

Abstract
Perforin plays a key role in the immune system via pore formation at the target cell membrane in the elimination of virus-infected and transformed cells. A vast number of observed mutations in perforin impair this mechanism resulting in a rare but fatal disease, familial hemophagocytic lymphohistiocytosis type 2 (FHL2). Here we report a comprehensive in silico structural analysis of a collection of 76 missense perforin mutations based on a proposed pore model. In our model, perforin monomers oligomerize having cyclic symmetry in consistent with previously found experimental constraints yet having flexibility in the size of the pore and the number of monomers involved. Clusters of the mutations on the model map to three distinct functional regions of the perforin. Calculated stability (free energy) changes show that the mutations mainly destabilize the protein structure, interestingly however, A91V polymorphism, leads to a more stable one. Structural characteristics of mutations help explain the severe functional consequences on perforin deficient patients. Our study provides a structural approach to the mutation effects on the perforin oligomerization and impaired cytotoxic function in FHL2 patients.
Keywords: perforin gene mutations, FHL2, familial hemophagocytic lymphohistiocytosis, structural analysis, FHL2 patients
Introduction
Familial hemophagocytic lymphohistiocytosis (FHL) is an autosomal recessive disorder in which five types corresponding to five causative loci have been described so far: FHL1 at 9q21.3–q22 (unknown gene), FHL2 at 10q22.1–q22 (perforin1, PRF1 gene), FHL3 at 17q25.1 (UNC13D gene), FHL4 at 6q24.2 (STX11 gene), and FHL5 at 19p13.3–13.2 (STXBP2 gene). However, approximately 10% of FHL cases still lack a genetic basis, one or more genes yet unidentified might be involved. Each of these genes encodes for a protein that is charged in various steps of secretory granule-mediated death pathway. FHL2 is a rare but lethal disorder characterized by fever, hepatosplenomegaly, cytopenia, hyperferritinemia, hypertriglyceridemia and/or hypofibrinogenemia, decreased natural killer (NK) cell activity, increased CD25 level, and hemophagocytosis. Patients suffering from FHL2 show a variety of phenotypes that are often associated with severe clinical symptoms resulting in death if not treated with limited treatment capability (HLH–2004 protocol1 and bone marrow transplantation). Sequencing of PRF1 gene in these patients proved the link between the disease and the perforin mutations.2
Perforin is involved in one of the primary mechanisms of lymphocyte-mediated cytolysis. Immune response of cytotoxic T cells or natural killer (NK) cells against viruses and pathogenic agents are initiated by the secretion of cytolytic granules including perforin and granzyme B onto the target membranes of the infected cells. The defect in this mechanism caused by perforin mutations leads to the symptoms of familial hemophagocytic lymphohistiocytosis type 2 (FHL2).2–4 Absence/reduction of functional perforin proves its indispensable role in the immune system. Consequently, patients face infections caused by viruses or pathogens due to the insufficient killing of the infected cells, which triggers the overproduction of activated cytokines.5,6
Perforin mutations have been reported to account for up to 40% of total FHL cases.2,7 More than 100 mutations in perforin were observed so far, of which the significant majority were reported as disease-linked.8 These mutations can be in homozygous, heterozygous, or compound heterozygous form, where the last one results in varying phenotypes. Genotype–phenotype studies show that there is a strong correlation between the genetic defect and the function of perforin.9–11 The impact of the mutations appears as absent or reduced perforin activity, which in turn drive severe clinical symptoms in the patients. Although FHL2 patients were diagnosed in several countries, some single mutations have higher incidences in some certain populations (such as W374X in Turkish population,12 L17X in African-Americans, and L364X in Japanese people10) which might be due to the presence of consanguineous marriage or founder effect. Although deletion, insertion, nonsense, and missense type of mutations have been described in functional domains of perforin, the mechanism underlying the genotype–phenotype correlation still remains to be elusive.3,7,13–17
Perforin is a 67-kDa multidomain protein and it oligomerizes to form a pore on the target cell membrane as the key step to its cytotoxic function. Perforin is a thin key-shaped molecule made of three domains: an amino-terminal membrane attack complex perforin like (MACPF)/cholesterol-dependent cytolysin (CDC) domain, an epidermal growth factor (EGF) domain, and a Ca2+-dependent membrane binding C-terminal C2 domain. The sequence similarity between perforin and complement components C6-C9 of the membrane attack complex suggests a common mechanism of transmembrane channel formation (MAC). Lately, the data from structural studies revealed that perforin has also homologous domains in bacterial CDCs.18–21 Although perforin utilizes MACPF domain for defense as opposite to the attacking function as in bacteria, it was strongly suggested that they share a common mechanism, until the differences between two have been unraveled22 showing the flexibility of the protein. The essentiality of perforin for cytotoxic lymphocyte function has been known for decades, however, the molecular and structural bases for membrane binding and pore formation have been recently revealed.22,23 Observed mutations in perforin are well documented so far, but the structure–function relationship of the protein leading pathogenicity remains to be deciphered. A number of studies published so far are limited to present novel mutations and the clinical findings on FHL2. In this study, we collect all available mutation data on perforin and their associated phenotypic effects. We model the three-dimensional structure of the pore-forming perforin complex. We then investigate the possible functional consequences of perforin mutations based on this model structure and the resulting effect on phenotypes of FHL2 patients. We find that a large collection of missense perforin mutations map to three distinct functional regions of perforin where Ca binding, membrane insertion, and oligomerization occur. The mutations occupy critical positions on the structure in terms of stability. The majority of them cause an increase in the total free energy leading to the instability of the protein. However, A91V is an exception to this, which is widely accepted as a neutral polymorphism. Here, we also propose a model for the initial state of perforin pore, which is consistent with the previously known intermolecular interactions. The analysis of the clinical data of the FHL2 patients suggests the existence of the relationship between the structural defects and the observed symptoms. The phenotype of the patient is related to the characteristics of the corresponding mutation. Our study, to our knowledge, is the first example that associates the present clinical data at such large scale with a novel structural analysis.
Results
Perforin is a protein having many lethal mutations mostly spread on its functional domains. In order to acquire a complete understanding of the role of the mutated residues in protein function, we included all known mutations in our analysis. In HGMD,8 currently a total of 103 reported mutations is given including missense, nonsense, deletion, and insertion types of mutations (Table I). Here we analyzed the 76 missense perforin mutations that we collected from the public databases and the reported cases in the literature (see Supporting Information Table S1). Among these, the majority (58 residues) is found in the large MACPF domain (which is composed of 349 residues) whereas another significant portion (13) resides in the Ca-dependent lipid-binding domain C2 (83 residues). It should be noted that the total number of different mutation sites is 65, indicating that more than one mutation occurred at some residues. Large number of mutations with their high proportion in functional domains particularly enabled us to infer substantial associations regarding the mutation–disease relationship, providing a broader outlook compared to previous studies where smaller groups of mutations were considered.
Table I.
Mutation Types and Corresponding Number of Mutations in Perforin as Currently Given in HGMDa (http://www.hgmd.cf.ac.uk/ac/gene.php?gene=PRF1)
| Mutation type | Number of mutations |
|---|---|
| Missense/nonsense | 86 |
| Small deletions | 13 |
| Small insertions | 4 |
| Total 103 |
The Human Gene Mutation Database.
The perforin model we created has its basis on a recent experimental observation on murine perforin22; however, in our study, we started with human perforin (homology model) to build up our pore model. With this step, we aimed to avoid the vicarious inference from the murine perforin pore that may possibly differ from the human perforin pore, although there is no such evidence reported so far, expecting to have a direct assessment on the functional impact of mutations that are observed in human. Our model is a representative of experimental data that is known about pore formation in human (Fig. 1), where 20 monomers are present (C20 symmetry) having ∼14 nm diameter.
Figure 1.

