, Hellingwerf KJ, Kaptein R. Solution structure and backbone dynamics of the photoactive yellow protein. Biochemistry. 1998;37:12689–12699. doi: 10.1021/bi9806652. [DOI] [PubMed] [Google Scholar]
- 219.Borgstahl GE, Williams DR, Getzoff ED. 1.4 A structure of photoactive yellow protein, a cytosolic photoreceptor: unusual fold, active site, and chromophore. Biochemistry. 1995;34:6278–6287. doi: 10.1021/bi00019a004. [DOI] [PubMed] [Google Scholar]
- 220.Gustin MC, Zhou XL, Martinac B, Kung C. A mechanosensitive ion channel in the yeast plasma membrane. Science. 1988;242:762–765. doi: 10.1126/science.2460920. [DOI] [PubMed] [Google Scholar]
- 221.Corry B, Hurst AC, Pal P, Nomura T, Rigby P, Martinac B. An improved open-channel structure of MscL determined from FRET confocal microscopy and simulation. J Gen Physiol. 2010;136:483–494. doi: 10.1085/jgp.200910376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Gsponer J, Futschik ME, Teichmann SA, Babu MM. Tight regulation of unstructured proteins: from transcript synthesis to protein degradation. Science. 2008;322:1365–1368. doi: 10.1126/science.1163581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Grimmler M, Wang Y, Mund T, Cilensek Z, Keidel EM, Waddell MB, Jakel H, Kullmann M, Kriwacki RW, Hengst L. Cdk-inhibitory activity and stability of p27Kip1 are directly regulated by oncogenic tyrosine kinases. Cell. 2007;128:269–280. doi: 10.1016/j.cell.2006.11.047. [DOI] [PubMed] [Google Scholar]
- 224.Iakoucheva LM, Radivojac P, Brown CJ, O'Connor TR, Sikes JG, Obradovic Z, Dunker AK. The importance of intrinsic disorder for protein phosphorylation. Nucleic Acids Res. 2004;32:1037–1049. doi: 10.1093/nar/gkh253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Paliy O, Gargac SM, Cheng Y, Uversky VN, Dunker AK. Protein disorder is positively correlated with gene expression in Escherichia coli. J Proteome Res. 2008;7:2234–2245. doi: 10.1021/pr800055r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Fernandez A, Scheraga HA. Insufficiently dehydrated hydrogen bonds as determinants of protein interactions. Proc Natl Acad Sci USA. 2003;100:113–118. doi: 10.1073/pnas.0136888100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Fernandez A. Keeping dry and crossing membranes. Nat Biotechnol. 2004;22:1081–1084. doi: 10.1038/nbt0904-1081. [DOI] [PubMed] [Google Scholar]
- 228.Kelly JW. The alternative conformations of amyloidogenic proteins and their multi-step assembly pathways. Curr Opin Struct Biol. 1998;8:101–106. doi: 10.1016/s0959-440x(98)80016-x. [DOI] [PubMed] [Google Scholar]
- 229.Dobson CM. Protein misfolding, evolution and disease. Trends Biochem Sci. 1999;24:329–332. doi: 10.1016/s0968-0004(99)01445-0. [DOI] [PubMed] [Google Scholar]
- 230.Bellotti V, Mangione P, Stoppini M. Biological activity and pathological implications of misfolded proteins. Cell Mol Life Sci. 1999;55:977–991. doi: 10.1007/s000180050348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Uversky VN, Talapatra A, Gillespie JR, Fink AL. Protein deposits as the molecular basis of amyloidosis. I. Systemic amyloidoses. Med Sci Monitor. 1999;5:1001–1012. [Google Scholar]
- 232.Uversky VN, Talapatra A, Gillespie JR, Fink AL. Protein deposits as the molecular basis of amyloidosis. II. Localized amyloidosis and neurodegenerative disordres. Med Sci Monitor. 1999;5:1238–1254. [Google Scholar]
