

Abstract
The essential metabolic enzyme biotin protein ligase (BPL) is a potential target for the development of new antibiotics required to combat drug-resistant pathogens. Staphylococcus aureus BPL (SaBPL) is a bifunctional protein, possessing both biotin ligase and transcription repressor activities. This positions BPL as a key regulator of several important metabolic pathways. Here, we report the structural analysis of both holo- and apo-forms of SaBPL using X-ray crystallography. We also present small-angle X-ray scattering data of SaBPL in complex with its biotin-carboxyl carrier protein substrate as well as the SaBPL:DNA complex that underlies repression. This has revealed the molecular basis of ligand (biotinyl-5′-AMP) binding and conformational changes associated with catalysis and repressor function. These data provide new information to better understand the bifunctional activities of SaBPL and to inform future strategies for antibiotic discovery.
Keywords: biotin protein ligase, biotin-carboxyl carrier protein, DNA-binding, bacterial enzyme, biotinylation, small-angle X-ray scattering
Introduction
Biotin protein ligase (BPL) is a key enzyme in biotin biology that is responsible for specifically attaching the prosthetic group biotin onto biotin-dependent enzymes. In some organisms, it also plays a role in the regulation of biotin biosynthesis. Biotin (Vitamin H or B7) is an important micronutrient found throughout the biosphere. All living organisms use biotin as a prosthetic group for a family of enzymes that perform carboxylation, decarboxylation, and transcarboxylation of substrates in metabolic pathways. For example, acetyl CoA carboxylase catalyzes the first committed step in fatty acid biosynthesis, and pyruvate carboxylase is required to replenish the tricarboxylic acid cycle with oxaloacetate. Biotin is particularly important in bacterial pathogenesis. During infection, bacteria have a high demand for the micronutrient, and respond by increasing the expression of proteins required for de novo biotin synthesis and uptake.1–5 There is thus great interest in understanding the metabolic adaption processes that allow bacteria to survive during infection, in particular, the role of BPL.6–8
The ligation reaction that BPL catalyzes is well characterized,9 requiring biotin and ATP and proceeding through an adenylated intermediate, biotinyl-5′-AMP, before transfer of the biotin moiety to the biotin receiving enzymes at their biotin-carboxyl carrier protein (BCCP) domain. BPL isozymes fall into three structural classes. Class I enzymes are simple modules with the sole function of biotin attachment. High-resolution crystal structures have been reported for Pyrococcus horikoshii OT3 (PhBPL), Aquifex aeolicus, and Mycobacterium tuberculosis.10–12 These proteins exist as homodimers in both solution and in their crystallized forms. Other Class I enzymes, from both M. tuberculosis and A. aeolicus, have been reported to be monomeric, even in the presence of ligands.12,13 The two other classes contain the conserved catalytic module of Class I but differ in their N-terminal regions. Class II enzymes have an N-terminal DNA-binding domain, enabling dual functionality. Upon ligand binding, these BPLs homodimerize, facilitating a co-operative interaction with specific DNA sequences14,15 as exemplified by the Escherichia coli BPL (EcBPL, also known as BirA) (reviewed16). The dimers of Class I and Class II BPLs are structurally distinct as they use different surfaces for homodimerization. The Class III enzymes (which include human BPL) are monomeric and contain an N-terminal domain unrelated to the DNA-binding domain of Class II BPLs—the function of which has yet to be resolved. However, it has been shown to be required for substrate recognition during catalysis using a mechanism that is distinct from Class I and II enzymes.17–19 These key differences point to a plausible application for bacterial BPL as a target for novel antibiotics.
Bacteraemia caused by drug-resistant strains of Staphylococcus aureus is a major threat to health worldwide. Common bacterial infections are becoming more difficult and expensive to treat.20,21 To address this, research efforts have been devoted to identifying and validating novel drug targets. For example, pathways required for virulence, such as biotin metabolism, provide an untapped source of potential new targets.4,22,23 We have recently provided the first data to demonstrate it is possible to selectively inhibit SaBPL over both EcBPL and their human counterpart, holocarboxylase synthetase (HCS).24 Here, structural biology played an important role in the design of small molecule inhibitors with antibacterial activity against S. aureus. However, a greater understanding of how these inhibitors impact on SaBPL regulated pathways at the molecular and cellular level is required.
The BPL target in S. aureus (SaBPL) belongs to the bifunctional Class II enzymes. Genome annotation studies have revealed that it is required for the activation of two biotin-dependent enzymes, namely acetyl CoA carboxylase and pyruvate carboxylase. Additionally, it has been proposed that it functions as a transcriptional repressor of the biotin biosynthetic operon operator (bioO) and the biotin transporter bioY.25 By combining biotin ligase and transcriptional repressor activity in a single protein, BPL is uniquely placed as the key regulator of biotin metabolism in S. aureus. How these two activities are co-ordinated by SaBPL is still unclear. However, through extensive genetic and biochemical studies of the closely related E. coli BPL (EcBPL) it has been proposed that the homodimerization and protein substrate binding interfaces on BPL are mutually exclusive.26 Crystal structures of the EcBPL homodimer and PhBPL in complex with the BCCP subunit of methylmalonyl-CoA decarboxylase have been reported,27,28 providing some molecular detail for these protein:protein interactions. However, the Class I PhBPL enzyme does not undergo the monomer–dimer transition in response to biotin binding as seen with EcBPL,29 and also uses a distinctly different surface for homodimerization. Furthermore, as the Class I enzymes lack DNA-binding activity they fail to provide enough detail to elucidate the complex lifecycle of the Class II BPLs. The absence of any structural data of a Class II BPL in complex with DNA has also restricted attempts to delineate this system further.
Here, we report three X-ray crystal structures of SaBPL (1) in complex with biotin, (2) with the adenylated reaction intermediate biotinyl-5′-AMP, and (3) apo-SaBPL. The holo-structures reveal the details of the dimer formation as facilitated by the binding of the biotinyl ligand. We also present a detailed investigation of the intermolecular interactions of SaBPL with binding partners SaBCCP and the biotin biosynthetic operon operator (bioO) that is the DNA target of SaBPL using small-angle X-ray scattering (SAXS). This analysis illustrates the mode of DNA binding of SaBPL and confirms that the two protein:protein interactions, that is, homodimer and heterodimer, indeed compete for a common surface on SaBPL. These data provide a molecular mechanism that underlies the dual functions of this protein, paving the way for the development of a new class of antibiotics.
Results
Structural overview of apo- and holo-forms of SaBPL
To characterize the interaction between SaBPL and its native ligand biotinyl-5′-AMP, and its impact on SaBPL dimerization, we solved its crystal structure in apo-form, in complex with biotin alone (Bt-SaBPL) and in complex with biotinyl-5′-AMP (Btyl-SaBPL) (Table I). SaBPL was observed as a crystallographic dimer when either biotin or the reaction intermediate biotinyl-5′-AMP was bound to the catalytic site. In contrast, apo-SaBPL was a monomeric molecule with an overall fold analogous to the monomeric unit of dimeric SaBPL. Similarly to E. coli BPL the protein can be divided into three discrete domains [Fig. 1(A)]. The N-terminal domain (residues 1–61) forms a winged helix-turn-helix DNA-binding domain. The central catalytic domain (residues 68–274) forms an α–β structure containing eight β-sheets that form a major face of the catalytic site and seven α-helices that buttress against the β-sheet scaffold. The third, C-terminal domain (residues 282–323) is characterized by a fold similar to an SH3 domain31 and caps the catalytic site. Two short random coil linkers (residues 62–67 and 275–281) link these three domains. In the case of apo-SaBPL only, no electron density is visible for the loop that forms over the bound ligand, known as the biotin-binding loop ([BBL] residues 118–129 [β5–β6 loop]) and residue 288, due to disorder in the absence of ligand.