A model for perforin pore. Each monomer is shown in a different color (20 monomers are in the model). The model is consistent with the three experimental constraints: i) Perforin monomers interact via the opposite flat faces of MACPF domain.23 ii) Arg214 (positively charged) and Glu344 (negatively charged) undergo a direct ionic interaction between two adjacent monomers, as well as Asp192 (negative) is important for oligomerization.23 iii) The pore is symmetric where orientation of MACPF domain is opposite to that of in CDCs22 (rotational cyclic symmetry of C20 is shown).
Clustering mutations that are close in the 3D structure resulted in three large clusters where each includes at least two pairs of “connected” mutations. Cluster 1, Cluster 2, and Cluster 3 consist of 34, 13, and 10 mutations, respectively (Table II). Other than these, one cluster including one pair of connected mutations and six clusters including single mutations were found, which were disregarded to be discussed further since the number of mutations included are not sufficient to be associated with a common feature. Cluster 1 and Cluster 2 are located in the MACPF domain whereas Cluster 3 sits at the tip of the C2 domain. Mapping the mutation clusters on the perforin structure reveals three distinct regions where each of them depicts a different functional group of perforin (Fig. 2). Cluster 1 maps the region where perforin monomers interact with each other during oligomerization, including nearby mutations to residues Asp192, Arg214, and Glu344, which have been previously shown to undergo ionic interactions required for the pore assembly.23 Cluster 2 maps the region where the two small clusters of α helices (CH1 and CH2) are located that they refold into membrane-spanning amphipathic β strands which is a crucial step for the membrane insertion. Last, Cluster 3 maps the region where C2 domain exerts its function, Ca-binding membrane attachment, which is a prerequisite for the pore formation. Mutations within the same cluster are expected to have similar overall effect in contribution to the disease phenotype rather than the ones in different clusters. Therefore each of these three cluster-functional region associations suggests a distinct possible explanation for the disruption of perforin function at structural base.
Table II.
Clusters of Missense Perforin Mutations and Their Functional Assignment on the Protein Structure
| Cluster no | Domain | Region | Function |
|---|---|---|---|
| Cluster 1 | MACPF | Interface of perforin monomers | Oligomerization |
| Cluster 2 | MACPF | Small clusters of α helices | Membrane-spanning |
| Cluster 3 | C2 | Ca binding domain | Membrane attachment |
Residue numbers in clusters:Cluster 1: 38, 39, 45, 50, 54, 70, 73, 89, 91, 95, 102, 119, 123, 126, 149, 177, 183, 193, 198, 201, 215, 220, 221, 222, 224, 225, 232, 305, 306, 334, 345, 356, 357, 361Cluster 2: 157, 168, 170, 240, 252, 253, 261, 262, 279, 298, 299, 313, 317Cluster 3: 426, 429, 430, 435, 438, 450, 459, 481, 486, 491
Figure 2.

Three main clusters of perforin mutation regions shown on the model structure.
The structural effects of mutations are strongly associated with their location on protein structure. Mutations which occur on the surface of the protein may disrupt the binding site whereas those occur in the core region distort the protein conformation. We classified the mutated residues in perforin as buried, exposed, and residues with intermediate exposure according to their relative solvent-accessible surface areas (RASA) (Table III). Our analysis shows that perforin mutations are uniformly distributed to the surface and core regions of the protein.
Table III.
The Number of Mutations in Three Groups Classified According to Relative Accessible Surface Area
| RASAa | ||||
|---|---|---|---|---|
| <5% | Between 5% and 30% | >30% | ||
| Number of mutations | 20 (113) | 28 (184) | 17 (234) | |
The numbers in brackets show the total number of residues with the given RASA thresholds for each group. <5%: buried, between 5% and 30%: intermediate exposure, >30%: exposed.
Relative accessible surface area. RASA values for each mutated residue are given in Table VI.
Mutations are often associated with the stability of protein structure, resulting in free energy changes. As a result, the native state of the protein may be totally lost as in the case of nonsense or frame shift mutations, or may be partially altered as in our case of missense mutations. To have further insight on these mutations, we analyzed in detail the total energy changes on the perforin caused by the 76 observed missense mutations. To assess the specificity, not only the 76 observed mutations but also all the possible missense mutations over the entire sequence (531 × 20 = 10,620 where n is the total residue number) were computationally studied. A statistical comparison between three samples was performed, where the first sample is the 65 clinically observed different mutation sites with the total number of destabilizing (ΔΔG > 2 kcal/mol) mutations per site. The second and the third samples are the entire sequence (n = 531) and the non-mutation sites (n = 466), respectively. We concluded that observed mutations occupy critical positions for perforin stability, having significantly greater energy changes than that in the random case (P = 0.008 and SD = 7.05% between sample 1 (n = 65, mean = 8.91) and sample 2 (n = 531, mean = 6.36); P = 0.002 and SD = 6.99% between sample 1 and sample 3 (n = 466, mean = 6.00)).
Further stability analysis was carried on the types of mutations and the relative accessible surface areas of the corresponding residues. The mutations were classified according to the residue types into four groups as charged → charged (8%), charged → neutral (30%), neutral → charged (20%), and neutral → neutral (42%) (Supporting Information Table S2). Among these ΔΔG values of the third group (neutral → charged) were particularly high showing that charge contribution of the mutated residues greatly lowers the overall stability of the protein as expected. It also points that the mutations of this group are located at such functionally critical regions of the perforin that addition of charge groups cannot be tolerated. Moreover, individual analysis of the mutations revealed that there is a good inverse correlation between the ΔΔG and the total relative accessible surface area. The residues having very low RASA resulted in significantly high ΔΔG values, suggesting that the mutations in the core region might cause instability (maybe leading to an unfolded conformation) or displacement of charge groups that stabilize the protein (Table IV). On the other hand, the majority of the stabilizing mutations have large relative accessible surface areas falling into the “exposed” category.
Table IV.
The Correlation Between Total RASA and ΔΔG of Perforin Mutations
| ΔΔG (in kcal/mol) | |||||
|---|---|---|---|---|---|
| <0 | Between 0 and 2 | >2 | Total | ||
| Total RASA | Buried (<5%) | 1a | 4 | 21 | 26 |
| Intermediate exposure (between 5% and 30%) | 2 | 14 | 15 | 31 | |
| Exposed (>30%) | 8 | 9 | 2 | 19 | |
| Total | 11 | 27 | 38 | 76 | |
Numbers show the total number of mutations for a given RASA and ΔΔG range.
A91V polymorphism.
Mutations are usually expected to destabilize the protein structure. Among the 76 missense mutations we studied, half of them fall into this category having a total energy change greater than 2 kcal/mol, yet other 27 have less than 2 kcal/mol but still positive values. However, a group of 11 mutations resulted in negative numbers, which by definition shows that these mutations contribute to the stability of the protein. These results were in consistent with outcomes of another energy calculation method and two prediction tools for functional impact of amino acid substitution (see Supporting Information Table S3 for Polyphen2,24 see Supporting Information Table S4 for SIFT,25 see Supporting Information Table S5 for PoPMuSiC26), where Polyphen2 and SIFT are based on phylogenetic approach which takes position conservation into account. One mutation with the greatest negative energy change among the last group was remarkable, A91V, which is discussed further in the “Discussion” section.
Perforin monomers have a large interface along two alpha helices protruding from the MACPF domain where they oligomerize (Fig. 3 and Supporting Information Fig. S1). At this interface, a total of 38 residues were predicted to be in the interaction site between two perforin molecules based on a given distance threshold (the sum of van der Waals radii of two atoms + 0.5 Å). Six of these residues on either face of the MACPF domain are among the observed 65 mutation sites (Supporting Information Table S6). Out of 38 interface residues 16 of them are predicted as hot spots (the residues which account for the majority of the binding energy at the interfaces28), and three of them are the mutation sites. These three mutation sites that are hot spots at the interface might show that the absent or reduced cytotoxicity in particular patients carrying them is due to the disruption of perforin oligomerization. Corroboratively, in a patient with homozygous R225W mutation (one of our predicted hot spot residues), although perforin expression was at normal range (91%), natural killer cell function was reported as significantly reduced.29 However, more experimental setups are required to verify the characteristics of interactions between individual residues at the interface.
