

Abstract
Chalcones 1~8 and 5-deoxyflavonoids 9~22 were synthesized in good yields by aldol condensation, Algar-Flynn-Oyamada reaction, glycosidation, and deacetylation reaction, respectively, starting from 2-acetyl phenols substituted by methoxy or methoxymethoxy group and appropriately benzaldehydes substituted by methoxy, methoxymethoxy group, or chlorine. Among them, 13 and 17~22 are new compounds. The cytotoxicity bioassays of these chalcones and 5-deoxyflavonoids were screened using the sulforhodamine B (SRB) protein staining method, and the results showed that compounds 2, 4, 5, 6, 10, 15, and 19 exhibited moderate cytotoxicity against the cancer cell line of MDA-MB-231, U251, BGC-823, and B16 in comparison with control drugs (HCPT, Vincristine, and Taxol).
1. Introduction
Chalcones are a class of natural compounds that widely exist in a variety of plant species. Chemically, they consist of open-chain flavonoids in which the two aromatic rings are joined by a three carbon α, β-unsaturated carbonyl system. The flexible structure of chalcones makes them have a large number of biological activities including antitumor [1], antifungal [2], antiprotozoal [3], antimitotic [4], and antivirus [5] properties. The 5-deoxyflavonoids are also one of the main classes of natural flavonoids; they possess antitumor [6], antiviral, and antibiotic effects [7].
Despite lots of recent impressive reports on chalcones [8, 9] and 5-deoxyflavonoids [10], the full potential of such class of compounds is yet to be realized in terms of both more new molecules as drugs and varied biological activity. This situation is largely due to their simple chemical structure and useful template. It has recently become more apparent that most of the important classes of drugs, especially those derived from natural products, are glycosides having a sugar moiety linked to an aglycon through an O- or C-glycosidic bond. In our continued efforts to use natural products only as synthetic templates and thereby replace the original plant sources with synthetic ones and investigate structure-activity relationship, herein, we wish to report the synthesis and cytotoxicity bioassays of a series of chalcones 1~8 and 5-deoxyflabonoids 9~16 as well as their glycoside derivatives 17~22. Among them, 13 and 17~22 are new compounds.
2. Results and Discussion
Scheme 1 outlines the synthesis of chalcones 1~8 and 5-deoxyflavonoids 9~22, starting from appropriate benzaldehydes 23~27, 33, 34, and acetyl phenols substituted by methoxy or methoxymethoxy, which were purchased or prepared with an improved traditional method in good yields. In the synthesis process, the methoxymethoxy group was chosen to protect the OH group, because it is stable in basic environment and easy to deprotect. Chalcones 28~32 were prepared by using aldol condensation of appropriate substituted benzaldehydes 23~27, 33, 34, and acetyl phenols in KOH/EtOH and deprotection reaction. The aldol condensation was very sensitive to modification of reaction parameters. A significant excess of KOH (10~15 equiv) was required to force the reaction to completion. Flavonols 9~16 were prepared by classic Algar-Flynn-Oyamada reaction treating the corresponding chalcones with 15% H2O2 and 16% NaOH (aq) and deprotection reaction.
Scheme 1.