Table I.
Data collection and refinement statistics (molecular replacement)
| apo-SaBPL | Biotin-SaBPL | Btyl-SaBPL | |
|---|---|---|---|
| Data collectiona | |||
| Wavelength (Å) | 0.95364 | 0.99810 | 1.5418 (Cu Kα) |
| Space group | P21 | P42212 | P42212 |
| Cell dimensions | |||
| a, b, c (Å) | 50.1, 51.4, 67.6 | 94.2, 94.2, 130.9 | 93.6, 93.6, 130.7 |
| α, β, γ (°) | 90, 108, 90 | 90, 90, 90 | 90, 90, 90 |
| Resolution (Å) | 35.0–2.1 (2.16–2.1) | 40–3.2 (3.45–3.2) | 20.0–2.6 (2.67–2.6) |
| Rsym or Rmerge | 11.3 (43.3) | 5.2 (44.1) | 5.7 (36.2) |
| I/σI | 8.8 (2.8) | 11.1 (2.0) | 12.7 (2.1) |
| Completeness (%) | 96.1 (96.3) | 98.7 (92.6) | 98.3 (94.7) |
| Redundancy | 3.8 (3.9) | 4.6 (4.3) | 4.1 (3.7) |
| Refinement | |||
| Resolution (Å) | 35.0–2.1 | 30–3.2 | 20.0–2.6 |
| No. reflections | 17,599 | 9929 | 18,509 |
| Rwork/Rfree | 19.6/24.9 | 19.2/23.9 | 19.9/25.7 |
| No. atoms | |||
| Protein | 2484 | 2602 | 2602 |
| Ligand/ion | 0 | 16 | 38 |
| Water | 212 | 0 | 305 |
| B-factors | |||
| Protein | 32.1 | 57.6 | 50.8 |
| Ligand/ion | – | 58.2 | 55.2 |
| Water | 36.8 | – | 65.2 |
| R.M.S. deviations | |||
| Bond lengths (Å) | 0.019 | 0.015 | 0.022 |
| Bond angles (°) | 1.9 | 1.9 | 1.9 |
| % in most favored regions of a Ramachandran plot | 94.0 | 95.0 | 97.1 |
Diffraction data were collected from one crystal for each structure.
Figure 1.
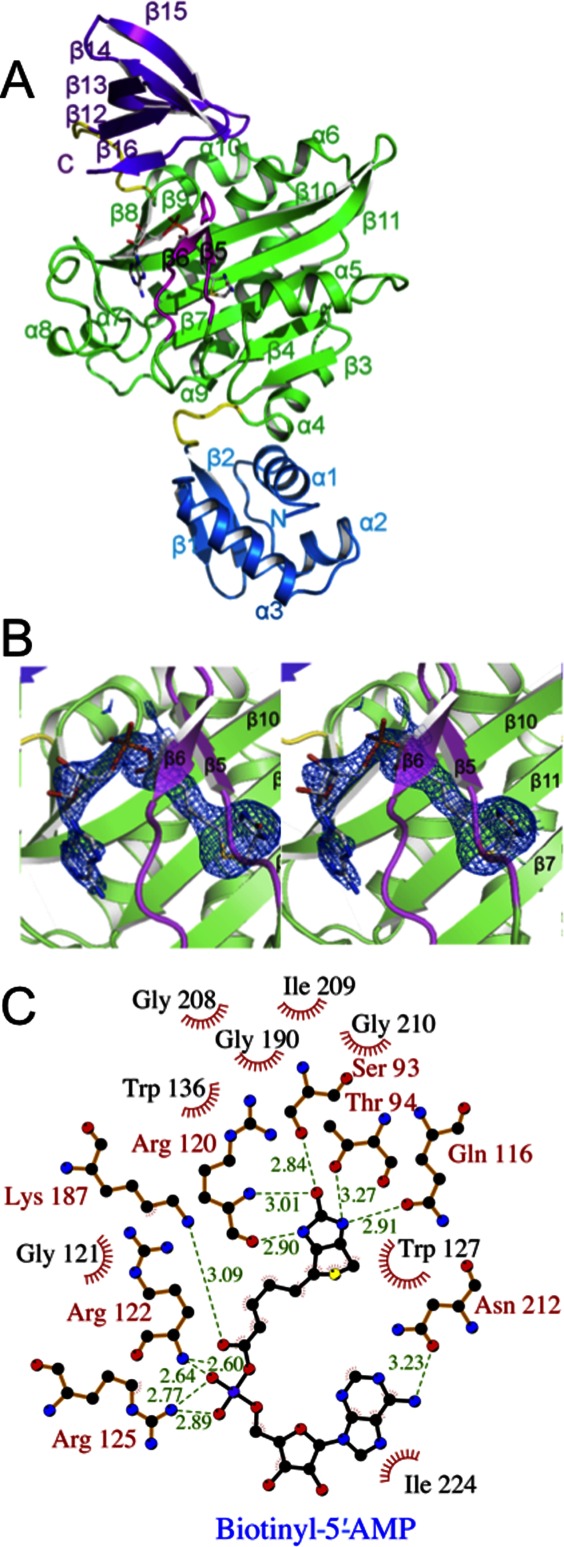
Crystal structure of SaBPL. (A) Cartoon schematic of Btyl-SaBPL monomer showing the N-terminal DNA-binding domain, residues 2–60 (blue), the central catalytic domain residues 75–268 (green), and the C-terminal cap domain 282–323 (purple). The random coil linkers residues 62–67 and 275–281 that connect the domains are in yellow. The corepressor biotinyl-5′-AMP is shown in stick representation. The BBL is highlighted in pink. (B) Cartoon schematic of the catalytic site of Btyl-SaBPL and the final 2Fo-Fc map (determined to 2.6 Å resolution and contoured at the 1 σ level) showing the positioning of biotinyl-5′-AMP (stick representation). (C) A schematic representation of contacts made to biotinyl-5′-AMP within the active site of SaBPL made using LIGPLOT.30 Residues involved in hydrogen bonding are annotated in red and hydrophobic interactions are annotated in black with red fray. Biotinyl-5′-AMP is shown with purple bonds and hydrogen bonding distance shown in Ångstroms by green dashed lines.
Structural details of biotinyl-5′-AMP and biotin interactions in holo-SaBPL
Biotinyl-5′-AMP (abbreviated “Btyl” when SaBPL complex is referred to) binds with a tight “V” shape geometry and a buried surface area of 727 Å2. Figure 1(B) shows the electron density around biotinyl-5′-AMP bound in the active site of SaBPL. The key interactions between SaBPL and biotinyl-5′-AMP are summarized in Figure 1(C). These interactions are consistent with those observed in the complex of SaBPL and a chemical analogue of biotinyl-5′-AMP, biotinol-5′-AMP (PDBID 3RKW24). The biotin moiety is bound in a predominantly hydrophobic pocket of SaBPL with the carbon chain of the valeric acid group and the tetrahydrothiophene component of biotin surrounded by Gly121, Trp127, Trp 136, Gly208, Gly 190, Ile209, and Gly210. Hydrogen bonds stabilize the interactions with sidechains from Ser93, Thr94, Gln116, and mainchain hydrogen bonds from Arg120 to the polar ureido head group of biotin. The oxygen atom of the tetrahydrothiophene is solvent exposed. The BBL is positioned over the biotin-binding pocket making multiple interactions with the ligand. The basic sidechains of Arg122 and Arg125 in the BBL interact with the carboxyl group of the valeric acid tail. The adenosine-binding site is formed by residues of β10, including Lys187, Ser128, Asn212, and the loop between α7 and α8, including Ile224, Arg227, and Ala228 (known as the adenosine-binding loop [ABL]), while Trp127 positions the adenylate moiety via π–π stacking interactions.