Figure 3.
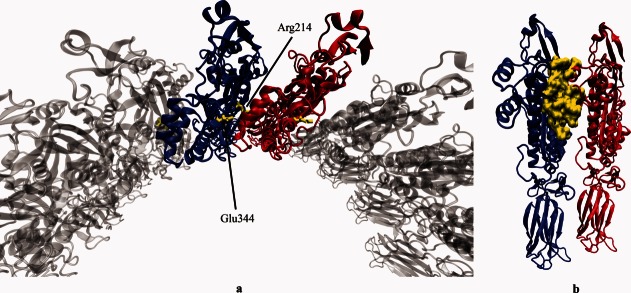
Interface between perforin monomers. (a) The ionic interaction between Arg214 and Glu344.23 (b) The highlighted interface residues predicted by Hotpoint Webserver.27
Discussion
Perforin has remained implicit for decades after its role in cytotoxicity has been first described in 1985.30,31 Nevertheless since then it was well known that perforin acts via pore formation in synergy with granzyme B, however, the exact mechanism of the pore formation remained elusive because of the lack of sufficient evidence. For decades, an analogy was made between perforin and CDCs. Recently, the determination of the X-ray crystal structure of the murine perforin elucidated the mechanism of perforin pore formation.22 However, the number of functional studies on perforin mutations is very few. In this study, we investigated the human perforin (homology model) with a structural perspective, redounding the understanding of severe consequences of perforin deficiency due to mutations observed on FHL2 patients.
The perforin pore model we created is consistent with the previously proposed classical pore models; differently, our model is based on human perforin where the homology model is used, which is relevant since the associated clinical data refers to human. Additionally, it suggests flexibility for the size of the pore and the number of monomers involved, both of which are indeed observed to vary in vivo by cryoelectron microscopy.22,23 However, perforin undergoes a large conformational change during its rearrangement to insert into the membrane, forming a large amphipathic transmembrane β-barrel.32 Since the contact regions between monomers are subject to change during this transition, our interface analysis is mainly valid for the initial pore state. As a result, our in silico functional analysis primarily addresses the postsynaptic defects of perforin mutations. Further experimental studies revealing structure information of the complete pore state will help us better understand these defects.
Mapping of the mutations onto the protein structure shows valuable information about the potential characteristics of the mutations. Perforin is functional only in the presence of high concentration of Ca2+ ions, which bind to the C2 domain thereby initiate the membrane attachment. It was shown that mutagenesis of a number of conserved aspartate residues at this region including two of the mutated residues in our Cluster 3 (D430A and D486A) caused a total loss of cytotoxic activity of perforin.33 In the same study, two more mutations in this cluster (G429E and T435M) that are close to critical aspartate residues including these two for Ca binding were characterized and the former was found to significantly reduce the cytotoxicity whereas the latter, surprisingly, had no effect on perforin activity, as opposed to the prediction in two other reports.16,34
Proteins interact with each other via interface residues. Disruption of the interaction is the primary target responsible for dysfunction of multipartner protein complexes, so any structural change that occurs at the interface residues has the potential to render the protein less active or inactive. Likewise, having a large interface between perforin monomers used for oligomerization prior to the pore formation may help to explain the fatal consequences of perforin mutations, particularly those which are found at or nearby the interface (Cluster 1). As a matter of fact, it was previously shown that charge reversal or substitution to different type of residues of three interface residues dramatically decreased cytotoxicity (D192K, R214E, and E344R).23 Moreover, our interface analysis suggests that there are more residues involved in the perforin–perforin interaction, as we predicted in total 38 residues of which only above three were experimentally demonstrated so far. The characteristics of the interface being such large and strong require many more individual interactions that keep the monomers intact during the pore formation. These yet-unidentified residues contributing to the perforin–perforin interaction have a high potential to be in our prediction list, as we correctly predicted the previously known three residues as well, by using the same algorithm on our webserver.27,28 With this analysis, we for the first time provide a list of candidate perforin residues that might be useful to be primarily investigated in further studies aiming to reveal perforin–perforin interaction.
Perforin mutations obviously have a heterogeneous character. Three types of perforin mutations in terms of genetic form are observed: homozygous, heterozygous, and compound heterozygous. Although the significance of homozygous mutations is relatively straightforward to interpret, heterozygous ones contribute to the complexity of the disease, such as late onset cases. Since an important question about perforin mutations is whether they are distinguishable in their resulting effects (i.e., perforin expression, NK cell activity, cytotoxicity, severity, and onset of the disease), different genetic forms of the same mutation might be useful to solve it. Indeed, there are few examples in which mutations are found both in homozygous and compound heterozygous form (V50M,7,29,35 G149S,29,35–37 G220S,3,13 R225W,3,13,29 and H222Q37,38). Although it is possible to study the structural effects of these mutations separately, the lack of the sufficient knowledge about the underlying mechanisms of perforin function keeps our question alive. Nevertheless our detailed analysis of the missense perforin mutations reveals important clues about the perforin deficiency in FHL2 patients. More clinical reports about FHL2 patients and functional studies about perforin mutations will further clarify this question, as well as the one why some patients show late onset of the disease.
A novel assignment of the missense perforin mutations as “stabilizing” or “destabilizing” and the comparison with introduced random mutations led us to uncover that the perforin mutations are not randomly distributed over the protein structure, but rather occupy critical positions in terms of stability, although they are virtually found anywhere on the structure. However, the stability analysis is mainly valid for the postsynaptic mutations, those attain a native fold and then undergoes conformational change. In relevant analyses throughout this study, it is noteworthy that we used the complete dataset of mutations including presynaptic and postsynaptic types, as it is not possible to study only postsynaptic mutations since such an agreed classification of perforin mutations currently is not available, due to the lack of knowledge on particular impact of each individual mutation.
The prediction results on stability change upon mutation we collect here should be evaluated carefully. Although we present data from several effective methods for comparison, experimental knowledge on individual mutations will potentially have further useful information wherever available. Illustratively, a previous study shows that several perforin mutations are temperature-sensitive, correlating with late onset cases, suggesting another mechanistic dimension to perforin cytotoxicity.16 Yet a very recent report of a novel missense mutation (D49N) demonstrates a gain of a glycosylation site leading to protein misfolding and instability.39 These findings provide invaluable additional information to our prediction results.
FHL2 patients carrying A91V change show variety in phenotype that makes it difficult to determine if it is a neutral polymorphism or a pathogenic one. Previously it was reported that A91V change was found in healthy populations with high allele frequency presenting no signs of the disease.24,40 Later it was shown that A91V change causes decreased level of perforin expression and partial loss of lytic capacity.13 Likewise, perforin harboring A91V was not folded properly41 and was also temperature-sensitive.16 Some other studies revealed late onset cases associated with it.35,42 Recently, an experimental study demonstrated that A91V change causes conformational change and reduces the production of the active perforin, resulting in defective cytotoxic function.4 Moreover, A91V change was observed in a FHL2 patient together with tuberculosis, suggesting its potential contribution to the pathophysiology and possible synergistic role in the late onset of FHL2.43 All of these diverse findings bring A91V change to the focus of perforin mutations, which actually might be important to understand the structure–phenotype relationship between perforin and FHL2 if studied in detail. In our stability analysis, we found that A91V change decreased the total energy of the protein structure significantly (ΔΔG = −1.07 kcal/mol) although it was buried in the core region of MACPF domain (RASA = 3%). Therefore, A91V change particularly stabilizes the protein structure. Furthermore, the conserved alanine residue at position 91 does not directly take place at the interface of perforin monomers in our pore model, also is far from the small clusters of membrane-spanning α helices. In addition, both having small neutral side chains, change of alanine to valine might be tolerated for the sake of chemical resemblance. Hence our results supports that A91V polymorphism is rather a neutral or “milder” class mutation17 from the structural point of view. However, severe pathogenic cases of A91V change primarily suggests that pathogenicity might arise from the second allele (in compound heterozygous cases). Alternatively, other factors such as epigenetic regulation44 or temperature sensitivity45 might be involved which makes the link between the mutation and the perforin deficiency more complex than the assumption of a direct cause-and-effect relationship, such as differential levels of protein expression or cytotoxicity. In a broader context, FHL can also be triggered by a combination of mutations that occurs in several proteins involved in the secretory granule-dependent pathway. The impact of these mutations will stay hidden unless the patients are also sequenced for these proteins. In such a case, the exact contribution of the particular mutations will remain unclear.