- 233.Rochet JC, Lansbury PT., Jr Amyloid fibrillogenesis: themes and variations. Curr Opin Struct Biol. 2000;10:60–68. doi: 10.1016/s0959-440x(99)00049-4. [DOI] [PubMed] [Google Scholar]
- 234.Uversky VN, Fink AL. Conformational constraints for amyloid fibrillation: the importance of being unfolded. Biochim Biophys Acta. 2004;1698:131–153. doi: 10.1016/j.bbapap.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 235.Uversky VN, Fink AL. Pathways to amyloid fibril formation: partially folded intermediates in fibrillation of unfolded proteins. In: Sipe JD, editor. Amyloid proteins: the beta pleated sheet conformation and disease. Weinheim, Germany: Wiley-VCH, Verlag GmbH & Co. KGaA; 2005. pp. 247–265. [Google Scholar]
- 236.Glenner GG, Wong CW. Alzheimer's disease and Down's syndrome: sharing of a unique cerebrovascular amyloid fibril protein. Biochem Biophys Res Commun. 1984;122:1131–1135. doi: 10.1016/0006-291x(84)91209-9. [DOI] [PubMed] [Google Scholar]
- 237.Masters CL, Multhaup G, Simms G, Pottgiesser J, Martins RN, Beyreuther K. Neuronal origin of a cerebral amyloid: neurofibrillary tangles of Alzheimer's disease contain the same protein as the amyloid of plaque cores and blood vessels. EMBO J. 1985;4:2757–2763. doi: 10.1002/j.1460-2075.1985.tb04000.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Lee VM, Balin BJ, Otvos L, Jr, Trojanowski JQ. A68: a major subunit of paired helical filaments and derivatized forms of normal Tau. Science. 1991;251:675–678. doi: 10.1126/science.1899488. [DOI] [PubMed] [Google Scholar]
- 239.Ueda K, Fukushima H, Masliah E, Xia Y, Iwai A, Yoshimoto M, Otero DA, Kondo J, Ihara Y, Saitoh T. Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc Natl Acad Sci USA. 1993;90:11282–11286. doi: 10.1073/pnas.90.23.11282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Wisniewski KE, Dalton AJ, McLachlan C, Wen GY, Wisniewski HM. Alzheimer's disease in Down's syndrome: clinicopathologic studies. Neurology. 1985;35:957–961. doi: 10.1212/wnl.35.7.957. [DOI] [PubMed] [Google Scholar]
- 241.Dev KK, Hofele K, Barbieri S, Buchman VL, van der Putten H. Part II: alpha-synuclein and its molecular pathophysiological role in neurodegenerative disease. Neuropharmacology. 2003;45:14–44. doi: 10.1016/s0028-3908(03)00140-0. [DOI] [PubMed] [Google Scholar]
- 242.Prusiner SB. Shattuck lecture—neurodegenerative diseases and prions. N Engl J Med. 2001;344:1516–1526. doi: 10.1056/NEJM200105173442006. [DOI] [PubMed] [Google Scholar]
- 243.Zoghbi HY, Orr HT. Polyglutamine diseases: protein cleavage and aggregation. Curr Opin Neurobiol. 1999;9:566–570. doi: 10.1016/S0959-4388(99)00013-6. [DOI] [PubMed] [Google Scholar]
- 244.Okazawa H. Polyglutamine diseases: a transcription disorder? Cell Mol Life Sci. 2003;60:1427–1439. doi: 10.1007/s00018-003-3013-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Cummings CJ, Zoghbi HY. Fourteen and counting: unraveling trinucleotide repeat diseases. Hum Mol Genet. 2000;9:909–916. doi: 10.1093/hmg/9.6.909. [DOI] [PubMed] [Google Scholar]
- 246.Gusella JF, MacDonald ME. Molecular genetics: unmasking polyglutamine triggers in neurodegenerative disease. Nat Rev Neurosci. 2000;1:109–115. doi: 10.1038/35039051. [DOI] [PubMed] [Google Scholar]