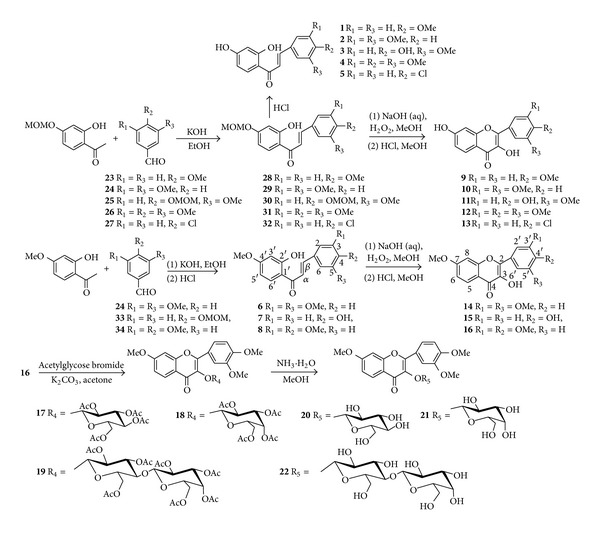
It is well know that sugar moiety could enhance water solubility and improve the targeting activity of bioactive molecules [11]. For example, lactose can be recognized by the hepatic asialoglycoprotein receptor (ASGP-R), and ASGP-R localized to liver cells provides an efficient entry point for lactose-modified molecules [12]. The modification of 5-deoxyflavonoids with lactose may be possible to specifically target molecules to liver cells, facilitating application of bioactive 5-deoxyflavonoids to the treatment of hepatitis B, hepatitis C, and liver cancer. On the other hand, the largely hydrophobic character of 5-deoxyflavonoids makes it poorly soluble in aqueous media which in some cases limits their therapeutic efficacy, and this has a strong influence on their pharmacokinetic properties. Then, we turned our attention to the introduction of glucosyl, galactosyl, or lactosyl moiety into 5-deoxyflavonoids, and compound 16 was, respectively, condensed with α-acetylglucose bromide, α-acetylgalactose bromide, or α-acetyllactose bromide in the presence of anhydrous K2CO3 in a solvent of acetone at room temperature to yield the protected 5-deoxyflavonoids glycosides 17~19. Careful deprotection of the acetyl groups under mildly alkaline condition (NH3·H2O in MeOH) at room temperature afforded the desired three novel 5-deoxyflavonoids-3-O-β-D-glycosides 20~22. The glycosylation selectively affords β-products by taking the advantage of 2-OAc neighboring participation effects to secure the 1, 2-trans glycosylation of each sugar residue. In the 1H NMR spectra of compounds 20~22, the chemical shift of the C1-H in the glycosyl ring appeared downfield (δ 5.5~5.8) with a coupling constant J 1,2 = 7.3 ~ 8.0 Hz, which confirmed their β-anomeric configuration [13].
The 22 designed target chalcones 1~8 and 5-deoxyflavonoids 9~22 were exposed to four human cancer cell lines MDA-MB-23 (human breast cancer cell), U251 (human glia cancer cell), BGC-823 (human stomach cancer cell), and B16 (mouse melanoma cell), respectively, for 48 h using the sulforhodamine B (SRB) protein staining method with the Hydroxycamptothecin (HCPT), Vincristine, and Taxol as positive control. It appeared that these closely related molecules displayed a wide range of inhibitory activities against MDA-MB-23, U251, BGC-823, and B16 cancer cells lines at the maximum concentration of 10 μg/mL as shown in Table 1. Compounds 2, 4, 5, 6, 10, 15, and 19 showed moderate cytotoxic activity against four cancer cell lines with IC50 values ranging from 2.37 to 9.71 μg/mL.
Table 1.
IC50 value (μg/mL) of chalcones and deoxyflavonoids on the cancer cell lines.
| Compound | MDA-MB-231 | U251 | BGC-823 | B16 |
|---|---|---|---|---|
| HCPTa | 1.18 | |||
| Vincristinb | 0.81 | |||
| Taxolc | 0.002 | 0.109 | ||
| 2 | 4.97 | 2.37 | 3.42 | >10 |
| 4 | >10 | >10 | 3.87 | 2.88 |
| 5 | >10 | 6.53 | 3.55 | 2.66 |
| 6 | 6.17 | 3.37 | 3.99 | >10 |
| 10 | 4.21 | 8.33 | 3.92 | >10 |
| 15 | 8.68 | 4.95 | 3.71 | >10 |
| 19 | 5.49 | 9.71 | 4.33 | 4.77 |
a,b,cUsed for positive control.