Up to now, numerous clinical studies have reported the clinical manifestations of FHL2 patients carrying a variety of perforin mutations (Table V). On the other hand, several studies addressed the structure of homologous partners of perforin or MACPF domain,18–20 where a huge step was recently taken by the determination of the crystal structure of monomeric murine perforin. Our study combines these two methods, providing a comprehensive analysis of perforin mutations from a structural perspective.
Table V.
Clinical Data Collection of 89 FHL2 Patients Harboring Perforin Mutations Reported in the Literature
| Mutationa | Patient/case ID as in the reference | Nucleotide sequence alteration | Country of origin | Consanguinity/family history | Age at diagnosis (d: day, m: month, y: year)/sex | CNS | F | S | C | H/H | H | Treatment | BMT status/outcome/survival after diagnosis | Reference (PMID) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Trp374Stop | 1 | 1122 G→A | Turkey | +/+ | 2 m/M | − | + | + | + | + | + | Vp/Cs/CsA | No BMT/dead | 11179007 |
| 7 | 1122 G→A | Turkey | −/+ | 1 m/F | + | + | + | + | Vp/Cs | No BMT/dead | 11179007 | |||
| 11 | 1122 G→A | Turkey | +/+ | 39 m/M | + | + | + | + | + | HLH–94 | BMT/alive and well | 11179007 | ||
| 17 | 1122 G→A | Turkey | +/+ | 10 m/F | + | + | + | + | + | No treatment | No BMT/dead | 11179007 | ||
| Case 1 | Turkey | 1st/− | 7 d/M | − | No treatment | 2 d | 18190960 | |||||||
| Case 2 | Turkey | 2nd/+ | 4 m/M | − | Steroid | 7 d | 18190960 | |||||||
| Case 3 | Turkey | 1st/+ | 6 m/F | + | HLH–94 | 1 m | 18190960 | |||||||
| Patient 21 | 1122 G→A | Turkey | +/+ | 12060139 | ||||||||||
| Patient 34 | 1122 G→A | Turkey | +/+ | 12060139 | ||||||||||
| Patient 62 | 1123 G→A | Turkey | +/+ | 12060139 | ||||||||||
| Case 7 | 1123 G→A | Turkey | +/+ | 3 m/M | − | + | + | + | BMT/alive | 11565555 | ||||
| Case 8 | 1123 G→A | Turkey | +/+ | 2.5 m/F | + | + | + | + | BMT/alive | 11565555 | ||||
| Case 10 | 1123 G→A | Turkey | +/− | 2 m/M | − | + | + | + | BMT/alive | 11565555 | ||||
| Ile224Asp | 3 | 671 T→A | Sweden | +/− | 58 m/F | + | + | + | + | + | Vp/Cs/CsA | BMT/alive, mild retardation | 11179007 | |
| Tyr219Stop/Val50Met | 9 | 657 C→A/148 G→A | Turkey | −/− | 4 m/M | + | + | + | + | + | HLH–94 | No BMT/dead | 11179007 | |
| Lys285Del | 33 | 853–855 del AAG | Turkey | −/− | 3 m/F | + | + | + | + | + | No treatment | No BMT/dead | 11179007 | |
| Gly149Ser | Case 4 | Turkey | 2nd/+ | 26 d/M | + | HLH–94 | 10 m | 18190960 | ||||||
| Case 5 | Turkey | 3rd/+ | 14 m/F | + | HLH–2004 | 8 m | 18190960 | |||||||
| Val50Met | Case 6 | Turkey | 1st/− | 16 y/M | + | HLH–2004 | Alive | 18190960 | ||||||
| Ala91Val | Case 7 | Turkey | 1st/− | 13 y/F | + | HLH–2004 | Alive | 18190960 | ||||||
| Case 8 | Turkey | 3rd/+ | 12 y/M | − | HLH–2004 | 1.5 y | 18190960 | |||||||
| II-2 | 272 C→T | Spain | −/ | 49 y/M | − | + | + | 16956828 | ||||||
| Ala523Asp | Case 9 | Turkey | 1st/+ | 36 m/F | + | HLH–2004 | 2 m | 18190960 | ||||||
| Pro89Thr | Family 1-Case A | 265 C→A | Oman | +/+ | 1 m/F | − | + | + | + | + | HLH–94 | BMT/alive | 14578030 | |
| Family 7-Case A | 265 C→A | Oman | +/− | 2 m/F | − | + | + | + | + | HLH–94 | BMT/alive | 14578030 | ||
| Gly45Arg/Arg54Ser | P10 | 133 G→A/160 C→T | Hispanic | 3 y/F | 11756153, 14757862 | |||||||||
| Pro39His/Gly149Ser | P1 | 116 C→A/445 G→A | White | 10 y/F | 11756153, 14757862 | |||||||||
| Ser150Stop/Arg225Trp | P12 | 449 C→A/673 C→T | White | 5 y/F | 11756153, 14757862 | |||||||||
| Leu17Stop | P9 | 50delT | African/American | 3 m/F | 11756153, 14757862 | |||||||||
| P11 | 50delT | African/American | 8 m/M | 11756153, 14757862 | ||||||||||
| Patient 4 | 50delT | Algeria | +/+ | 12060139 | ||||||||||
| Patient 92 | 50delT | Haiti | +/+ | 12060139 | ||||||||||
| P21 | 50delT | African/American | 2 m/M | 14757862 | ||||||||||
| P30 | 50delT | African/American | 2 m/M | 14757862 | ||||||||||
| P59 | 50delT | African/American | 3 m/M | 14757862 | ||||||||||
| P26 | 50delT | African/American | 6 m/F | 14757862 | ||||||||||
| P31 | 50delT | African/American | 6 m/F | 14757862 | ||||||||||
| P56 | 50delT | African/American | 14757862 | |||||||||||
| Case 1 | 50delT | Ghana | +/− | 2 m/M | − | + | + | + | BMT/alive | 11565555 | ||||
| Val38Leu/??? | Middle East | 18710388 | ||||||||||||
| Glu46Stop/Ala91Val+Arg119Trp | 4-year-old girl | Russia (German descent) | −/− | 4 y/F | HLH–94 | BMT/alive | 17328077 | |||||||
| Tyr450Met | Case 1 | 1349 C→T | Japan | 7 y/F | + | + | + | + | + | + | HLH–94,CHOP | BMT/alive | 12716377, 15632205, 17266056 | |
| Met1Val/Gly317Arg | Case 2 | 1 A→G/949 G→A | Japan | /+ | 11 y/F | + | + | + | + | + | - | HLH–94 | No BMT/alive | 12716377, 15632205, 17266056 |
| Arg410Trp/Leu364fs | Case 3 | 1228 C→T/1090.91delCT | Japan | 12/F | − | + | + | + | + | + | PSL, CsA | 12716377, 15632205, 17266056 | ||
| Met1I/Asp62fsX12 | Patient 2 | 3 G→A/185_195del11 | Germany | −/ | 2 m/ | 16278825 | ||||||||
| Val38Met/Ala91Val | Patient 19 | 112 G→A/272 C→T | Morocco | +/ | 6 m/ | 16278825 | ||||||||
| Gly149Ser/Ala262fsX22 | Patient 3 | 445 G→A/786_801del16 | Germany | −/ | 2 m/ | 16278825 | ||||||||
| Gly149Ser | Patient 16 | 445 G→A | Turkey | +/ | 3 m/ | 16278825 | ||||||||
| P50 | 445 G→A | Hispanic | 6 m/M | 14757862 | ||||||||||
| Pro201Thr/Lys285Del | Patient 17 | 601 C→A/853_855delAAG | Turkey | −/ | 10 y/ | 16278825 | ||||||||