- 247.Orr HT. Beyond the Qs in the polyglutamine diseases. Genes Dev. 2001;15:925–932. doi: 10.1101/gad.888401. [DOI] [PubMed] [Google Scholar]
- 248.Fischbeck KH. Polyglutamine expansion neurodegenerative disease. Brain Res Bull. 2001;56:161–163. doi: 10.1016/s0361-9230(01)00577-9. [DOI] [PubMed] [Google Scholar]
- 249.Lee H, Mok KH, Muhandiram R, Park KH, Suk JE, Kim DH, Chang J, Sung YC, Choi KY, Han KH. Local structural elements in the mostly unstructured transcriptional activation domain of human p53. J Biol Chem. 2000;275:29426–29432. doi: 10.1074/jbc.M003107200. [DOI] [PubMed] [Google Scholar]
- 250.Adkins JN, Lumb KJ. Intrinsic structural disorder and sequence features of the cell cycle inhibitor p57Kip2. Proteins. 2002;46:1–7. doi: 10.1002/prot.10018. [DOI] [PubMed] [Google Scholar]
- 251.Chang BS, Minn AJ, Muchmore SW, Fesik SW, Thompson CB. Identification of a novel regulatory domain in Bcl-X(L) and Bcl-2. EMBO J. 1997;16:968–977. doi: 10.1093/emboj/16.5.968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Campbell KM, Terrell AR, Laybourn PJ, Lumb KJ. Intrinsic structural disorder of the C-terminal activation domain from the bZIP transcription factor Fos. Biochemistry. 2000;39:2708–2713. doi: 10.1021/bi9923555. [DOI] [PubMed] [Google Scholar]
- 253.Sunde M, McGrath KC, Young L, Matthews JM, Chua EL, Mackay JP, Death AK. TC-1 is a novel tumorigenic and natively disordered protein associated with thyroid cancer. Cancer Res. 2004;64:2766–2773. doi: 10.1158/0008-5472.can-03-2093. [DOI] [PubMed] [Google Scholar]
- 254.Ng KP, Potikyan G, Savene RO, Denny CT, Uversky VN, Lee KA. Multiple aromatic side chains within a disordered structure are critical for transcription and transforming activity of EWS family oncoproteins. Proc Natl Acad Sci USA. 2007;104:479–484. doi: 10.1073/pnas.0607007104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Erkizan HV, Uversky VN, Toretsky JA. Oncogenic partnerships: EWS-FLI1 protein interactions initiate key pathways of Ewing's sarcoma. Clin Cancer Res. 2010;16:4077–4083. doi: 10.1158/1078-0432.CCR-09-2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Anderson CW, Appella E. Signaling to the p53 tumor suppressor through pathways activated by genotoxic and nongenotoxic stress. In: RA Bradshaw, Dennis EA., editors. Handbook of cell signaling. New York: Academic Press; 2004. pp. 237–247. [Google Scholar]
- 257.Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 mutations in human cancers. Science. 1991;253:49–53. doi: 10.1126/science.1905840. [DOI] [PubMed] [Google Scholar]
- 258.Balint EE, Vousden KH. Activation and activities of the p53 tumour suppressor protein. Br J Cancer. 2001;85:1813–1823. doi: 10.1054/bjoc.2001.2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Chene P. Inhibition of the p53-hdm2 interaction with low molecular weight compounds. Cell Cycle. 2004;3:460–461. doi: 10.4161/cc.3.4.791. [DOI] [PubMed] [Google Scholar]
- 260.Chene P. Inhibition of the p53-MDM2 interaction: targeting a protein-protein interface. Mol Cancer Res. 2004;2:20–28. [PubMed] [Google Scholar]
- 261.Uversky VN, Oldfield CJ, Dunker AK. Intrinsically disordered proteins in human diseases: introducing the D2 concept. Annu Rev Biophys. 2008;37:215–246. doi: 10.1146/annurev.biophys.37.032807.125924. [DOI] [PubMed] [Google Scholar]