3. Experimental
Melting points were measured on a XRC-1 apparatus and were uncorrected. IR spectra were recorded on a Bruker Tensor-27 spectrometer. 1HNMR spectra were recorded on a Bruker AM-500 or Bruker AM-400 instrument, using tetramethylsilane as an internal standard, chemical shifts (δ) in ppm, and coupling constants (J) in Hz. Mass spectra were determined with ZAB-HS spectrometer by the EI or FAB method. Elemental analyses were carried out on a PerkinElmer 240B microanalyser. All solvents were dried by standard procedures. α-Acetylglucose bromide, α-acetylgalactose bromide, and α-acetyllactose bromide were prepared as described in detail [14, 15].
3.1. General Procedure for the Synthesis of Chalcones 1~8
To a stirred solution of KOH (9.3 g, 165 mmol) in EtOH (40 mL) cooled in an ice bath was added dropwise a solution of the corresponding acetophenone (12.9 mmol) and aldehyde (12.9 mmol) in EtOH (40 mL). The mixture was kept at 0°C for 0.5 h and then room temperature for 22 h. The mixture was poured into ice water (20 mL), adjusted to pH 3~4 with 1 mol·L−1 HCl, filtered, and then recrystallized from EtOH to obtain the desired products 1~8, respectively.
2′,4′-Hydroxy-4-methoxy chalcone (1): light-yellow needles, yield 91%, and m.p. 178~180°C (lit. [16]: 168~170°C). 1H NMR (400 MHz, DMSO-d 6): δ 3.83 (3H, s, OCH3), 6.29 (1H, d, J = 2.4 Hz, H-3′), 6.42 (1H, dd, J = 9.2, 2.4 Hz, H-5′), 7.03(2H, d, J = 8.8 Hz, H-3,5), 7.76-7.77 (2H, d, J = 16.0 Hz, H-α, β), 7.78 (2H, d, J = 8.6 Hz), 8.20 (1H, d, J = 9.2 Hz, H-6′), 10.71 (1H, s, 4′-OH), and 13.56 (1H, s, 2′-OH); Anal. Calcd for C16H14O4: C, 71.10; H, 5.22. Found: C, 71.32; H, 5.17.
2′,4′-Dihydroxy-3,5-dimethoxychalcone (2): yellow needles, yield 92%, and m.p. 96~97°C (lit. [17]: 97~98°C); IR (KBr) ν/cm−1: 3235, 2967, 1629, 1552, 1281, 1217, 1141, 1059, and 969; 1H NMR (500 MHz, acetone-d 6): δ 3.83 (6H, s, 2OCH3), 6.37 (1H, d, J = 2.3 Hz, H-3′), 6.46 (1H, dd, J = 8.8, 2.3 Hz, H-5′), 6.57 (1H, s, H-4), 7.01 (2H, s, H-6), 7.78 (1H, d, J = 15.4 Hz, H-β), 7.92 (1H, d, J = 15.4 Hz, H-α), 8.12 (1H, d, J = 8.8 Hz, H-6′), 9.50 (1H, s, 2′-OH), and 13.20 (1H, s, 4′-OH); MS (FAB+) m/z: 301 [M+H]+.
2′,4′,4-Trihydroxy-3-methoxychalcone (3): yellow needles, yield 82%, and m.p. 192~194°C (lit. [16]: 192~194°C); 1H NMR (400 MHz, DMSO-d 6): δ 3.88 (3H, s, OCH3), 6.28 (1H, d, J = 2.4 Hz, H-3′), 6.42 (1H, dd, J = 8.4, 2.4 Hz, H-5′), 6.83 (1H, d, J = 8.4 Hz, H-6′), 7.28 (1H, dd, J = 8.8, 1.6 Hz, H-6), 7.53 (1H, d, J = 1.6 Hz, H-5), 7.73 (1H, d, J = 15.2 Hz, H-β), 7.79 (1H, d, J = 15.2 Hz, H-α), 8.20 (1H, d, J = 8.8 Hz, H-2), and 9.74~13.65 (3H, s, OH-2′, 4′, 4); MS (FAB+) m/z: 287 [M+H]+.
