

Abstract
An increasing number of cryo-electron microscopy (cryo-EM) density maps are being generated with suitable resolution to trace the protein backbone and guide sidechain placement. Generating and evaluating atomic models based on such maps would be greatly facilitated by independent validation metrics for assessing the fit of the models to the data. We describe such a metric based on the fit of atomic models with independent test maps from single particle reconstructions not used in model refinement. The metric provides a means to determine the proper balance between the fit to the density and model energy and stereochemistry during refinement, and is likely to be useful in determining values of model building and refinement metaparameters quite generally.
Keywords: cryo-electron microscopy, protein structure, model validation, model refinement
Introduction
There has been considerable progress over the past decade in the generation of subnanometer cryo-electron microscopy (cryo-EM) density maps of protein complexes,1 with an increasing number of maps sufficiently resolved to recover a backbone trace.2–4 In parallel, computational methods have been developed to generate and refine models using subnanometer resolution cryo-EM density.5–8 However, while there are a number of ways of assessing the reliability9 and resolution10,11 of single-particle reconstructions, there is no standard method by which models built and refined into such reconstructions may be independently validated. Recent studies (e.g.12) use some combination of density correlation and protein structure geometry13 to assess model accuracy.
Validation criteria are very important both for assessing the quality of structure models and for guiding the modeling process itself as illustrated by the extensive use of the free R factor to assess and guide crystallographic model refinement.14 Indeed, development of independent validation criteria is essential to progress in building detailed atomic models based on cryo-EM data using methods such as Rosetta.7 Decisions that arise during modeling such as when to incorporate explicit side-chain representations, the extent of flexibility to allow for backbone and side-chain conformations, the extent of refinement against the density, and the balance between the density and the physical chemistry implicit in the force field used in modeling all require an independent measure of model quality. The pitfall in using the fit of a model to the density data used to guide model building as a validation criterion is that the higher the weight on the density term the better the overall fit.
In this paper, we explore the use of independent reconstructions for model validation. We split a large set of particle images into two independent sets and build density reconstructions from each sets. One of these maps-the “training map”—is used for model building and refinement, and the other—the “testing map”—is used for cross validation.
Results
To evaluate this approach to cross validation, we focused on the single-particle reconstruction of wild-type Mm-cpn in the ATP/AlFx induced closed state. The 616 CCD frames used in the original 4.3 Å resolution reconstruction2 were divided into two separate image data sets. The contrast transfer function parameters were independently determined for each frame. The training map was reconstructed using 22,571 particles boxed out from one set of 308 CCD frames while the testing map was reconstructed using 22,446 particles boxed out from another set of 308 CCD frames. Two different initial density models, generated independently from program startcsym in EMAN115 were used to initiate the refinement. While the resultant independently reconstructed maps appear to have consistent backbone connectivity, some of the protruding densities which likely correspond to sidechains differ, probably due to the limited map resolution exacerbated by use of only half of the data-set (Supporting Information Movie 1). One of these reconstructions, the training map, was used for model refinement, while the other, the testing map, was held aside and only used after refinement for model validation (analogous to the crystallographic Rfree reflections16).
We used the Rosetta structure prediction methodology to build and refine models into the training map by adding a score term to the Rosetta energy function assessing agreement between model and map.7 A weighing term wa controls the contribution of the experimental data relative to the Rosetta all atom energy. Models were built from the homologous thermosome KS-1 structure (PDB accession code 1Q3Q), and refined into the training set density using 12 different values of wa. The Rosetta symmetric refinement protocol17 was used to model the entire D8 symmetric complex [Fig. 1(B)]. All modeling used a voxel spacing of 1.30 Å, which is different from the previous value of 1.33 Å.2 This recalibration reduced the Rosetta all atom energy and improved the fit of models to both the testing and training maps.
Figure 1.
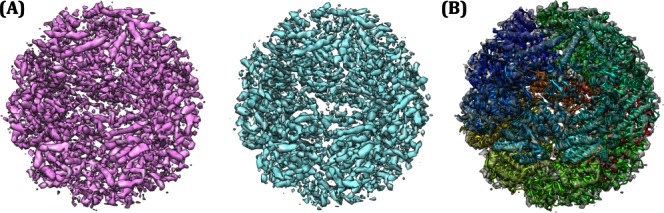
A: Independent cryo-EM reconstructions of Mm-Cpn. The training map is in magenta and the testing map in cyan. B: A model refined into the training set density (at wa = 0.1).
The Rosetta all atom energy of the models is shown as a function of wa in the green dotted line in Figure 2. The energy of the models is roughly constant as wa increases to 0.2, and then rises steeply. The steep increase suggests that models above this point may be overfit to the training map. Molprobity validation13 (not shown) similarly suggests a sharp decrease in model quality above wa ≍ 0.2.
Figure 2.