- 262.Uversky VN, Oldfield CJ, Midic U, Xie H, Xue B, Vucetic S, Iakoucheva LM, Obradovic Z, Dunker AK. Unfoldomics of human diseases: linking protein intrinsic disorder with diseases. BMC Genomics. 2009;10(Suppl 1):S7. doi: 10.1186/1471-2164-10-S1-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Garner E, Romero P, Dunker AK, Brown C, Obradovic Z. Predicting binding regions within disordered proteins. Genome Inform Ser Workshop Genome Inform. 1999;10:41–50. [PubMed] [Google Scholar]
- 264.Cheng Y, Oldfield CJ, Meng J, Romero P, Uversky VN, Dunker AK. Mining alpha-helix-forming molecular recognition features with cross species sequence alignments. Biochemistry. 2007;46:13468–13477. doi: 10.1021/bi7012273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Oldfield CJ, Cheng Y, Cortese MS, Romero P, Uversky VN, Dunker AK. Coupled folding and binding with alpha-helix-forming molecular recognition elements. Biochemistry. 2005;44:12454–12470. doi: 10.1021/bi050736e. [DOI] [PubMed] [Google Scholar]
- 266.Chen CT, Wagner H, Still WC. Fluorescent, sequence-selective peptide detection by synthetic small molecules. Science. 1998;279:851–853. doi: 10.1126/science.279.5352.851. [DOI] [PubMed] [Google Scholar]
- 267.Kussie PH, Gorina S, Marechal V, Elenbaas B, Moreau J, Levine AJ, Pavletich NP. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science. 1996;274:948–953. doi: 10.1126/science.274.5289.948. [DOI] [PubMed] [Google Scholar]
- 268.Vassilev LT. Small-molecule antagonists of p53-MDM2 binding: research tools and potential therapeutics. Cell Cycle. 2004;3:419–421. [PubMed] [Google Scholar]
- 269.Klein C, Vassilev LT. Targeting the p53-MDM2 interaction to treat cancer. Br J Cancer. 2004;91:1415–1419. doi: 10.1038/sj.bjc.6602164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, Fotouhi N, Liu EA. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 271.Cheng Y, LeGall T, Oldfield CJ, Mueller JP, Van YY, Romero P, Cortese MS, Uversky VN, Dunker AK. Rational drug design via intrinsically disordered protein. Trends Biotechnol. 2006;24:435–442. doi: 10.1016/j.tibtech.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 272.Arkin MR, Wells JA. Small-molecule inhibitors of protein–protein interactions: progressing towards the dream. Nat Rev Drug Discov. 2004;3:301–317. doi: 10.1038/nrd1343. [DOI] [PubMed] [Google Scholar]
- 273.Arkin M. Protein–protein interactions and cancer: small molecules going in for the kill. Curr Opin Chem Biol. 2005;9:317–324. doi: 10.1016/j.cbpa.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 274.Cochran AG. Antagonists of protein-protein interactions. Chem Biol. 2000;7:R85–R94. doi: 10.1016/s1074-5521(00)00106-x. [DOI] [PubMed] [Google Scholar]
- 275.Hammoudeh DI, Follis AV, Prochownik EV, Metallo SJ. Multiple independent binding sites for small-molecule inhibitors on the oncoprotein c-Myc. J Am Chem Soc. 2009;131:7390–7401. doi: 10.1021/ja900616b. [DOI] [PubMed] [Google Scholar]
- 276.Uversky VN. Intrinsically disordered proteins may escape unwanted interactions via functional misfolding. Biochim Biophys Acta. 2011;1814:693–712. doi: 10.1016/j.bbapap.2011.03.010. [DOI] [PubMed] [Google Scholar]
- 277.Uversky VN. Intrinsically disordered proteins and novel strategies for drug discovery. Expert Opin Drug Discov. 2012;7:475–488. doi: 10.1517/17460441.2012.686489. [DOI] [PubMed] [Google Scholar]