2′,4′-Dihydroxy-3,4,5-trimethoxychalcone (4): light-yellow needles, yield 94%, and m.p. 188~190°C (lit. [18]: 199~200°C); IR (KBr) ν/cm−1: 3320, 2912, 2887, 2821, 1619, 1575, 1492, 1455, 1365, 1320, and 1182; 1H NMR (400 MHz, DMSO-d 6): δ 3.91 (3H, s, OCH3), 3.94 (6H, s, 2OCH3), 5.79 (1H, s, 4′-OH), 6.44~6.46 (2H, m, H-3′,5′), 6.87 (2H, s, H-2,6), 7.45 (1H, d, J = 15.2 Hz, H-α), 7.82 (1H, d, J = 15.2 Hz, H-β), and 13.38 (1H, s, 2′-OH); MS (FAB+) m/z: 331 [M+H]+.
2′,4′-Dihydroxy-4-chlorochalcone (5): yellow needles, yield 93%, m.p. 154~156°C (lit. [16]: 156~158°C); 1H NMR (400 MHz, CDCl3): δ 6.33 (1H, d, J = 2.4 Hz, H-3′), 6.43 (1H, dd, J =8.8, 2.0 Hz, H-5′), 7.54 (2H, d, J = 8.8 Hz, H-3, 5), 7.78 (1H, d, J = 15.2 Hz, H-β), 8.01 (1H, d, J = 15.2 Hz, H-α), 8.21 (1H, d, J = 9.2 Hz, H-6′), and 10.79 (1H, s, 4′-OH); Anal. Calcd for C15H11ClO3: C, 65.58; H, 4.04. Found: C, 65.45; H, 4.10.
2′-Hydroxy-3,5,7′-trimethoxychalcone (6): yellow needles, yield 83%, and m.p. 147~149°C; IR (KBr) ν/cm−1: 3460, 3009, 2940, 2306, 1639, 1602, 1456, 1370, 1158, and 839; 1H NMR (400 MHz, CDCl3): δ 3.85 (6H, s/each, 3,5-OCH3), 3.87 (3H, s, 4′-OCH3), 6.48 (1H, d, J = 2.8 Hz, H-3′), 6.50 (1H, dd, J = 8.4, 2.8 Hz, H-5′), 6.54 (1H, t, J = 2.4 Hz, H-4), 6.79 (2H, d, J = 2.4 Hz, H-2, 6), 7.53 (1H, d, J = 15.6 Hz, H-β), 7.80 (1H, d, J = 15.6 Hz, H-α), and 7.83 (1H, d, J = 8.4 Hz, H-6′); MS (EI) m/z: 314(M+, 100), 297 (10), 283 (13), 177 (60), 164(18), and 151(46).
2′,4-Dihydroxy-4′-methoxy chalcone (7): yellow needles, yield 86%, and m.p. 152~153°C; 1H NMR (400 MHz, DMSO-d 6): δ 3.95 (3H, s, OCH3-4′), 6.28 (1H, d, J = 2.0 Hz, H-3′), 6.41 (1H, dd, J = 9.2, 2.4 Hz, H-5′), 6.84 (2H, d, J = 8.6 Hz, H-3, 5), 7.75~7.77 (4H, m, H-α, H-β, H-2, 6), 8.17 (1H, d, J = 9.2 Hz, H-6′), 10.15 (1H, s, 4′-OH), 10.70 (1H, s, 4-OH), and 13.61 (1H, s, 2′-OH); MS (FAB+) m/z: 271 [M+H]+.