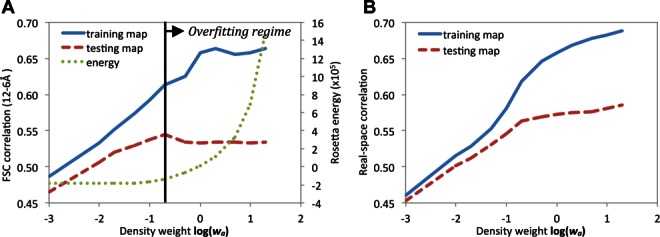
A: Assessment of the extent of model overfitting with increasing wa. Solid line: average model Fourier shell correlation (12–6 Å) with the training map; dashed line: correlation with testing map; dotted line: Rosetta all atom energy. The black vertical line indicates “inflection point” around w = 0.1–0.2, above which the training map correlation increases, the testing map correlation decreases, and the Rosetta energy increases markedly, suggesting overfitting of models to noise in the data. B: The same data plotted using real-space correlation instead; above the inflection point the correlation with the testing map continues to increase, albeit much more slowly than the correlation with the training map. [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
The fit of the refined models to the training map and the independent testing map was assessed using the Fourier shell correlation (FSC) in the highest resolution shells (12–6 Å). As expected, the fit of the refined models to the training map increases with increasing wa during model refinement [Fig. 2(A), blue solid line]. The fit of the refined models to the testing map [Fig. 2(A), red dashed line] increases in parallel with the fit to the training map for wa < 0.1. However, above 0.1–0.2 there is an “inflection point,” where the fit of the refined models to the training map continues to increase but the fit to the testing map decreases, suggesting overfitting to the training map. The overfitting is not as pronounced when the density correlation rather than the FSC is used to assess the fit between the models and maps [Fig. 2(B)]; while a similar inflection point is observed around wa = 0.1–0.2, the agreement to the testing map continues to increase, albeit slowly, as wa increases. The individual model-versus-map FSC curves (Supporting Information Fig. 2) suggest that this residual correlation arises from the lowest resolution Fourier shells-in these shells, the models agree less well with either dataset than the datasets agree with one another. This may be due to missing features in modeling, such as bulk solvent.
It is notable that the steep increase in Rosetta energy, and maximal agreement to the independent test map, both occur in the same wa range (from 0.1 to 0.2). Taken together, the increase in fit to the testing map and the lack of increase in Rosetta energy as wa varies from 0 to 0.1 suggests that model accuracy increases with increasing wa over this range. Above wa = 0.2, the sharp increase in Rosetta energy and the decreasing agreement to the testing map suggest that model quality decreases in this region due to overfitting to the training map.
The two independent maps also provide insight into the issue of resolution assessment in cryo-EM maps. The resolution of a cryo-EM map has been commonly estimated from the Fourier Shell Correlation (FSC) between maps computed from split odd and even sets of image data during the last iteration of map refinement. Using this criterion, the resolutions of each of the two reconstructed density maps [Fig. 1(A)] were assessed to be 4.6 Å and 4.7 Å, respectively, at a 0.5 threshold (Supporting Information Fig. 1). However, the FSC between the maps independently reconstructed from two halves of data split from the onset of the refinement indicates a resolution of only 6.7 Å, at 0.143 threshold10 (Supporting Information Fig. 4). The actual resolution is likely somewhere between these two values: the former estimate is likely overly optimistic18 and the latter estimate is overly pessimistic. The resolvability of the Mm-cpn map using the entire data set is adequate to resolve β-strands in the equatorial domains of the subunit and to trace the polypeptide backbone.2 The low value of the FSC correlation between the independent maps may reflect significant structural variation in the apical domains and loop regions (Supporting Information Fig. 5). Development of a more informative resolution definition for cryo-EM maps remains an open area of investigation.
Discussion
The development of metrics for cryo-EM model validation will be a critical step towards robustly generating high accuracy all-atom models from cryo-EM density. The split dataset approach explored here provides a metric that should be immediately useful: the Fourier correlation between the refined model and an independently reconstructed density map over shells with resolution higher than 12 Å. This metric can be used to determine the balance between the fit to the map and model energy and stereochemistry during refinement, as described in this paper, as well as any other metaparameter(s) related to model building and refinement.
The overall density correlation is less sensitive to overfitting than the FSC estimate at subnanometer resolution range because at very low resolution the training and test maps are more similar to each other than to the models. The source of the discrepancy between model and map at low resolution is not clear; it could reflect inaccuracies in the physical model (failure to treat model heterogeneity, bulk solvent effects, etc.), incorrect modeling of the contrast transfer function, or biases in the reconstruction process itself. Whatever the source, the problem can be avoided by discarding the lowest resolution shell during cross validation.
Splitting of the data-set into two halves inevitably limits the resolution and the quality of the reconstructed maps; this problem can be overcome by using the split data set approach for weight and other modeling parameter selection, after which a final refinement could be carried out using all of the data. It also might be possible to use individual particle images directly for validation provided alignment of the images to the model can be carried out sufficiently rapidly and accurately; this could require much less independent data and would have the added benefit of reducing possible bias introduced in the reconstruction process. Such an approach may be applicable for large virus particles, which may have sufficient inherent contrast for individual particle orientation and conformation assessment.