The Bt-SaBPL structure (containing biotin only) was resolved to the moderate resolution of 3.2 Å. Although not providing details of the position of some sidechains, it does confirm that there are no major differences in the positions of the domains of SaBPL between the holo-forms. The R.M.S.D. of the biotin-SaBPL structure compared to Btyl-SaBPL is 0.4 Å for 320 Cα atoms. Further, the mainchain of the ABL loop is in the same position even in the absence of the adenosyl moiety (the sidechains of this loop were not defined in the electron density). These structures, together with a recently reported structure of SaBPL with biotin-based ligands bound in the active site,32 show that any biotin ligand is sufficient to stabilize the BBL and to result in the formation of an SaBPL dimer.
The dimerization interface of Btyl-SaBPL
The dimerization interface of Btyl-SaBPL consists of α5-helix, the β5–β6 BBL, the β10–β11 loop including regions of these β-strands, the C-terminal end of the α6-helix, and β15–β16 loop from the C-terminal domain [Fig. 2(A)]. Together these form a buried surface of 1028.9 Å2.34 Central to the dimer interaction is the antiparallel β-strand arrangement of the β10-strand from one monomer to its counterpart on the other monomer to create an extended, albeit distorted, β-sheet joining the two monomers. The β10–β11 loop is juxtaposed against the opposing monomer and all the observed hydrogen bond and electrostatic interactions emanate from residues 194–203 to their partners on the opposing monomer. The α5-helix contributes to dimer stability via an antiparallel interaction with the α5-helix on the opposing monomer. The DNA-binding domains make no contact with each other and are spaced at their closest point (Lys3) 63.6 Å apart.
Figure 2.

SaBPL dimer. Cartoon schematic of the holo-SaBPL dimer. (A) The regions of secondary structure involved in forming the dimer interface are colored according to the program PROFACE33 and shown from two angles. The central cluster including β10– β11, β15– β16, and α5 are green and elements at the rim of the interface, including Lys118, α6, and the BBL loop, are colored red and yellow. (B) Contacts stabilized by the biotinyl-5′-AMP ligand are shown in context of the SaBPL dimer. One monomer is colored using the same scheme as in Figure 1, while the other monomer is gray. The ligand is in red stick figure representation. Residues involved in hydrogen bonding are shown in stick figure representation. Hydrogen bonds are indicated as dotted lines.
Of particular importance for its activity as a transcriptional repressor is the manner in which the dimer is stabilized by the bound biotin or biotinyl-5′-AMP ligand. Both the C-terminal domain and the BBL, which form direct contacts with the ligand, also make contact at the dimer interface. In particular, the β15–β16 loop of the C-terminal cap domain (residues 317–319) and residues Arg122 and Phe123 of the BBL reside in the dimer interface [Fig. 2(B)]. Interestingly, residues from both of these elements (Ser318 from the C-terminal domain and Arg122 from the BBL) form a hydrogen bond with Asp200 emanating from the end of the β10–β11 loop from the opposing monomer. Further, upon ligand binding and dimer formation, Asp320 forms a salt bridge with Arg125 in the catalytic core. This interaction appears to stabilize the loops between β5–β6 and the cap domain that forms the biotinyl-5′-AMP binding site.
Comparison between apo and ligand bound SaBPL
A comparison of the monomeric apo-SaBPL structure and dimeric holo-SaBPL reveals the structural perturbations that take place concomitant with ligand binding and the monomer–dimer transition. Apo- and holo-SaBPL structures display highly similar folds; the R.M.S.D of apo-SaBPL superposed with holo-SaBPL is 1.2 Å (between 302 Cα atoms). Apo-SaBPL, however, occurs as a monomer, with the BBL completely undefined (residues 118–129 are not defined by electron density). This is in contrast with holo-SaBPL where the BBL is positioned through contacts with biotin in the catalytic site, forming an interaction surface for dimerization. Other small differences occur in the position of loops between β7–α6, β10–β11, α7–α8, and β12–β13, respectively.
Structural analysis of SaBPL in complex with its target DNA
It has previously been predicted, but not shown, that dimeric SaBPL binds to the S. aureus bioO.25 To verify this binding interaction, we identified a 44-bp sequence containing an imperfect inverted repeat, and tested binding to holo-SaBPL using electrophoretic mobility shift assay. Binding of holo-SaBPL to this sequence (and not to a control 44-bp dsDNA of an unrelated sequence) was confirmed (Supporting Information Fig. S2 and Supporting Information Methods). The affinity of holo-SaBPL for the Sa-bioO DNA sequence was approximated to be nanomolar (KD = 100 nM) by EMSA titration.
To obtain structural information for this interaction, we subjected a complex of holo-SaBPL and the 44-bp DNA (henceforth referred to as Sa-bioO) to SAXS. SAXS data were collected for Btyl-SaBPL, the Sa-bioO, and the Btyl-SaBPL/Sa-bioO complexes. The experimental and calculated scattering curves are shown in Figure 3(A), the pair-wise distance distribution function, P(r), is shown in Figure 3(B) and structural parameters are reported in Table II. The radius of gyration (Rg) for Btyl-SaBPL was determined by Guinier analysis to be 30.7 Å and the pair-wise distribution P(r) for Btyl-SaBPL exhibits a bimodal distribution indicative of a dimer. The Rg and maximum dimension of the particle (Dmax) derived from the function (P(r)) profile were 30.2. and 97.0 Å, respectively. These values are consistent with those calculated from the crystal structure of Btyl-SaBPL. The Dmax derived from the P(r) profile for Sa-bioO was 125 Å, and the P(r) profile showed significant tailing towards a higher Dmax that is consistent with an elongated molecule such as dsDNA. Interestingly, this Dmax is shorter than the expected maximal dimension of an elongated 44-mer dsDNA (145 Å) suggesting that the dsDNA may be bent. This is possible as the Sa-bioO sequence contains a large number of A–T base pairs in its centre, which can increase the propensity of the dsDNA to bend. The P(r) profile for the Btyl-SaBPL/Sa-bioO complex exhibited both a bimodal distribution as well as tailing towards higher Dmax values. The Dmax of this complex was estimated to be 145 Å indicative of non-bent Sa-bioO upon binding of SaBPL.
Figure 3.

SAXS analysis of apo- and holo-SaBPL, SaBPL/DNA, and SaBPL/BCCP. (A) Scattering data for the six SaBPL, Sa-bioO, and SaBCCP samples (black crosses) with the predicted scattering curves (gray solid line) derived from models of these molecules and complexes (a.u. corresponds to arbitrary units). (B) Pair-wise distribution (P(r)) profiles for holo-SaBPL, Sa-bioO, and SaBPL/Sa-bioO complexes. (C) Ab initio reconstruction of the six samples overlaid with models derived from crystallographic structural data and modeling. (D) Pair-wise distribution (P(r)) profiles for apo-SaBPL, SaBCCP, and the SaBCCP/SaBPL complex. Note that each of the six scattering curves, P(r) profiles, and ab initio models are correlated by a Roman numeral label.