2′-Hydroxy-3,4,4′-trimethoxy chalcone (8): yellow needles, yield 74%, and m.p. 145~147°C; 1H NMR (400 MHz, CDCl3): δ 3.95 (9H, s, 3OCH3), 6.48(1H, s, H-3′), 6.51(1H, d, J = 2.8 Hz, H-5′), 6.91 (1H, d, J = 8.4 Hz, H-5), 7.16 (1H, d, J = 1.6 Hz, H-6), 7.25 (1H, d, J = 1.6 Hz, H-2), 7.44 (1H, d, J = 15.2 Hz, H-α), 7.84 (1H, s, H-6′), 7.86 (1H, d, J = 16.8 Hz, H-β), and 13.54 (1H, s, OH); MS (FAB+) m/z: 315 [M+H]+.
3.2. General Procedure for the Synthesis of 5-Deoxyflavonoids 9~16
To a solution of chalcones 6~8, 28~32 (0.3 mmol) in methanol (5.5 mL) was, respectively, added 16% NaOH (aq) (0.6 mL), 15% H2O2 (0.3 mL). The mixture was stirred at room temperature for 24 h, adjusted to pH 3~4 with 1 mol·L−1 HCl, filtered and then recrystallized from ethanol to obtain the corresponding 5-deoxyflavonols 9~16, respectively.
3,7-Dihydroxy-4′-methoxyflavonol (9): yellow solid, yield: 68%, and m.p. 289~290°C; 1H NMR (400 MHz, DMSO-d 6): δ 3.83 (3H, s, OCH3-4′), 6.92 (1H, dd, J = 2.1, 9.1 Hz, H-6), 6.97 (1H, d, J = 2.0 Hz, H-8), 7.03 (2H, d, J = 8.8 Hz, H-3′,5′), 7.62 (2H, d, J = 8.5 Hz, H-2′,6′), 9.15 (1H, s, OH-7), and 10.74 (1H, s, OH-3); MS (FAB+) m/z: 285 [M+H]+.
3,7-Dihydroxy-3′,5′-dimethoxyflavonol (10): white solid, yield 93.2%, and m.p. 100~101°C; IR (KBr) ν/cm−1: 3333, 3123, 1741, 1617, 1562, 1274, 1204, 1156, 1064, and 849; 1H NMR (500 MHz, DMSO-d 6): δ 3.80 (6H, s, 2OCH3), 6.63 (1H, s, H-8), 6.91 (1H, d, J = 8.8 Hz, H-6), 6.97 (1H, s, H-4′), 7.33 (2H, d, J = 1.3 Hz, H-2′,6′), 7.93 (1H, d, J = 8.8 Hz, H-5), 9.39 (1H, s, 3-OH), and 10.8 (1H, s, 7-OH); MS (FAB+) m/z: 315 [M+H]+.
7,4′-Dihydroxy-3′-methoxyflavonol (11): yellow solid, yield 93%, and m.p. 274~275°C; 1H NMR (400 MHz, DMSO-d 6): δ 3.85 (3H, s, OCH3-3′), 6.91 (1H, dd, J = 9.2, 2.0 Hz, H-6), 6.94 (1H, d, J = 8.4 Hz, H-5′), 6.98 (1H, d, J = 2.0 Hz, H-8), 7.70 (1H, dd, J = 8.4, 2.0 Hz, H-6′), 7.77 (1H, d, J = 2.0 Hz, H-2′), 7.93 (1H, d, J = 9.2 Hz, H-5), 9.13, 9.67 (2H, s/each, OH-4′, 7), and 10.74 (1H, s, OH-3); MS (FAB+) m/z: 317[M+H]+.
3,7-Dihydroxy-3′,4′,5′-trimethoxyflavonol (12): yellow solid, yield 85%, and m.p. 120~122°C; 1H NMR (400 MHz, DMSO-d 6): δ 3.74 (3H, s, OCH3-4′), 3.86 (6H, s, OCH3-5′, 6′), 6.92 (1H, d, J = 6.8, 2.0 Hz, H-8), 7.02 (1H, dd, J =8.8, 2.0 Hz, H-6), 7.50 (2H, s, H-2′,6′), 7.93 (1H, d, J = 8.8 Hz, H-5), 9.36 (1H, s, OH-7), and 10.78 (1H, s, OH-3); MS (FAB+) m/z: 345 [M+H]+.