Acknowledgments
The authors thank Drs. Steve J. Ludtke, Michael F. Schmid, and Matthew L. Baker for discussions, and Cathy Lawson for her comments on the paper. They also thank an anonymous reviewer for valuable suggestions.
The cryo-EM maps have been deposited to EMDB (accession numbers EMD-5645 and EMD-5646). The model was deposited to PDB (id: 3j3x).
Glossary
- cryo-EM
cryo-electron microscopy
- FSC
Fourier shell correlation.
Supplementary material
Additional Supporting Information may be found in the online version of this article.
References
- 1.Chiu W, Baker ML, Jiang W, Dougherty M, Schmid MF. Electron cryomicroscopy of biological machines at subnanometer resolution. Structure. 2005;13:363–372. doi: 10.1016/j.str.2004.12.016. [DOI] [PubMed] [Google Scholar]
- 2.Zhang J, Baker ML, Schröder GF, Douglas NR, Reissmann S, Jakana J, Dougherty M, Fu CJ, Levitt M, Ludtke SJ, Frydman J, Chiu W. Mechanism of folding chamber closure in a group II chaperonin. Nature. 2010;463:379–383. doi: 10.1038/nature08701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang X, Settembre E, Xu C, Dormitzer PR, Bellamy R, Harrison SC, Grigorieff N. Near-atomic resolution using electron cryomicroscopy and single-particle reconstruction. Proc Natl Acad Sci U S A. 2008;105:1867–1872. doi: 10.1073/pnas.0711623105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang X, Jin L, Fang Q, Hui WH, Zhou ZH. 3.3 Å cryo-EM structure of a nonenveloped virus reveals a priming mechanism for cell entry. Cell. 2010;141:472–482. doi: 10.1016/j.cell.2010.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Trabuco LG, Villa E, Mitra K, Frank J, Schulten K. Flexible fitting of atomic structures into electron microscopy maps using molecular dynamics. Structure. 2008;16:673–683. doi: 10.1016/j.str.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schröder GF, Brunger AT, Levitt M. Combining efficient conformational sampling with a deformable elastic network model facilitates structure refinement at low resolution. Structure. 2007;15:1630–1641. doi: 10.1016/j.str.2007.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.DiMaio F, Tyka MD, Baker ML, Chiu W, Baker D. Refinement of protein structures into low-resolution density maps using rosetta. J Mol Biol. 2009;392:181–190. doi: 10.1016/j.jmb.2009.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baker ML, Abeysinghe SS, Schuh S, Coleman RA, Abrams A, Marsh MP, Hryc CF, Ruths T, Chiu W, Ju T. Modeling protein structure at near atomic resolutions with Gorgon. J Struct Biol. 2011;174:360–373. doi: 10.1016/j.jsb.2011.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van Heel M, Schatz M. Fourier shell correlation threshold criteria. J Struct Biol. 2005;151:250–262. doi: 10.1016/j.jsb.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 10.Rosenthal PB, Henderson R. Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J Mol Biol. 2003;333:721–745. doi: 10.1016/j.jmb.2003.07.013. [DOI] [PubMed] [Google Scholar]
- 11.Henderson R, Chen S, Chen JZ, Grigorieff N, Passmore LA, Ciccarelli L, Rubinstein JL, Crowther RA, Stewart PL, Rosenthal PB. Tilt-pair analysis of images from a range of different specimens in single-particle electron cryomicroscopy. J Mol Biol. 2011;413:1028–1046. doi: 10.1016/j.jmb.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baker ML, Zhang J, Ludtke SJ, Chiu W. Cryo-EM of macromolecular assemblies at near-atomic resolution. Nature Protoc. 2010;5:1697–1708. doi: 10.1038/nprot.2010.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen VB, Arendall WB, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Cryst. 2010;D66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Read RJ, Adams PD, Arendall WB, Brunger AT, Emsley P, Joosten RP, Kleywegt GJ, Krissinel EB, Lutteke T, Otwinowski Z, Perrakis A, Richardson JS, Sheffler WH, Smith JL, Tickle IJ, Vriend G, Zwart PH. A new generation of crystallographic validation tools for the protein data bank. Structure. 2011;19:1395–1412. doi: 10.1016/j.str.2011.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ludtke SJ, Baldwin PR, Chiu W. EMAN: semiautomated software for high-resolution single-particle reconstructions. J Struct Biol. 1999;128:82–97. doi: 10.1006/jsbi.1999.4174. [DOI] [PubMed] [Google Scholar]
- 16.Brünger AT. Free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature. 1992;355:472–475. doi: 10.1038/355472a0. [DOI] [PubMed] [Google Scholar]
- 17.DiMaio F, Leaver-Fay A, Bradley P, Baker D, André I. Modeling symmetric macromolecular structures in Rosetta3. PLoS One. 2011;6:e20450. doi: 10.1371/journal.pone.0020450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Scheres SH, Chen S. Prevention of overfitting in cryo-EM structure determination. Nature Meth. 2012;9:853–854. doi: 10.1038/nmeth.2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.