- 278.Stamm S, Ben-Ari S, Rafalska I, Tang Y, Zhang Z, Toiber D, Thanaraj TA, Soreq H. Function of alternative splicing. Gene. 2005;344:1–20. doi: 10.1016/j.gene.2004.10.022. [DOI] [PubMed] [Google Scholar]
- 279.Oparin AI. The origin of life (in Russian) Moscow: Moscow Worker publisher; 1924. [Google Scholar]
- 280.Haldane JBS. The origin of life. In: Watts CA, editor. The rationalist annual for the year 1929. London: Watts & Co; 1929. pp. 3–10. [Google Scholar]
- 281.Miller SL. A production of amino acids under possible primitive earth conditions. Science. 1953;117:528–529. doi: 10.1126/science.117.3046.528. [DOI] [PubMed] [Google Scholar]
- 282.Miller SL, Urey HC. Organic compound synthesis on the primitive earth. Science. 1959;130:245–251. doi: 10.1126/science.130.3370.245. [DOI] [PubMed] [Google Scholar]
- 283.Crick FH. The origin of the genetic code. J Mol Biol. 1968;38:367–379. doi: 10.1016/0022-2836(68)90392-6. [DOI] [PubMed] [Google Scholar]
- 284.Wong JT. A co-evolution theory of the genetic code. Proc Natl Acad Sci USA. 1975;72:1909–1912. doi: 10.1073/pnas.72.5.1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Jukes TH. Possibilities for the evolution of the genetic code from a preceding form. Nature. 1973;246:22–26. doi: 10.1038/246022a0. [DOI] [PubMed] [Google Scholar]
- 286.Trifonov EN. Consensus temporal order of amino acids and evolution of the triplet code. Gene. 2000;261:139–151. doi: 10.1016/s0378-1119(00)00476-5. [DOI] [PubMed] [Google Scholar]
- 287.Poole AM, Jeffares DC, Penny D. The path from the RNA world. J Mol Evol. 1998;46:1–17. doi: 10.1007/pl00006275. [DOI] [PubMed] [Google Scholar]
- 288.Jeffares DC, Poole AM, Penny D. Relics from the RNA world. J Mol Evol. 1998;46:18–36. doi: 10.1007/pl00006280. [DOI] [PubMed] [Google Scholar]
- 289.Treiber DK, Williamson JR. Beyond kinetic traps in RNA folding. Curr Opin Struct Biol. 2001;11:309–314. doi: 10.1016/s0959-440x(00)00206-2. [DOI] [PubMed] [Google Scholar]
- 290.Cristofari G, Darlix JL. The ubiquitous nature of RNA chaperone proteins. Prog Nucleic Acid Res Mol Biol. 2002;72:223–268. doi: 10.1016/s0079-6603(02)72071-0. [DOI] [PubMed] [Google Scholar]
- 291.Gilbert W. Origin of life—the RNA world. Nature. 1986;319:618–618. [Google Scholar]
- 292.Csermely P. Proteins, RNAs and chaperones in enzyme evolution: a folding perspective. Trends Biochem Sci. 1997;22:147–149. doi: 10.1016/s0968-0004(97)01026-8. [DOI] [PubMed] [Google Scholar]
- 293.Doolittle WF. Uprooting the tree of life. Sci Am. 2000;282:90–95. doi: 10.1038/scientificamerican0200-90. [DOI] [PubMed] [Google Scholar]
- 294.Theobald DL. A formal test of the theory of universal common ancestry. Nature. 2010;465:219–222. doi: 10.1038/nature09014. [DOI] [PubMed] [Google Scholar]
- 295.Chemes LB, Glavina J, Alonso LG, Marino-Buslje C, de Prat-Gay G, Sanchez IE. Sequence evolution of the intrinsically disordered and globular domains of a model viral oncoprotein. PLoS One. 2012;7:e47661. doi: 10.1371/journal.pone.0047661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Chen SC, Chuang TJ, Li WH. The relationships among microRNA regulation, intrinsically disordered regions, and other indicators of protein evolutionary rate. Mol Biol Evol. 2011;28:2513–2520. doi: 10.1093/molbev/msr068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Lin YS, Hsu WL, Hwang JK, Li WH. Proportion of solvent-exposed amino acids in a protein and rate of protein evolution. Mol Biol Evol. 2007;24:1005–1011. doi: 10.1093/molbev/msm019. [DOI] [PubMed] [Google Scholar]