7-Hydroxy-4′-chloroflavonol (13): yellow solid, yield 58%, and m.p. 145~146°C; 1H NMR (400 MHz, CDCl3): δ 6.50 (1H, s, OH-3), 6.52~6.54 (2H, d, J = 8.4 Hz, H-3,5), 7.09~7.13 (2H, m, H-6,8), 7.60 (2H, d, J = 8.4 Hz, H-2,6), and 8.20 (1H, d, J = 8.8 Hz, H-5); Anal. Calcd for C15H9ClO4: C, 62.41; H, 3.14. Found: C, 62.22; H, 3.19.
3-Hydroxy-7,3′,5-trimethoxyflavonol (14): yellow needles, yield 49%, and m.p. 197~198°C; IR (KBr) ν/cm−1: 3449, 3009, 1603, 1556, 1410, 1381, 1263, 1215, 1154, 1120, and 829; 1H NMR (400 MHz, CDCl3): δ 3.89 (6H, s, 3′,5′-OCH3), 3.95 (3H, s, 7-OCH3), 6.58 (1H, s, H-8), 6.96~7.01 (3H, m, H-6,4′,OH), 7.42 (2H, s, H-2′,6′), and 8.14 (1H, d, J = 7.6 Hz, H-5); MS (EI) m/z: 328 (M+, 100), 313 (10), 297 (19), 285 (30), 178 (10), 149 (23), 122 (16), and 107 (26).
3,4′-Dihydroxy-7-methoxyflavonol (15): yellow solid, yield 93%, and m.p. 295~297°C (lit. [19]: 270~272°C); 1H NMR (400 MHz, DMSO-d 6): δ 3.91 (3H, s, 7-OCH3), 6.92~6.96 (2H, m, H-3′,5′), 7.03 (1H, dd, J = 8.8, 2.4 Hz, H-6), 7.26 (1H, d, J = 2.0 Hz, H-8), 7.98 (1H, d, J = 8.8 Hz, H-5), 8.10 (2H, d, J = 8.8 Hz, H-2′,6′), 9.23 (1H, s, 3-OH), and 10.07 (1H, s, 4-OH); MS (FAB+) m/z: 285 [M+H]+.
3′,4′,7-Trimethoxyflavonol (16): yellow solid, yield 86%, and m.p. 178~180°C (lit. [20]: 185°C); 1H NMR (400 MHz, CDCl3): δ 4.03~3.98 (9H, s/each, 3 OCH3), 6.99 (1H, d, J = 2.2 Hz, H-6), 7.02 (1H, d, J = 2.3 Hz, H-8), 7.06 (1H, s, OH), 7.04 (1H, d, J = 3.3 Hz, H-5′),8.17 (1H, d, J = 8.9 Hz, H-5), 7.89 (1H, dd, J = 8.5, 2.0 Hz, H-6′), and 7.86 (1H, d, J = 1.9 Hz, H-2′); MS (FAB+) m/z: 329 [M+H]+.