| His222Gln/Arg232His | Patient 18 | 666 C→A/695 G→A | Italy | −/ | >10 y/ | 16278825 | ||||||||
| Arg240Gly | Patient 20 | 718 C→G | Greece | +/ | 12 m/ | 16278825 | ||||||||
| Leu526fsX87 | Patient 1 | 1576delT | Germany | +/ | 10 m/ | 16278825 | ||||||||
| Arg225Trp/Gly429Glu | Patient 5 | 673 C→T/1286 G→A | Africa | −/+ | 12060139 | |||||||||
| Pro345Leu | Patient 6 | 1034 C→T | Africa | +/+ | 12060139 | |||||||||
| Cys279Tyr/Val183Gly | Patient 11 | 836 G→A/548 T→G | France | −/− | 12060139 | |||||||||
| Asn252Ser/? | Patient 25 | 755 A→G/? | Italy | −/+ | 12060139 | |||||||||
| Arg232His/? | Patient 27 | 695 G→A/? | Italy | −/+ | 12060139 | |||||||||
| Gln64Stop | Patient 29 | 190 C→T | Algeria | +/− | 12060139 | |||||||||
| Met1Ile | Patient 30 | 3 G→A | Lebanon | +/+ | 12060139 | |||||||||
| Gln261Lys | Patient 82 | 781 G→A | Algeria | +/− | 12060139 | |||||||||
| Gly220Ser/Leu17Stop | Patient 95 | 658 G→A/50delT | Algeria | −/− | 12060139 | |||||||||
| Trp94Arg | Case 2 | 283 T→C | Italy | +/− | 3 m/M | − | + | + | + | BMT/alive | 11565555 | |||
| Tyr219Stop | Case 3 | 657 C→A | Italy | +/+ | 1.5 m/M | − | + | + | + | BMT/alive | 11565555 | |||
| Gly220Ser | Case 4 | 658 G→A | Italy | +/− | 1.5 m/F | + | + | + | + | Dead | 11565555 | |||
| Thr221Ile/Arg225Trp | Case 5 | 662 C→T/673 C→T | Italy | −/− | 6 m/F | + | + | + | + | Dead | 11565555 | |||
| Arg232Cys/Gly394Fs | Case 6 | 694 C→T/1182insT | Italy | −/− | 6 y/M | − | + | + | + | Dead | 11565555 | |||
| Phe193Leu/Arg410Pro | 577 T→C/1229 G→C | Caucasian | /− | 7 y/M | − | + | + | + | + | + | 14576041 | |||
| Val50Met | P35 | 148 G→A | Russia | 7 y/M | 14757862 | |||||||||
| Met1Ile/Asp70Tyr | P58 | 3 G→A/208 G→T | White | 8 y/M | 14757862 | |||||||||
| His222Arg/ | P22 | 665 A→G | White/Asian | 1 m/M | 14757862 | |||||||||
| Phe157Val/fs | P23 | 469 T→G/1628insT | White | 2 m/F | 14757862 | |||||||||
| Leu17Stop/Gln481Pro | P33 | 50delT/1442 A→C | Hispanic | 2 m/F | 14757862 | |||||||||
| Leu17Stop/Cys73Arg | P32 | 50delT/217 T→C | African/American | 4 m/M | 14757862 | |||||||||
| Leu17Stop/Gly45Glu | P34 | 50delT/134 G→A | African/American | 8 m/F | 14757862 | |||||||||
| His222Gln/fs | P28 | 666 C→A/1636delC | White | 3 m/M | 14757862 | |||||||||
| Arg225Trp | P57 | 673 C→T | Filipino | 2 y/M | 14757862 | |||||||||
| Gly149Ser/Arg299Cys | P25 | 445 G→A/895 C→T | Portugal | 2 y/M | 14757862 | |||||||||
| Gly149Ser/Arg361Trp | P24 | 445 G→A/1081 A→T | Hispanic | 4 y/F | 14757862 | |||||||||
| Arg225Trp | P29 | 673 C→T | White | 5 y/F | 14757862 | |||||||||
| Met1Ile/Asp313Val | P27 | 3 G→A/938 A→T | White | 14757862 | ||||||||||
| Ala91Val-Arg231His/Ala91Val | Proband (twin) | 272 C→T-695 G→A/272 C→T | ? | /− | 13/F | − | + | + | + | + | + | 14739222 | ||
| Ala91Val/Trp374Stop | Sibling 1 | 272 C→T/1122 G→A | Italy | 27 y/M | + | + | + | + | + | No BMT/alive | 12229880 | |||
| Sibling 2 | 272 C→T/1122 G→A | Italy | 25.5/F | + | + | + | + | + | BMT/alive | 12229880 | ||||
| Pro459Leu | Patient 1 | 1376 C→T | Algeria | +/− | 9 m/M | + | − | + | − | Dead | 15598808 | |||
| Patient 2 | 1376 C→T | Algeria | +/− | 33 m/M | + | + | + | + | + | Dead | 15598808 | |||
| Thr435Met | Patient W. | 1304 C→T | 12599189 | |||||||||||
| His222Gln | Patient 1 | 666 C→A | Netherlands | +/ | 2 m/F | + | + | + | + | + | SCT/alive and well | 17525286 | ||
| Glu317Arg/Asp430Tyr | Patient 2 | 949 G→A/1288 G→T | Sweden | 0 m/M | + | + | + | + | + | Dead | 17525286 |
If single mutation is shown, it is homozygous; otherwise mutations are compound heterozygous.
+, finding is present; −, finding is not present; M, male; F, female; fs, frameshift; ins, insertion; BMT, bone marrow transplantation; SCT, stem cell transplantationSymptoms: CNS, central nervous system involvement; F, fever; S, splenomegaly; C, cytopenia; H/H, hypertriglyceridemia and/or hypofibrinogenemia; H, hemophagocytosis.
Methods
Homology modeling
A homology model of human perforin was obtained from Swiss-Model Webserver46 based on mouse perforin as the template (PDB ID: 3NSJ,22 resolution: 2.75Å, RMSD: 2.28, sequence identity: 68%, residues modeled: 22–552), which is minimized by the server at the end of the modeling procedure. The model fully covers all three domains of perforin, which are MACPF, EGF-like, and C2, where MACPF is a large conserved domain among bacterial cytolysins and complement components. All residue numbers throughout this study are for human perforin. UniProtKB47 was used for sequence and domain assessment of the human perforin (ID: P14222).
Data collection
A total of 76 observed missense perforin mutations were collected from the public mutation databases: The Human Gene Mutation Database (HGMD),8 Leiden Open Variation Database (LOVD),48 and the reported cases in the literature, belonging to 65 different mutation sites on the model protein sequence. Clinical data of 89 FHL2 patients harboring perforin mutations were manually curated from the literature where available. The complete list of mutations is given in the Supporting Information Table S1 and the curated clinical data is given in Tables V and VI. The clinical data include patient/case ID, nucleotide sequence alteration, country of origin, consanguinity/family history, age at diagnosis, sex, central nervous system involvement, symptoms, treatment, and BMT status and its outcome.
Table VI.