Table II.
Structural parameters calculated from SAXS data and model of SaBPL and complexes
| Molecule | Rg (Å)a (Guinier) | Rg (Å)b (P(r)) | Dmax (Å)c | χ2d | NSDe |
|---|---|---|---|---|---|
| Holo-SaBPL | |||||
| SAXS | 30.7 | 30.2 | 97.0 | 0.78 | 0.89 |
| Model (i) | 30.7 | 99.8 | |||
| Sa-bioO | |||||
| SAXS | 35.6 | 36.3 | 125.0 | 6.10 | 0.65 |
| Model (ii) | 36.7 | 145.0 | |||
| Sa-bioO/SaBPL | |||||
| SAXS | 44 | 44.4 | 145 | 0.6 | 0.81 |
| Model (iii) | 40.7 | 153.0 | |||
| Apo-SaBPL | |||||
| SAXS | 22.0 | 22.8 | 90 | 0.56 | 0.51 |
| Model (iv) | 22.3 | 86.3 | |||
| SaBCCP | |||||
| SAXS | 16.9 | 16.7 | 57 | 0.77 | 0.60 |
| Model (v) | 16.7 | 54.1 | |||
| SaBCCP/SaBPL | |||||
| SAXS | 22.5 | 22.8 | 75 | 0.98 | 0.69 |
| Model (vi) | 23.6 | 86.1 | |||
Rg—radius of gyration given by Guinier approximation.
Rg—estimated from pair-wise distribution function.
Dmax—maximum molecular dimension from P(r) function.
Goodness of fit of theoretical end experimental scattering curves calculated using FOXS.35
Normalized spatial discrepancy for ab initio SAXS models calculated from DAMAVER.36
Ab initio SAXS structures are displayed in Figure 3(C). Molecular models constructed from crystallographic structures are superimposed with the solution structure and the predicted scattering curve from this model was overlaid with the raw scattering data [Fig. 3(A)]. The ab initio SAXS structure for holo-SaBPL correlates remarkably well to the crystallographically determined Sa-BPL structure confirming the suitability of the system for SAXS analysis. In the case of Sa-bioO, the SAXS data indicate a solution structure that approximates the model based on extended B-form dsDNA [Fig. 3(C(ii))]. Third, an ab initio SAXS structure was calculated for the Btyl-SaBPL/Sa-bioO complex. Molecular coordinates were positioned into the SAXS structure using the program SITUS37 which resulted in the recognition helices (residues 20–40) of the N-terminal domain of SaBPL positioned in adjacent major grooves of the DNA. This gave good consistency between the SAXS structure and the model [Fig. 3(C(iii))] with dimeric SaBPL straddled across the DNA at an approximately 45° angle.
Interaction of SaBPL with SaBCCP
SAXS was also used to further characterize the interaction between Btyl-SaBPL and SaBCCP. Data were collected for both apo-SaBPL and biotinylated-SaBCCP separately and together in a heterodimeric complex. The experimental and calculated scattering curves are shown in Figure 3(A), the P(r) profiles shown in Figure 3(D) and structural parameters are reported in Table II. The apo-SaBPL SAXS data are in excellent agreement with the crystal structure. The Rg of 22.0 Å confirms the monomeric nature of this species. Likewise, the Rg and Dmax values for the biotinylated-SaBCCP are consistent with the size and shape expected, including a biotinyl-lysine group that is extended away from SaBCCP, in a conformation ideally suited for insertion into the intact carboxylase for catalysis. This is reflected in the P(r) plot as a small shoulder on the high Dmax side. Notably, upon the addition of SaBCCP to BtOH-SaBPL (i.e., SaBPL in complex with biotinol-5′-AMP, a nonhydrolysable analogue of biotinyl-5′-AMP), the SAXS data indicate the absence of dimeric SaBPL and instead report a shape that is slightly larger than apo-SaBPL. The P(r) plot closely resembles that of monomeric SaBPL; however, the Guinier Rg of 22.5 Å is slightly larger than that determined for apo-SaBPL.
The ab initio SAXS structure for apo-SaBPL is highly comparable to the crystallographically determined SaBPL monomer [Fig. 3(C(iv))]. The ab initio SAXS structure for the biotinylated-SaBCCP also accommodates the crystallographically determined SaBCCP structure (PDB ID: 3BG5) with an extrusion from one side that we suggest represents the biotinyl-lysine group [Fig. 3(C(v))]. A molecular model of SaBPL/BCCP was built using a monomer of holo-SaBPL in complex with the SaBCCP structure by analogy to the P. horikoshii BPL/BCCP complex (PDB ID: 2EJG). This required the repositioning of the C-terminal cap region of SaBPL relative to the catalytic domain by 5°, to open up the binding site to accommodate the SaBCCP (Supporting Information Fig. S3). The SaBPL/BCCP model is comparable to the ab initio SAXS structure [Fig. 3(C(vi))] although the fit of the scattering data calculated from this model with the experimental data was not ideal. This could have arisen from a number of sources. The juxtaposition of SaBCCP to the SaBPL could be subtly different to that observed for the Pyrococcus complex, and/or, the SAXS data may reflect the presence of small amounts of dimeric SaBPL or free SaBCCP. Importantly, it is clear that the SaBCCP disrupted the dimeric SaBPL.
Comparison with human BPL
Together the X-ray crystallographic and SAXS data verify the interactions of SaBPL with its DNA and BCCP binding partners. With this understanding of the surfaces of SaBPL important for its function, we considered the potential for SaBPL to be targeted by specific inhibitors designed to inhibit SaBPL function. In particular, we compared the SaBPL molecular surface to that predicted for human BPL (also known as human HCS). Figure 4(A) illustrates the predicted surface similarity of SaBPL and HCS, based upon a sequence alignment of catalytic and C-terminal cap domains [Fig. 4(B)]. Although the ligand-binding site itself contains conserved residues that are involved in ligand binding [identical residues are colored blue in Fig. 4(A)], a significant number of residues that immediately surround the site show considerable variation. In particular, the adjacent ABL and BBL provide distinct side-chain functionalities that could form the basis of selectivity in inhibitor molecules targeted to the ligand-binding site. Additionally, the proposed BCCP binding site identified in this study also represents a region of poor sequence similarity that could be exploited in SaBPL inhibitor development.
Figure 4.
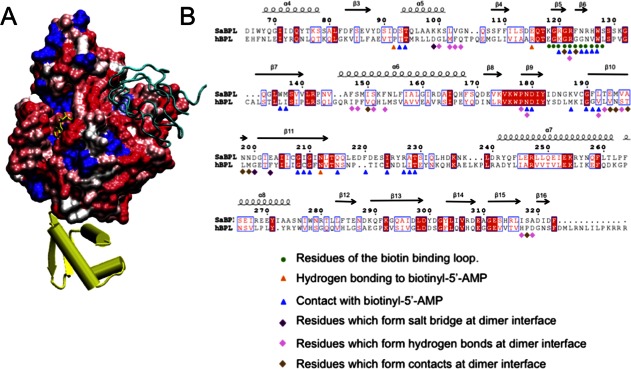
SaBPL as a potential drug target. (A) Similarity of surface amino acid residues of human BPL mapped to the surface of SaBPL. Identical amino acids are shown in blue. Sequence similarity is shown on a ramped scale from blue through white to red scored using the BLOSUM60 matrix where white indicates sequence similarity, pink and dark pink represent lesser similarity, and red represents the highest level of dissimilarity. The N-terminal DNA-binding domain is completely unrelated to the N-terminal domain of human BPL and is shown in yellow. SaBCCP modeled in its bound position is shown in cyan ribbons. (B) SaBPL sequence alignment with human SaBPL used for the surface comparison. Indicated are SaBPL amino acid residues involved at the catalytic site or the dimer interface of SaBPL.