- 298.Shaiu WL, Hu T, Hsieh TS. The hydrophilic, protease-sensitive terminal domains of eucaryotic DNA topoisomerases have essential intracellular functions. Pac Symp Biocomput. 1999:578–589. doi: 10.1142/9789814447300_0057. [DOI] [PubMed] [Google Scholar]
- 299.Sayers EW, Gerstner RB, Draper DE, Torchia DA. Structural preordering in the N-terminal region of ribosomal protein S4 revealed by heteronuclear NMR spectroscopy. Biochemistry. 2000;39:13602–13613. doi: 10.1021/bi0013391. [DOI] [PubMed] [Google Scholar]
- 300.Wissmann R, Baukrowitz T, Kalbacher H, Kalbitzer HR, Ruppersberg JP, Pongs O, Antz C, Fakler B. NMR structure and functional characteristics of the hydrophilic N terminus of the potassium channel beta-subunit Kvbeta1.1. J Biol Chem. 1999;274:35521–35525. doi: 10.1074/jbc.274.50.35521. [DOI] [PubMed] [Google Scholar]
- 301.Brown CJ, Johnson AK, Dunker AK, Daughdrill GW. Evolution and disorder. Curr Opin Struct Biol. 2011;21:441–446. doi: 10.1016/j.sbi.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Nilsson J, Grahn M, Wright AP. Proteome-wide evidence for enhanced positive Darwinian selection within intrinsically disordered regions in proteins. Genome Biol. 2011;12:R65. doi: 10.1186/gb-2011-12-7-r65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Breydo L, Wu JW, Uversky VN. Alpha-synuclein misfolding and Parkinson's disease. Biochim Biophys Acta. 2012;1822:261–285. doi: 10.1016/j.bbadis.2011.10.002. [DOI] [PubMed] [Google Scholar]
- 304.George JM. The synucleins. Genome Biol. 2002;3:REVIEWS3002. doi: 10.1186/gb-2001-3-1-reviews3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Vonderviszt F, Kanto S, Aizawa S, Namba K. Terminal regions of flagellin are disordered in solution. J Mol Biol. 1989;209:127–133. doi: 10.1016/0022-2836(89)90176-9. [DOI] [PubMed] [Google Scholar]
- 306.Chen JW, Romero P, Uversky VN, Dunker AK. Conservation of intrinsic disorder in protein domains and families. I. A database of conserved predicted disordered regions. J Proteome Res. 2006;5:879–887. doi: 10.1021/pr060048x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Chen JW, Romero P, Uversky VN, Dunker AK. Conservation of intrinsic disorder in protein domains and families. II. functions of conserved disorder. J Proteome Res. 2006;5:888–898. doi: 10.1021/pr060049p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Xue B, Brown CJ, Dunker AK, Uversky VN. Intrinsically disordered regions of p53 family are highly diversified in evolution. Biochim Biophys Acta. 2013;1834:725–738. doi: 10.1016/j.bbapap.2013.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Daughdrill GW, Narayanaswami P, Gilmore SH, Belczyk A, Brown CJ. Dynamic behavior of an intrinsically unstructured linker domain is conserved in the face of negligible amino acid sequence conservation. J Mol Evol. 2007;65:277–288. doi: 10.1007/s00239-007-9011-2. [DOI] [PubMed] [Google Scholar]
- 310.Moesa HA, Wakabayashi S, Nakai K, Patil A. Chemical composition is maintained in poorly conserved intrinsically disordered regions and suggests a means for their classification. Mol Biosyst. 2012;8:3262–3273. doi: 10.1039/c2mb25202c. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.