3.3. Synthesis of 3′,4′,7-Trimethoxyflavonoid-3-O-β-D-Acetylglucoside (17)
Anhydrous K2CO3 (150 mg, 1.09 mmol) was added to the mixture of compound 16 (70 mg, 0.2 mmol) and dry acetone (20 mL); then,α-acetylglucose bromide (200 mg, 0.49 mmol) was added to the mixture with stirring. After stirring for 12 h at room temperature, the acetone was removed under reduced pressure. The residual was chromatographed on silica gel with petroleum ether/ethyl acetate (3 : 1, volume ratio) as eluent to afford a yellow solid, yield 86%, and m.p. 145~146°C; 1H NMR (400 MHz, CDCl3): δ 2.12~1.89 (12H, s/each, COCH3), 3.66~3.61 (1H, m, H-6′′), 3.93 (3H, s, OCH3), 3.97 (3H, s, OCH3),4.03 (3H, s, OCH3), 5.08 (1H, t, J = 9.6 Hz, H-4′′), 5.23~5.17 (1H, m, H-5′′), 5.28 (1H, d, J = 9.4 Hz, H-3′′), 5.74 (1H, d, J = 7.9 Hz, H-1′′), 6.92 (1H, d, J = 2.2 Hz, H-2′′), 7.01~6.96 (2H, m, H-6,8), 7.41~7.29 (1H, m, H-5′), 7.67 (1H, dd, J = 8.6, 2.0 Hz, H-6′), 7.73 (1H, d, J = 2.0 Hz, H-2′), and 8.12 (1H, d, J = 8.9 Hz, H-5); Anal. Calcd for C32H34O15: C, 58.36; H, 5.20. Found: C, 58.61; H, 5.13.
3.4. Synthesis of 3′,4′,7-Trimethoxyflavonoid-3-O-β-D-Acetylgalactoside (18)
Compound 18 was prepared from 16 and α-acetylgalactose bromide as described for the preparation of compound 17 from compound 16 and α-acetylglucose bromide. Yellow solid, yield 74%, and m.p. 136~138°C; 1H NMR (400 MHz, CDCl3): δ 1.91~2.16 (12H, s/each, COCH3), 3.90~3.98 (9H, 3s, OCH3), 4.07 (3H, s, H-2, 4′′, 6′′), 5.15 (1H, dd, J = 10.5, 3.5 Hz, H-5′′), 5.39~5.45 (2H, m, H-2′′, 3′′), 5.74 (1H, d, J = 8.0 Hz, H-1′′), 6.91 (1H, d, J = 2.3 Hz, H-8), 6.97~7.02 (2H, m, H-5′, 6), 7.68 (1H, dd, J = 8.6, 2.1 Hz, H-6′), 7.96 (1H, d, J = 2.1 Hz, H-2′), and 8.07~8.13 (1H, m, H-5); MS (FAB+) m/z: 659 [M+H]+.
3.5. Synthesis of 3′,4′7-Trimethoxyflavonoid-3-O-β-D-Acetyllactoside (19)
Compound 19 was prepared from 16 and α-acetyllactose bromide as described for the preparation of compound 17 from compound 16 and α-acetylglucose bromide. Yellow solid, yield: 42%, and m.p. 151~152°C. IR (KBr) ν/cm−1: 3548, 3414, 3139, 1749, 1637, 1618, 1514, 1400, 1237, 1135, 1063, 956, 838, 780, 620, 541, and 484. 1H NMR (400 MHz, CDCl3): δ 1.85~2.15 (21H, s/each, COCH3), 3.53~3.55 (1H, m, H-sugar), 3.77~3.92 (3H, m, H-sugar), 3.93~4.01 (9H, s/each, 3OCH3), 4.05 (1H, d, J = 7.5 Hz, H-sugar), 4.08 (1H, d, J = 7.5 Hz, H-sugar), 4.14 (1H, dd, J = 6.2, 11.1 Hz, H-sugar), 4.28 (1H, dd, J = 12.0, 2.0 Hz, H-sugar), 4.43 (1H, d, J = 7.9 Hz, H-1′′′), 4.92 (1H, dd, J = 10.4, 3.4 Hz, H-sugar), 5.05~5.13 (2H, m, H-sugar), 5.25 (1H, t, J = 9.3 Hz, H-sugar), 5.33 (1H, d, J = 2.5 Hz, H-sugar), 5.65 (1H, d, J = 7.9 Hz, H-1′′), 6.91 (1H, d, J = 2.3 Hz, H-6), 6.95~7.01 (2H, m, H-5′, 8), 7.62~7.68 (2H, m, H-2′, 6′), and 8.11 (1H, d, J = 8.9 Hz, H-5); MS (FAB+) m/z: 947 [M+H]+.