Furthermore, the N-terminal domains of S. aureus and human BPL show no sequence homology at all. The N-terminal domains of eukaryotic BPLs are thought to perform an entirely different function that does not involve DNA binding.38 Truncations and mutations to the N-terminal of human BPL render an inactive enzyme, indicating an underlying reliance on this region for catalytic function that is not seen for prokaryotic BPLs.9,19,38,39 Together, these key differences in the isozymes from S. aureus and human provide a structural basis for the selective inhibition of BPL for antibiotic discovery.
Discussion
We have investigated the molecular detail of BPL from the important human pathogen S. aureus. The crystallographically determined structures of apo-SaBPL and SaBPL in complex with biotin and biotinyl-5′-AMP reveal that the enzyme undergoes a monomer to dimer transition upon ligand binding. Residues 118–129, that form the BBL, become ordered, as do specific loops creating part of the surface necessary for protein:protein interaction. Upon homodimer formation, the two N-terminal DNA binding domains are optimally positioned for an interaction with the major grooves at the inverted repeat sequences of the palindromic Sa-bioO DNA binding site.
This study confirms experimentally that SaBPL binds to the Sa-bioO DNA sequence and provides the first experimentally determined structure of the SaBPL/DNA complex using SAXS. By analogy to EcBPL, this complex will repress the transcription of genes containing the recognition motif in the promoter region. E. coli BPL controls expression of an operon containing the genes that encode the biotin biosynthetic enzymes, whereas in S. aureus, it is proposed that this operon as well as the gene encoding the biotin transport protein BioY are all regulated by BPL.25 Our SAXS derived structure of the holo-SaBPL/Sa-bioO complex reveals the arrangement of SaBPL straddled across the dsDNA. The relative positioning of the molecules differs from previous models proposed for EcBPL bound to dsDNA.27 This is due to slight differences in the dimer interface of holo-SaBPL and the positioning of the N-terminal domains revealed by X-ray crystallography compared to that of EcBPL (Supporting Information Fig. 4). Together these impact on their interaction at the major grooves of the DNA. Furthermore, the SAXS data reveal that the SaBPL dimer sits lower on the dsDNA than previous models have predicted, reflecting the highly complementary interface of the SaBPL dimer and target dsDNA.
This study also demonstrates that the presence of SaBCCP competes with SaBPL homodimer formation. SAXS data were consistent with the formation of a SaBPL/SaBCCP heterodimer, with no evidence of Btyl-SaBPL homodimer persisting in solution. This is consistent with a mutational study performed for EcBPL that tests the role of interface residues in homodimer and heterodimer formation.40 The data also suggest that the SaBPL/SaBCCP heterodimer forms analogously to the crystal structure of the P. horikoshii BPL/BCCP complex41 and as modeled for the EcBPL/BCCP.26 In our model of this complex, the reactive lysine of SaBCCP extends from the turn flanked between two β-strands. This strand-turn-strand structural element is positioned analogously to the β10-turn-β11 of the SaBPL partner in the dimeric form of SaBPL, albeit forming a parallel β-sheet interaction rather than antiparallel. This places the lysine towards the catalytic site where the biotin transfer can take place.
Together our data provide a molecular mechanism for biotin transfer in which the SaBCCP interacts at the dimer interface of holo-SaBPL (Fig. 5). Upon removal of the biotin from the catalytic site of SaBPL, the dimerization interface is destabilized—in particular by conformational changes to the BBL. This is predicted to free the biotinylated BCCP ready to perform its role in the biochemical pathway. In the absence of BCCP, holo-SaBPL forms a dimer and, with two DNA-binding domains held in position, is able to form a complex with the bioO dsDNA resulting in transcriptional repression. This article is the first report containing structural data to support this mechanism for a complex Class II BPL. Furthermore, this study shows that, in the case of SaBPL, it is the presence of apo-BCCP that determines whether SaBPL acts in its biotin ligase role or as a transcriptional repressor. Even in the presence of a biotin ligand, BCCP outcompetes homodimer formation and thus interferes with repressor activity.
Figure 5.
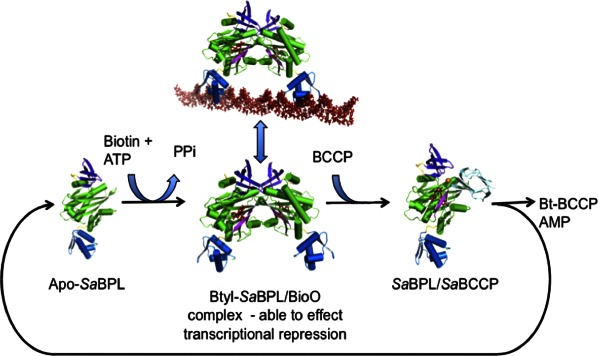
Schematic showing the regulation of SaBPL. The fate of SaBPL depends upon the availability of biotin for the conversion of apo-SaBPL to Btyl-SaBPL that is capable of effecting transcriptional repression and also the presence of BCCP that is able to compete for the SaBPL dimer surface becomes biotinylated resulting in the reformation of apo-SaBPL.
With the structural details for SaBPL, and its molecular interactions with binding partners, we have been able to assess the molecular surfaces of SaBPL and their suitability for the development of inhibitors. The biotinyl-5′-AMP binding site is clearly a critical site in SaBPL for the development of competitive inhibitors and, indeed, we have begun to exploit this site with the use of biotin analogues that bind tightly in the active site.24 Here, we have shown that this site, while clearly conserved between species for binding to biotinyl-5′AMP, possesses features that differ from human BPL that indicate the potential for the development of inhibitors specific for the bacterial BPL over the human form. Our work also confirms that the BCCP interface of SaBPL differs from that of human, suggesting it also as a site for potential inhibitor development. Third, the molecular details for the N-terminal domain of SaBPL that has no similarity at all to the N-terminal domain of human BPL may facilitate the development of yet another class of SaBPL inhibitor acting at the level of bioO derepression.
Because of the essential nature of BPL for survival, and S. aureus being one of the most clinically important pathogenic micro-organisms, our endeavor is to use the structural information reported here to target this protein for the development of new antibiotics. Targeting essential enzymes for which there are no pre-existing resistance mechanisms is a well-accepted strategy to combat the rise of drug-resistant bacteria. This data have provided a detailed understanding of the functional surfaces of SaBPL that will assist in the development of specific potent drugs against this class II BPL isoenzyme.
Materials and Methods
Preparation and crystallization of SaBPL
The cloning, purification, and crystallization of recombinant SaBPL have been previously reported.42 SaBPL was incubated with apo-BCCP for 1 h prior to final purification to ensure it was in the apo-form. Biotin or biotin+ATP+MgCl2 was added to apo-SaBPL prior to sample use to form holo-SaBPL. Both the apo- and holo-SaBPL crystals were grown from similar conditions.42
Preparation of SaBCCP
E. coli BL21 were transformed with a plasmid encoding GST-SaBCCP (residues 68–155 of acetyl-CoA carboxylase from S. aureus). The expression and purification of the biotin domain were performed as previously described.43
Structure determination of SaBPL
X-ray diffraction data for crystals of biotinyl-5′-AMP/SaBPL (Btyl-SaBPL) were collected in-house using a Rigaku RUH2R rotating copper anode X-ray source equipped with Osmic confocal optics, an R-Axis IV detector. Diffraction data for crystals of biotin/SaBPL (Bt-SaBPL) and apo-SaBPL were recorded at the MX1 beamline at the Australian Synchrotron using a MAR research 165 CCD detector. All data were collected with cryo-cooling using an Oxford Cryosystems 700 Series cryostream at 100 K.