Structure-phenotype relationship of missense perforin mutations on FHL2 patients. Mutations and their corresponding cluster ID (if available) are given in the first two columns. The third column shows the stability change upon mutation (ΔΔG) values calculated by FoldX. In the fourth column, the total relative accessible surface areas (RASA) are given for the original residue before mutation in the monomeric model structure. The fifth and the sixth columns show the clinical phenotypes of the patients carrying the corresponding mutations, natural killer cell activity and the percentage of perforin expressing natural killer cells, respectively. The last column shows if the mutation is observed in heterozygous state (yes), homozygous state (no) or compound heterozygous state (with the other mutation name). For references, see Table S1 in supplementary material. [Color table can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
 |
 |
Clustering mutations in the three-dimensional structure
Perforin has a vast number of missense mutations observed at 65 different sites, but not all of them are expected to cause the same structural impact on the protein. A link between groups of mutations and the functional regions of the perforin might be useful for predicting the structural cause of the resulting perforin deficiency. For this reason, the mutation sites in the three-dimensional structure were clustered. A distance matrix (65 × 65) was created from the Cα coordinates of 65 residues where the residues closer to each other than 10 Å were marked as “connected.” Any mutation site within this interval in the neighborhood was added to the same group. Each group having connected residues formed a cluster.
Stability analysis
The change in protein stability upon mutation was calculated by <PositionScan> command of FoldX49 version 3.0 beta 5.1 (http://foldx.crg.es/). Prior to energy calculation, the model structure was minimized using the <RepairPDB> command. Each of 531 residues in the model was mutated to other 20 amino acids (including itself). Then ΔΔG values were extracted from the FoldX output files. Any mutation resulting in ΔΔG < 0 kcal/mol and ΔΔG > 2 kcal/mol was considered as stabilizing and destabilizing, respectively. These cut-off values are used in accordance with the development of FoldX algorithm, obtained from correlations of ΔΔGexp and ΔΔGcalc values for 1088 mutants.50 The statistical significance of the observed mutations leading to the instability of the protein was measured with two-tailed Student's t-test, assuming unequal variance between two samples. The observed mutation sites with the total number of mutations per site which resulted in total energy changes greater than 2 kcal/mol according to the FoldX49 calculations comprised the first sample (n = 65 residues). The second sample consisted of either the entire sequence (n = 531 residues) or the non-mutation sites (n = 466 residues) with the same criteria.
Perforin pore model
The pore model was created in VMD51 from the monomeric homology model as the starting structure. Two copies of the homology model were loaded to form a homodimer complex (currently on top of each other). The initial and the new-loaded monomer will be referred as left partner and right partner of the complex from now on, respectively. First, the first three principal axes of the left partner were drawn originating from its center of mass [Fig. 4(a)]. Second, the complex was oriented so that the third principal axes were aligned to the z-axis; hence orthogonality of the complex to the xy plane was achieved [Fig. 4(b)]. Third, the right partner of the z-aligned complex was rotated about the z-axis by an arbitrary degree, which was used as a parameter to determine the number of monomers to be included in the pore, and translated along the y-axis by an arbitrary distance, which was used as a second parameter to determine the size of the pore [Fig. 4(c,d)]. Finally, the right partner of this z-aligned complex was superimposed with the left partner of another loaded complex by using <measure fit> command in VMD, this particular step was repeated until the desired number of monomers came together to obtain the pore model structure with a cyclic symmetry [Fig. 4(e,f)]. The redundant monomers, that is, all right partners, were finally deleted.
Figure 4.

Steps of perforin pore modeling. First row (a–d) shows the alignment of the homodimer model complex to z-axis along its three principal axes. Second row (e and f) shows the symmetrical growing of the monomers toward a complete pore. (a) The principal axes are drawn originating from the center of the mass of the protein. (b) The protein is oriented so that the third principal axis is aligned to the z-axis (both pointing downward). (c) The second monomer (the red one, initially on top of the blue one) is rotated about the z-axis by −18° and translated along the y-axis by 35 Å so that the homodimer model complex is created. (d) Top view of the aligned homodimer model complex in (c). (e) More monomers are added up by superimposing the homodimer model complexes. (f) Top view of (e).
Structural analysis
The interface between perforin monomers for the homodimer model complex was extracted by using Hotpoint Webserver27 with the default distance threshold, that is, the sum of van der Waals radii of two atoms + 0.5 Å. Some residues are attributed to be more important because of their higher contribution to the binding energy than others in protein interfaces. These residues are called hot spots. The hot spots between perforin monomers were predicted by the same server (see Supporting Information Fig. S1). The mutations observed at the hot spots are expected to have more dramatic changes for perforin oligomerization. The relative surface accessibility of the residues was calculated using PSAIA52 to check the distribution of the mutated residues on the protein structure.
Conclusion
This study presents the most extensive structural analysis of the missense perforin mutations in the literature where computational methods were used in silico. A pore model is created to see how the mutations interfere with the perforin's mechanism of action. In addition, clinical data from FHL2 patients are investigated to link the mutations to the resulting phenotypes. As a result, it is clearly shown that the mutations are closely associated with the functional regions of the perforin. Free energy calculations further confirm that the mutations occupy critical positions on the protein structure. Interface residues between perforin monomers play a direct role during oligomerization, which is the key step of the pore formation. The biochemical properties of the mutated residues reveal the underlying reasons for reduced or absent perforin activity. A special case, A91V, reflects that perforin deficiency is related to the stability of the protein structure. Heterozygous behavior of the mutations creates complexity in the resulting phenotypes. Overall, this study presents valuable findings useful for perforin biology and FHL disease. A network-based analysis might be promising to understand the perforin interactions in the secretory granule-mediated death pathway.
Acknowledgments
O. Keskin and A. Gurgey acknowledge Science Academy (Turkey) for the support.
Supplementary material
Additional Supporting Information may be found in the online version of this article.