The diffraction data were integrated using MOSFLM44 and the intensities were merged and scaled using the CPP4 suite of programs.45 Initial phases were determined for the Btyl-SaBPL crystal structure by molecular replacement using PHASER46 with the coordinates of the BPL from P. horikoshii OT3, PDB ID: 1WQ7, as the search model. The model was built with cycles of manual model building with COOT45 and refinement with REFMAC.47
The Btyl-SaBPL structure, with biotinyl-5′-AMP deleted, was used as the starting model for refinement of the Bt-SaBPL. Phases for the apo-SaBPL structure determination were determined by molecular replacement PHASER45 using holo-SaBPL as the search model. The quality of the final models was evaluated using MOLPROBITY.48 A final cycle of refinement was performed using the program PHENIX.49 Statistics for the data and refinement are reported in Table I.
Preparation of dsDNA Sa-bioO sequence
DNA of sequence: 5′-CCTTAAATGTAAACTTATTAATTATAAAAGTTTACATTTGGATT-3′ and its complementary strand were purchased (Geneworks) as single-stranded oligonucleotides in gel purified form. The oligonucleotides were resuspended in Tris-EDTA buffer. The annealing reaction was performed by mixing equal volumes of both strands of DNA at equimolar ratio. The mixture was heated to 95°C for 3 min, and slowly cooled down to room temperature over 1 h. The annealed Sa-bioO was then stored at 4°C. The dsDNA concentration was determined spectrophotometrically at λ = 260.
Preparation of SaBPL/SaBCCP and SaBPL/DNA samples for SAXS experiments
The SaBPL/DNA complex was formed by adding SaBPL and the Sa-bioO DNA together at 2:1 molar (dimeric SaBPL/DNA) concentrations (200 μM) in 50 mM Tris-HCl pH 7.0, 50 mM NaCl, 2 mM ATP, 2 mM biotin (or the nonhydrolysable analogue biotinol-5′-AMP), 10 mM MgCl2, and 5% glycerol. The SaBPL/SaBCCP complex was formed by adding the proteins at equimolar concentrations (200 μM) in presence of biotin and ATP (or biotinol-5′-AMP) in 50 mM Tris-HCl pH 7.0, 50 mM NaCl, and 5% glycerol. Just prior to the SAXS experiments all samples were buffer exchanged and concentrated in 3 K cutoff concentrators (Centricon) with the eluant used as the buffer blank. Sample concentrations were determined prior to complex formation spectrophotometrically at λ = 280 using experimentally determined extinction coefficients.
SAXS measurements and data reduction
SAXS was conducted for SaBPL and its complex with its cognate DNA (bioO) or SaBCCP as well as for the individual components of these complexes. SAXS measurements were made using the SAXS-WAXS beamline at the Australian Synchrotron, Melbourne, Australia, which is equipped with a Pilatus 1M-Detector. The scattering data were collected to provide an s range of 0.015–0.3 Å−1. Samples flowed past the X-ray beam in a 1.5 mm quartz capillary at room temperature during data collection. Scattering was collected over a range of concentrations, five concentrations between 0.2 and 2 mg/mL for each sample. The samples and matching buffer solutions were exposed to X-rays for 10 s, subdivided into 10 × 1 s exposures. The scattering images were integrated, averaged, and calibrated against water using software specific to the beamline. Scattering from the buffer and empty capillaries was subtracted after scaling scattering intensities to correspond to incident beam intensities. Data analysis was performed using the ATSAS suite of software. The program AUTOPOROD50 was used to yield the pair-wise distribution function (P(r)) and to determine Rg (from the second moment of the P(r) function) and Dmax. The Rg values calculated from the Guinear approximation and the P(r) were favorably comparable. The radius of gyration (Rg) did not vary significantly over the concentrations ranges of each species of molecule and all Guinear plots were linear for s.Rg 1.3.
Modeling of SAXS data
For each molecular species, the program DAMMIF51 was to generate 20 ab initio dummy atoms models. These models were superposed, merged, and filtered using the program DAMAVER.36 Theoretical scattering curves were calculated from the model coordinates and compared to the experimental data using FOXS.35
Molecular figures and sequence/structure analysis
Figures were prepared using PYMOL.52 LIGPLOT30 was used to generate Figure 1(C). The sequence alignment [Fig. 4(B)] was made using ESPript.53 Surface areas and contents were calculated using PISA.34 The surface of SaBPL shown in Figure 4(A) was colored using MULTISEQ (part of VMD54) according to sequence similarity based upon the BLOSUM60 matrix.
ACCESSION NUMBERS
Coordinates and structure factors have been deposited in the Protein Data Bank with accession numbers: Btyl-SaBPL PDBID = 3RIR, Bt-SaBPL PDBID = 3RKY, and, apo-SaBPL PDBID = 3RKX.
Acknowledgments
MCJW is an NHMRC Senior Research Fellow and acknowledges the support of the NHMRC and the Australian Research Council. NC was a Monash Centre for Synchrotron Science Fellow. Diffraction and scattering data were collected at the Australian Synchrotron MX1 and SAXS/WAXS beamlines. We also acknowledge the computer resources of the Victorian Partnership for Advanced Computing. We wish to thank Prof John Cronan (University of Illinois) for the kind gift of biotinol-5′-AMP.
Glossary
- BPL
biotin protein ligase
- BCCP
biotin-carboxyl carrier protein
- Btyl
biotinyl-5′-AMP
- SAXS
small-angle X-ray scattering
- ABL
adenosine-binding loop
- BBL
biotin-binding loop
- bioO
biotin biosynthetic operon operator
- BioY
biotin transporter
- HCS
human holocarboxylase synthetase.
Supplementary material
Additional Supporting Information may be found in the online version of this article.
Supplementary Figure 1. Comparison of apo (monomeric) and holo (dimeric) SaBPL. The cartoon representation of the dimer is coloured according to the similarity of the apo and holo SaBPL structures. Blue indicates that the structures are similar ie. the RMSD Cα atoms is less 2 Å. The colour is ramped through to red where the RMSD Cα is greater than 4 Å.
Supplementary Figure 2. Interaction of holo–SaBPL with the S. aureusbioO (Sa–bioO) DNA sequence. A) Electrophoretic mobility shift assay (EMSA) demonstrating that holo–SaBPL binds to the Sa–bioO DNA sequence but not to an unrelated dsDNA sequence. Lane 1: 0 μM Btyl–SaBPL+ 0.29 μM Sa–bioO; Lane 2: 0.29 μM Btyl–SaBPL+ 0.29 μM Sa–bioO; Lane 3: 0 μM Btyl–SaBPL+ 0.292 μM control non specific DNA; Lane 5: 0.29 μM Btyl–SaBPL + 0.292 μM control DNA. B) The region of the Staphylococcus aureus genome identified as Sa–bioO. Dashes are used to indicate the regions of the sequence that represent an inverted repeat and the bolded/boxed bases are the predicted regions of the sequence recognised by SaBPL.
Supplementary Figure 3. Model of the SaBPL/BCCP complex. Cartoon representation of the SaBCCP structure (PDB ID: 3BG5; coloured in yellow) positioned against one monomer of the SaBPL dimer by analogy to the BPL/BCCP complex from P. horikoshii (PDB ID: 2EJG). This required that the C–terminal cap (coloured light pink) be slightly repositioned to avoid clashes. BCCP interacts at the BPL homodimer interface, as seen from its overlap with the second SaBPL monomer (purple). Its reactive site lysine is positioned just above the BBL (dark pink) at the reactive site where biotin is transferred.
Supplementary Figure 4. Comparison of SaBPL dimer with EcBPL. SaBPL (top) and EcBPL (bottom: PDB ID: 1HXD) dimer structures are shown in cartoon representation. The SaBPL dimer interface differs from that of EcBPL resulting in a different angle of interaction of the catalytic domains (coloured green). Resultantly, the DNA binding domains (coloured light blue) are positioned differently in each model such that the α–helix 2 that binds at the major groove of target dsDNA are juxtaposed quite differently in the two models.
Supplementary Figure 5. SAXS analysis of apo– and holo–SaBPL, SaBPL/DNA and SaBPL/BCCP. Enlarged versions of the ab initio reconstruction of the six samples overlaid with models derived from crystallographic structural data and modelling as described in Figure 3.
Supplementary Figure 5. SAXS analysis of apo– and holo–SaBPL, SaBPL/DNA and SaBPL/BCCP. Enlarged versions of the ab initio reconstruction of the six samples overlaid with models derived from crystallographic structural data and modelling as described in Figure 3.
Supporting Information
References
- 1.Chiang SL, Mekalanos JJ. Use of signature-tagged transposon mutagenesis to identify Vibrio cholerae genes critical for colonization. Mol Microbiol. 1998;27:797–805. doi: 10.1046/j.1365-2958.1998.00726.x. [DOI] [PubMed] [Google Scholar]
- 2.Lee JM, Zhang S, Saha S, Santa Anna S, Jiang C, Perkins J. RNA expression analysis using an antisense Bacillus subtilis genome array. J Bacteriol. 2001;183:7371–7380. doi: 10.1128/JB.183.24.7371-7380.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sassetti CM, Rubin EJ. Genetic requirements for mycobacterial survival during infection. Proc Natl Acad Sci USA. 2003;100:12989–12994. doi: 10.1073/pnas.2134250100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bae T, Banger AK, Wallace A, Glass EM, Aslund F, Schneewind O, Missiakas DM. Staphylococcus aureus virulence genes identified by bursa aurealis mutagenesis and nematode killing. Proc Natl Acad Sci USA. 2004;101:12312–12317. doi: 10.1073/pnas.0404728101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bergman NH, Anderson EC, Swenson EE, Janes BK, Fisher N, Niemeyer MM, Miyoshi AD, Hanna PC. Transcriptional profiling of Bacillus anthracis during infection of host macrophages. Infect Immun. 2007;75:3434–3444. doi: 10.1128/IAI.01345-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eisenreich W, Dandekar T, Heesemann J, Goebel W. Carbon metabolism of intracellular bacterial pathogens and possible links to virulence. Nat Rev Microbiol. 2010;8:401–412. doi: 10.1038/nrmicro2351. [DOI] [PubMed] [Google Scholar]
- 7.Becher D, Hempel K, Sievers S, Zuhlke D, Pane-Farre J, Otto A, Fuchs S, Albrecht D, Bernhardt J, Engelmann S, Volker U, van Dijl JM, Hecker M. A proteomic view of an important human pathogen—towards the quantification of the entire Staphylococcus aureus proteome. PLoS One. 2009;4:e8176. doi: 10.1371/journal.pone.0008176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hecker M, Becher D, Fuchs S, Engelmann S. A proteomic view of cell physiology and virulence of Staphylococcus aureus. Int J Med Microbiol. 2010;300:76–87. doi: 10.1016/j.ijmm.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 9.Pendini NR, Bailey LM, Booker GW, Wilce MC, Wallace JC, Polyak SW. Microbial biotin protein ligases aid in understanding holocarboxylase synthetase deficiency. Biochim Biophys Acta. 2008;1784:973–982. doi: 10.1016/j.bbapap.2008.03.011. [DOI] [PubMed] [Google Scholar]
- 10.Bagautdinov B, Kuroishi C, Sugahara M, Kunishima N. Crystal structures of biotin protein ligase from Pyrococcus horikoshii OT3 and its complexes: structural basis of biotin activation. J Mol Biol. 2005;353:322–333. doi: 10.1016/j.jmb.2005.08.032. [DOI] [PubMed] [Google Scholar]
- 11.Gupta V, Gupta RK, Khare G, Salunke DM, Surolia A, Tyagi AK. Structural ordering of disordered ligand-binding loops of biotin protein ligase into active conformations as a consequence of dehydration. PLoS One. 2010;5:e9222. doi: 10.1371/journal.pone.0009222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tron CM, McNae IW, Nutley M, Clarke DJ, Cooper A, Walkinshaw MD, Baxter RL, Campopiano DJ. Structural and functional studies of the biotin protein ligase from Aquifex aeolicus reveal a critical role for a conserved residue in target specificity. J Mol Biol. 2009;387:129–146. doi: 10.1016/j.jmb.2008.12.086. [DOI] [PubMed] [Google Scholar]
- 13.Purushothaman S, Gupta G, Srivastava R, Ramu VG, Surolia A. Ligand specificity of group I biotin protein ligase of Mycobacterium tuberculosis. PLoS One. 2008;3:e2320. doi: 10.1371/journal.pone.0002320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abbott J, Beckett D. Cooperative binding of the Escherichia coli repressor of biotin biosynthesis to the biotin operator sequence. Biochemistry. 1993;32:9649–9656. doi: 10.1021/bi00088a017. [DOI] [PubMed] [Google Scholar]
- 15.Eisenstein E, Beckett D. Dimerization of the Escherichia coli biotin repressor: corepressor function in protein assembly. Biochemistry. 1999;38:13077–13084. doi: 10.1021/bi991241q. [DOI] [PubMed] [Google Scholar]
- 16.Beckett D. Biotin sensing at the molecular level. J Nutr. 2009;139:167–170. doi: 10.3945/jn.108.095760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hassan YI, Moriyama H, Olsen LJ, Bi X, Zempleni J. N- and C-terminal domains in human holocarboxylase synthetase participate in substrate recognition. Mol Genet Metabol. 2009;96:183–188. doi: 10.1016/j.ymgme.2008.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ingaramo M, Beckett D. Distinct amino termini of two human HCS isoforms influence biotin acceptor substrate recognition. J Biol Chem. 2009;284:30862–30870. doi: 10.1074/jbc.M109.046201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mayende L, Swift RD, Bailey LM, da Costa TP, Wallace JC, Booker GW, Polyak SW. A novel molecular mechanism to explain biotin-unresponsive holocarboxylase synthetase deficiency. J Mol Med (Berl) 2012;90:81–88. doi: 10.1007/s00109-011-0811-x. [DOI] [PubMed] [Google Scholar]
- 20.Deleo FR, Otto M, Kreiswirth BN, Chambers HF. Community-associated meticillin-resistant Staphylococcus aureus. Lancet. 2010;375:1557–1568. doi: 10.1016/S0140-6736(09)61999-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Turnidge JD, Kotsanas D, Munckhof W, Roberts S, Bennett CM, Nimmo GR, Coombs GW, Murray RJ, Howden B, Johnson PD, Dowling K. Staphylococcus aureus bacteraemia: a major cause of mortality in Australia and New Zealand. Med J Aust. 2009;191:368–373. doi: 10.5694/j.1326-5377.2009.tb02841.x. [DOI] [PubMed] [Google Scholar]