References
- 1.Henter JI, Horne A, Arico M, Egeler RM, Filipovich AH, Imashuku S, Ladisch S, McClain K, Webb D, Winiarski J, Janka G. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48:124–131. doi: 10.1002/pbc.21039. [DOI] [PubMed] [Google Scholar]
- 2.Stepp SE, Dufourcq-Lagelouse R, Le Deist F, Bhawan S, Certain S, Mathew PA, Henter JI, Bennett M, Fischer A, de Saint Basile G, Kumar V. Perforin gene defects in familial hemophagocytic lymphohistiocytosis. Science. 1999;286:1957–1959. doi: 10.1126/science.286.5446.1957. [DOI] [PubMed] [Google Scholar]
- 3.Clementi R, Stadt U, Savoldi G, Varoitto S, Conter V, De Fusco C, Notarangelo LD, Schneider M, Klersy C, Janka G, Danesino C, Arico M. Six novel mutations in the PRF1 gene in children with haemophagocytic lymphohistiocytosis. J Med Genet. 2001;38:643–646. doi: 10.1136/jmg.38.9.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Trambas C, Gallo F, Pende D, Marcenaro S, Moretta L, De Fusco C, Santoro A, Notarangelo L, Arico M, Griffiths GM. A single amino acid change, A91V, leads to conformational changes that can impair processing to the active form of perforin. Blood. 2005;106:932–937. doi: 10.1182/blood-2004-09-3713. [DOI] [PubMed] [Google Scholar]
- 5.Cetica V, Pende D, Griffiths GM, Arico M. Molecular basis of familial hemophagocytic lymphohistiocytosis. Haematologica. 2010;95:538–541. doi: 10.3324/haematol.2009.019562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arico M, Danesino C, Pende D, Moretta L. Pathogenesis of haemophagocytic lymphohistiocytosis. Br J Haematol. 2001;114:761–769. doi: 10.1046/j.1365-2141.2001.02936.x. [DOI] [PubMed] [Google Scholar]
- 7.Goransdotter Ericson K, Fadeel B, Nilsson-Ardnor S, Soderhall C, Samuelsson A, Janka G, Schneider M, Gurgey A, Yalman N, Revesz T, Egeler R, Jahnukainen K, Storm-Mathiesen I, Haraldsson A, Poole J, de Saint Basile G, Nordenskjold M, Henter J. Spectrum of perforin gene mutations in familial hemophagocytic lymphohistiocytosis. Am J Hum Genet. 2001;68:590–597. doi: 10.1086/318796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stenson PD, Mort M, Ball EV, Howells K, Phillips AD, Thomas NS, Cooper DN. The Human Gene Mutation Database: 2008 update. Genome Med. 2009;1:13. doi: 10.1186/gm13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Horne A, Ramme KG, Rudd E, Zheng C, Wali Y, Lamki Z, Gurgey A, Yalman N, Nordenskjold M, Henter JI. Characterization of PRF1, STX11 and UNC13D genotype-phenotype correlations in familial hemophagocytic lymphohistiocytosis. Br J Haematol. 2008;143:75–83. doi: 10.1111/j.1365-2141.2008.07315.x. [DOI] [PubMed] [Google Scholar]
- 10.Trizzino A, zur Stadt U, Ueda I, Risma K, Janka G, Ishii E, Beutel K, Sumegi J, Cannella S, Pende D, Mian A, Henter JI, Griffiths G, Santoro A, Filipovich A, Arico M. Genotype-phenotype study of familial haemophagocytic lymphohistiocytosis due to perforin mutations. J Med Genet. 2008;45:15–21. doi: 10.1136/jmg.2007.052670. [DOI] [PubMed] [Google Scholar]
- 11.Voskoboinik I, Smyth MJ, Trapani JA. Perforin-mediated target-cell death and immune homeostasis. Nat Rev Immun. 2006;6:940–952. doi: 10.1038/nri1983. [DOI] [PubMed] [Google Scholar]
- 12.Balta G, Okur H, Unal S, Yarali N, Gunes AM, Turker M, Guler E, Ertem M, Albayrak M, Patiroglu T, Gurgey A. Assessment of clinical and laboratory presentations of familial hemophagocytic lymphohistiocytosis patients with homozygous W374X mutation. Leuk Res. 2010;34:1012–1017. doi: 10.1016/j.leukres.2010.02.002. [DOI] [PubMed] [Google Scholar]
- 13.Feldmann J, Le Deist F, Ouachee-Chardin M, Certain S, Alexander S, Quartier P, Haddad E, Wulffraat N, Casanova JL, Blanche S, Fischer A, de Saint Basile G. Functional consequences of perforin gene mutations in 22 patients with familial haemophagocytic lymphohistiocytosis. Br J Haematol. 2002;117:965–972. doi: 10.1046/j.1365-2141.2002.03534.x. [DOI] [PubMed] [Google Scholar]
- 14.Suga N, Takada H, Nomura A, Ohga S, Ishii E, Ihara K, Ohshima K, Hara T. Perforin defects of primary haemophagocytic lymphohistiocytosis in Japan. Br J Haematol. 2002;116:346–349. doi: 10.1046/j.1365-2141.2002.03266.x. [DOI] [PubMed] [Google Scholar]
- 15.Ueda I, Morimoto A, Inaba T, Yagi T, Hibi S, Sugimoto T, Sako M, Yanai F, Fukushima T, Nakayama M, Ishii E, Imashuku S. Characteristic perforin gene mutations of haemophagocytic lymphohistiocytosis patients in Japan. Br J Haematol. 2003;121:503–510. doi: 10.1046/j.1365-2141.2003.04298.x. [DOI] [PubMed] [Google Scholar]
- 16.Chia J, Yeo KP, Whisstock JC, Dunstone MA, Trapani JA, Voskoboinik I. Temperature sensitivity of human perforin mutants unmasks subtotal loss of cytotoxicity, delayed FHL, and a predisposition to cancer. Proc Natl Acad Sci USA. 2009;106:9809–9814. doi: 10.1073/pnas.0903815106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Risma KA, Frayer RW, Filipovich AH, Sumegi J. Aberrant maturation of mutant perforin underlies the clinical diversity of hemophagocytic lymphohistiocytosis. J Clin Invest. 2006;116:182–192. doi: 10.1172/JCI26217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rosado CJ, Buckle AM, Law RH, Butcher RE, Kan WT, Bird CH, Ung K, Browne KA, Baran K, Bashtannyk-Puhalovich TA, Faux NG, Wong W, Porter CJ, Pike RN, Ellisdon AM, Pearce MC, Bottomley SP, Emsley J, Smith AI, Rossjohn J, Hartland EL, Voskoboinik I, Trapani JA, Bird PI, Dunstone MA, Whisstock JC. A common fold mediates vertebrate defense and bacterial attack. Science. 2007;317:1548–1551. doi: 10.1126/science.1144706. [DOI] [PubMed] [Google Scholar]
- 19.Hadders MA, Beringer DX, Gros P. Structure of C8alpha-MACPF reveals mechanism of membrane attack in complement immune defense. Science. 2007;317:1552–1554. doi: 10.1126/science.1147103. [DOI] [PubMed] [Google Scholar]
- 20.Xu Q, Abdubek P, Astakhova T, Axelrod HL, Bakolitsa C, Cai X, Carlton D, Chen C, Chiu HJ, Clayton T, Das D, Deller MC, Duan L, Ellrott K, Farr CL, Feuerhelm J, Grant JC, Grzechnik A, Han GW, Jaroszewski L, Jin KK, Klock HE, Knuth MW, Kozbial P, Krishna SS, Kumar A, Lam WW, Marciano D, Miller MD, Morse AT, Nigoghossian E, Nopakun A, Okach L, Puckett C, Reyes R, Tien HJ, Trame CB, van den Bedem H, Weekes D, Wooten T, Yeh A, Zhou J, Hodgson KO, Wooley J, Elsliger MA, Deacon AM, Godzik A, Lesley SA, Wilson IA. Structure of a membrane-attack complex/perforin (MACPF) family protein from the human gut symbiont Bacteroides thetaiotaomicron. Acta Cryst. 2010;F66:1297–1305. doi: 10.1107/S1744309110023055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Slade DJ, Lovelace LL, Chruszcz M, Minor W, Lebioda L, Sodetz JM. Crystal structure of the MACPF domain of human complement protein C8 alpha in complex with the C8 gamma subunit. J Mol Biol. 2008;379:331–342. doi: 10.1016/j.jmb.2008.03.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Law RH, Lukoyanova N, Voskoboinik I, Caradoc-Davies TT, Baran K, Dunstone MA, D'Angelo ME, Orlova EV, Coulibaly F, Verschoor S, Browne KA, Ciccone A, Kuiper MJ, Bird PI, Trapani JA, Saibil HR, Whisstock JC. The structural basis for membrane binding and pore formation by lymphocyte perforin. Nature. 2010;468:447–451. doi: 10.1038/nature09518. [DOI] [PubMed] [Google Scholar]
- 23.Baran K, Dunstone M, Chia J, Ciccone A, Browne KA, Clarke CJ, Lukoyanova N, Saibil H, Whisstock JC, Voskoboinik I, Trapani JA. The molecular basis for perforin oligomerization and transmembrane pore assembly. Immunity. 2009;30:684–695. doi: 10.1016/j.immuni.2009.03.016. [DOI] [PubMed] [Google Scholar]
- 24.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 26.Dehouck Y, Kwasigroch JM, Gilis D, Rooman M. PoPMuSiC 2.1: a web server for the estimation of protein stability changes upon mutation and sequence optimality. BMC Bioinform. 2011;12:151. doi: 10.1186/1471-2105-12-151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tuncbag N, Keskin O, Gursoy A. HotPoint: hot spot prediction server for protein interfaces. Nucleic Acids Res. 2010;38:W402–W406. doi: 10.1093/nar/gkq323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tuncbag N, Gursoy A, Keskin O. Identification of computational hot spots in protein interfaces: combining solvent accessibility and inter-residue potentials improves the accuracy. Bioinformatics. 2009;25:1513–1520. doi: 10.1093/bioinformatics/btp240. [DOI] [PubMed] [Google Scholar]
- 29.Molleran Lee S, Villanueva J, Sumegi J, Zhang K, Kogawa K, Davis J, Filipovich AH. Characterisation of diverse PRF1 mutations leading to decreased natural killer cell activity in North American families with haemophagocytic lymphohistiocytosis. J Med Genet. 2004;41:137–144. doi: 10.1136/jmg.2003.011528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Masson D, Tschopp J. Isolation of a lytic, pore-forming protein (perforin) from cytolytic T-lymphocytes. J Biol Chem. 1985;260:9069–9072. [PubMed] [Google Scholar]
- 31.Podack ER, Young JD, Cohn ZA. Isolation and biochemical and functional characterization of perforin 1 from cytolytic T-cell granules. Proc Natl Acad Sci USA. 1985;82:8629–8633. doi: 10.1073/pnas.82.24.8629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rosado CJ, Kondos S, Bull TE, Kuiper MJ, Law RH, Buckle AM, Voskoboinik I, Bird PI, Trapani JA, Whisstock JC, Dunstone MA. The MACPF/CDC family of pore-forming toxins. Cell Microbiol. 2008;10:1765–1774. doi: 10.1111/j.1462-5822.2008.01191.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Voskoboinik I, Thia MC, Fletcher J, Ciccone A, Browne K, Smyth MJ, Trapani JA. Calcium-dependent plasma membrane binding and cell lysis by perforin are mediated through its C2 domain: a critical role for aspartate residues 429, 435, 483, and 485 but not 491. J Biol Chem. 2005;280:8426–8434. doi: 10.1074/jbc.M413303200. [DOI] [PubMed] [Google Scholar]
- 34.McCormick J, Flower DR, Strobel S, Wallace DL, Beverley PC, Tchilian EZ. Novel perforin mutation in a patient with hemophagocytic lymphohistiocytosis and CD45 abnormal splicing. Am J Med Genet A. 2003;117A:255–260. doi: 10.1002/ajmg.a.10010. [DOI] [PubMed] [Google Scholar]
- 35.Okur H, Balta G, Akarsu N, Oner A, Patiroglu T, Bay A, Sayli T, Unal S, Gurgey A. Clinical and molecular aspects of Turkish familial hemophagocytic lymphohistiocytosis patients with perforin mutations. Leuk Res. 2008;32:972–975. doi: 10.1016/j.leukres.2007.11.033. [DOI] [PubMed] [Google Scholar]
- 36.Kogawa K, Lee SM, Villanueva J, Marmer D, Sumegi J, Filipovich AH. Perforin expression in cytotoxic lymphocytes from patients with hemophagocytic lymphohistiocytosis and their family members. Blood. 2002;99:61–66. doi: 10.1182/blood.v99.1.61. [DOI] [PubMed] [Google Scholar]
- 37.Zur Stadt U, Beutel K, Kolberg S, Schneppenheim R, Kabisch H, Janka G, Hennies HC. Mutation spectrum in children with primary hemophagocytic lymphohistiocytosis: molecular and functional analyses of PRF1, UNC13D, STX11, and RAB27A. Hum Mutat. 2006;27:62–68. doi: 10.1002/humu.20274. [DOI] [PubMed] [Google Scholar]
- 38.Bryceson YT, Rudd E, Zheng C, Edner J, Ma D, Wood SM, Bechensteen AG, Boelens JJ, Celkan T, Farah RA, Hultenby K, Winiarski J, Roche PA, Nordenskjold M, Henter JI, Long EO, Ljunggren HG. Defective cytotoxic lymphocyte degranulation in syntaxin-11 deficient familial hemophagocytic lymphohistiocytosis 4 (FHL4) patients. Blood. 2007;110:1906–1915. doi: 10.1182/blood-2007-02-074468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chia J, Thia K, Brennan AJ, Little M, Williams B, Lopez JA, Trapani JA, Voskoboinik I. Fatal immune dysregulation due to a gain of glycosylation mutation in lymphocyte perforin. Blood. 2012;119:1713–1716. doi: 10.1182/blood-2011-08-374355. [DOI] [PubMed] [Google Scholar]
- 40.Zur Stadt U, Beutel K, Weber B, Kabisch H, Schneppenheim R, Janka G. A91V is a polymorphism in the perforin gene not causative of an FHLH phenotype. Blood. 2004;104:1909–1910. doi: 10.1182/blood-2004-02-0733. [DOI] [PubMed] [Google Scholar]
- 41.Voskoboinik I, Sutton VR, Ciccone A, House CM, Chia J, Darcy PK, Yagita H, Trapani JA. Perforin activity and immune homeostasis: the common A91V polymorphism in perforin results in both presynaptic and postsynaptic defects in function. Blood. 2007;110:1184–1190. doi: 10.1182/blood-2007-02-072850. [DOI] [PubMed] [Google Scholar]
- 42.Busiello R, Adriani M, Locatelli F, Galgani M, Fimiani G, Clementi R, Ursini MV, Racioppi L, Pignata C. Atypical features of familial hemophagocytic lymphohistiocytosis. Blood. 2004;103:4610–4612. doi: 10.1182/blood-2003-10-3551. [DOI] [PubMed] [Google Scholar]
- 43.Mancebo E, Allende LM, Guzman M, Paz-Artal E, Gil J, Urrea-Moreno R, Fernandez-Cruz E, Gaya A, Calvo J, Arbos A, Duran MA, Canet R, Balanzat J, Udina MA, Vercher FJ. Familial hemophagocytic lymphohistiocytosis in an adult patient homozygous for A91V in the perforin gene, with tuberculosis infection. Haematologica. 2006;91:1257–1260. [PubMed] [Google Scholar]
- 44.Lu Q, Wu A, Ray D, Deng C, Attwood J, Hanash S, Pipkin M, Lichtenheld M, Richardson B. DNA methylation and chromatin structure regulate T cell perforin gene expression. J Immunol. 2003;170:5124–5132. doi: 10.4049/jimmunol.170.10.5124. [DOI] [PubMed] [Google Scholar]
- 45.Brennan AJ, Chia J, Trapani JA, Voskoboinik I. Perforin deficiency and susceptibility to cancer. Cell Death Differ. 2010;17:607–615. doi: 10.1038/cdd.2009.212. [DOI] [PubMed] [Google Scholar]
- 46.Bordoli L, Kiefer F, Arnold K, Benkert P, Battey J, Schwede T. Protein structure homology modeling using SWISS-MODEL workspace. Nat Protoc. 2009;4:1–13. doi: 10.1038/nprot.2008.197. [DOI] [PubMed] [Google Scholar]
- 47.Magrane M, Consortium U. 2011. UniProt Knowledgebase: a hub of integrated protein data. Database bar009.
- 48.Fokkema IF, Taschner PE, Schaafsma GC, Celli J, Laros JF, den Dunnen JT. LOVD v.2.0: the next generation in gene variant databases. Hum Mutat. 2011;32:557–563. doi: 10.1002/humu.21438. [DOI] [PubMed] [Google Scholar]
- 49.Schymkowitz J, Borg J, Stricher F, Nys R, Rousseau F, Serrano L. The FoldX web server: an online force field. Nucleic Acids Res. 2005;33:W382–W388. doi: 10.1093/nar/gki387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guerois R, Nielsen JE, Serrano L. Predicting changes in the stability of proteins and protein complexes: a study of more than 1000 mutations. J Mol Biol. 2002;320:369–387. doi: 10.1016/S0022-2836(02)00442-4. [DOI] [PubMed] [Google Scholar]
- 51.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 52.Mihel J, Sikic M, Tomic S, Jeren B, Vlahovicek K. PSAIA—protein structure and interaction analyzer. BMC Struct Biol. 2008;8:21. doi: 10.1186/1472-6807-8-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.