- 22.Liu CI, Liu GY, Song Y, Yin F, Hensler ME, Jeng WY, Nizet V, Wang AH, Oldfield E. A cholesterol biosynthesis inhibitor blocks Staphylococcus aureus virulence. Science. 2008;319:1391–1394. doi: 10.1126/science.1153018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heras B, Shouldice SR, Totsika M, Scanlon MJ, Schembri MA, Martin JL. DSB proteins and bacterial pathogenicity. Nat Rev Microbiol. 2009;7:215–225. doi: 10.1038/nrmicro2087. [DOI] [PubMed] [Google Scholar]
- 24.Soares da Costa TP, Tieu W, Yap MY, Pendini NR, Polyak SW, Sejer Pedersen D, Morona R, Turnidge JD, Wallace JC, Wilce MC, Booker GW, Abell AD. Selective inhibition of biotin protein ligase from Staphylococcus aureus. J Biol Chem. 2012;287:17823–17832. doi: 10.1074/jbc.M112.356576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rodionov DA, Mironov AA, Gelfand MS. Conservation of the biotin regulon and the BirA regulatory signal in Eubacteria and Archaea. Genome Res. 2002;12:1507–1516. doi: 10.1101/gr.314502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weaver LH, Kwon K, Beckett D, Matthews BW. Competing protein: protein interactions are proposed to control the biological switch of the E. coli biotin repressor. Protein Sci. 2001;10:2618–2622. doi: 10.1110/ps.32701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weaver LH, Kwon K, Beckett D, Matthews BW. Corepressor-induced organization and assembly of the biotin repressor: a model for allosteric activation of a transcriptional regulator. Proc Natl Acad Sci USA. 2001;98:6045–6050. doi: 10.1073/pnas.111128198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bagautdinov B, Matsuura Y, Bagautdinova S, Kunishima N. Protein biotinylation visualized by a complex structure of biotin protein ligase with a substrate. J Biol Chem. 2008;283:14739–14750. doi: 10.1074/jbc.M709116200. [DOI] [PubMed] [Google Scholar]
- 29.Wood ZA, Weaver LH, Brown PH, Beckett D, Matthews BW. Co-repressor induced order and biotin repressor dimerization: a case for divergent followed by convergent evolution. J Mol Biol. 2006;357:509–523. doi: 10.1016/j.jmb.2005.12.066. [DOI] [PubMed] [Google Scholar]
- 30.Wallace AC, Laskowski RA, Thornton JM. LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 1995;8:127–134. doi: 10.1093/protein/8.2.127. [DOI] [PubMed] [Google Scholar]
- 31.Noble MEM, Musacchio A, Saraste M, Courtneidge SA, Wierenga RK. Crystal structure of the SH3 domain in human Fyn—comparison of the 3-dimensional structures of SH3 domains in tyrosine kinases and spectrin. EMBO J. 1993;12:2617–2624. doi: 10.2210/pdb1shf/pdb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Soares Da Costa TP, Tieu W, Yap MY, Zvarec OJ, Bell JM, Turnidge JD, Wallace JC, Booker GW, Wilce MC, Abell AD, Polyak SW. Biotin analogues with antibacterial activity are potent inhibitors of biotin protein ligase. ACS Med Chem Lett. 2012;3:509–514. doi: 10.1021/ml300106p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Saha RP, Bahadur RP, Pal A, Mandal S, Chakrabarti P. ProFace: a server for the analysis of the physicochemical features of protein-protein interfaces. BMC Struct Biol. 2006;6:11. doi: 10.1186/1472-6807-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 35.Schneidman-Duhovny D, Hammel M, Sali A. FoXS: a web server for rapid computation and fitting of SAXS profiles. Nucleic Acids Res. 2010;38(Suppl):W540–544. doi: 10.1093/nar/gkq461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Volkov V, Svergun D. Uniqueness of ab initio shape determination in small-angle scattering. J Appl Crystallogr. 2003;36:860–864. doi: 10.1107/S0021889809000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wriggers W, Milligan RA, McCammon JA. Situs: a package for docking crystal structures into low-resolution maps from electron microscopy. J Struct Biol. 1999;125:185–195. doi: 10.1006/jsbi.1998.4080. [DOI] [PubMed] [Google Scholar]
- 38.Campeau E, Gravel RA. Expression in Escherichia coli of N- and C-terminally deleted human holocarboxylase synthetase. Influence of the N-terminus on biotinylation and identification of a minimum functional protein. J Biol Chem. 2001;276:12310–12316. doi: 10.1074/jbc.M009717200. [DOI] [PubMed] [Google Scholar]
- 39.Otsuka A, Abelson J. The regulatory region of the biotin operon in Escherichia coli. Nature. 1978;276:689–694. doi: 10.1038/276689a0. [DOI] [PubMed] [Google Scholar]
- 40.Adikaram PR, Beckett D. Functinal versatility of a single protein surface in two protein:protein interactions. J Mol Biol. 2012;419:223–233. doi: 10.1016/j.jmb.2012.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bagautdinov B, Matsuura Y, Bagautdinova S, Kunishima N. Protein biotinylation visualized by a complex structure of biotin protein ligase with a substrate. J Biol Chem. 2008;283:14739–14750. doi: 10.1074/jbc.M709116200. [DOI] [PubMed] [Google Scholar]
- 42.Pendini NR, Polyak SW, Booker GW, Wallace JC, Wilce MC. Purification, crystallization and preliminary crystallographic analysis of biotin protein ligase from Staphylococcus aureus. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2008;64:520–523. doi: 10.1107/S1744309108012244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chapman-Smith A, Turner DL, Cronan JE, Morris TW, Wallace JC. Expression, biotinylation and purification of a biotin-domain peptide from the biotin carboxy carrier protein of Escherichia coli acetyl-CoA carboxylase. Biochem J. 1994;302:881–887. doi: 10.1042/bj3020881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Leslie AG. Integration of macromolecular diffraction data. Acta Crystallogr D Biol Crystallogr. 1999;55:1696–1702. doi: 10.1107/s090744499900846x. [DOI] [PubMed] [Google Scholar]
- 45.The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 46.Lopez G, Valencia A, Tress ML. Firestar–prediction of functionally important residues using structural templates and alignment reliability. Nucleic Acids Res. 2007;35:W573–577. doi: 10.1093/nar/gkm297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 48.Lovell SC, Davis IW, Arendall WB, 3rd, de Bakker PI, Word JM, Prisant MG, Richardson JS, Richardson DC. Structure validation by Calpha geometry: phi,psi and Cbeta deviation. Proteins. 2003;50:437–450. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- 49.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Konarev P, Volkov V, AV S, Koch M, Svergun D. PRIMUS: a windows PC-based system for small-angle scattering data analysis. J Appl Cryst. 36:1277–1282. [Google Scholar]
- 51.Franke D, Svergun DI. DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering. J Appl Cryst. 42:342–346. doi: 10.1107/S0021889809000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.The PyMOL Molecular graphics system. 2002. (Version 1.5.0.2 Schrödinger, LLC.)
- 53.Gouet P, Robert X, Courcelle E. ESPript/ENDscript: extracting and rendering sequence and 3D information from atomic structures of proteins. Nucleic Acids Res. 2003;31:3320–3323. doi: 10.1093/nar/gkg556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Humphrey W, Dalke A, Schulten K. VMD—visual molecular dynamics. J Mol Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.