3.6. Synthesis of 3′,4′,7-Trimethoxyflavonoid-3-O-β-D-Glucoside (20)
Compound 17 (15 mg, 22.7 mmol) was added to a solution of 30% NH3·H2O (0.5 mL) in CH3OH (3 mL) with stirring. After stirring for 6 h at room temperature, the solvent was removed under reduced pressure. The residual was chromatographed on silica gel with ethyl acetate/EtOH (1 : 1, volume ratio) as eluent to afford a light-yellow solid 70 mg, yield 79%, and m.p. 110~111°C. 1H NMR (400 MHz, DMSO-d 6): δ 3.11~3.28 (4H, m, H-3′′, 4′′, 5′′, 6′′), 3.37~3.43 (1H, m, H-2′′), 3.57 (1H, dd, J = 11.4, 5.6 Hz, H-6′′), 3.85~3.92 (9H, s/each, OCH3), 4.38 (1H, t, J = 5.4 Hz, OH-6′′), 4.96 (1H, d, J = 4.1 Hz, OH-4′′), 5.09 (1H, d, J = 4.6 Hz, OH-3′′), 5.43 (1H, d, J = 4.2 Hz, OH-2′′), 5.65(1H, d, J = 7.3 Hz, H-1′′), 7.08 (1H, dd, J = 8.9, 2.3 Hz, H-6), 7.12 (1H, d, J = 8.7 Hz, H-8), 7.27 (1H, d, J = 2.3 Hz, H-5′), 7.68 (1H, dd, J =8.5, 1.9 Hz, H-6′), 7.96 (1H, d, J = 1.9 Hz, H-2′), and 7.98 (1H, d, J = 8.9 Hz, H-5); Anal. Calcd for C24H26O11: C, 58.77; H, 5.34. Found: C, 58.96; H, 5.28.
3.7. Synthesis of 3′,4′,7-Trimethoxyflavonoid-3-O-β-D-Galactoside (21)
Compound 21 was prepared from compound 18 as described for the preparation of compound 20 from compound 17. Light-yellow solid, yield 67%, and m.p. 135~137°C. IR (KBr) ν/cm−1: 3413, 3233, 1618, 1518, 1446, 1399, 1270, 1206, 1077, 1017, 883, 776, 620, and 482; 1H NMR (400 MHz, DMSO-d 6): δ 3.39~3.46 (3H, m, H-2′, 5′′, 6′′), 3.49 (1H, dd, J = 9.7, 4.6 Hz, H-3′′), 3.54~3.61 (1H, m, H-4′′), 3.68 (1H, t, J = 3.4 Hz, H-2′′), 3.85 (6H, s, OCH3), 3.92 (3H, s, OCH3), 4.50 (1H, d, J = 5.3 Hz, OH-6′′), 4.54 (1H, d, J = 3.7 Hz, OH-2′′), 4.91 (1H, d, J = 5.6 Hz, OH-4′′), 5.28 (1H, d, J = 4.3 Hz, OH-3′′), 5.57 (1H, d, J = 7.7 Hz, H-1′′), 7.09 (2H, dd, J = 13.7, 5.4 Hz, H-6,8), 7.28 (1H, d, J = 2.2 Hz, H-5′), 7.68 (1H, dd, J = 8.5, 1.9 Hz, H-6′), 7.98 (1H, d, J = 8.9 Hz, H-2′), and 8.04 (1H, d, J = 1.8 Hz, H-5); Anal. Calcd for C24H26O11: C, 58.77; H, 5.34. Found: C, 58.54; H, 5.26.
3.8. Synthesis of 3′,4′,7-Trimethoxyflavonoid-3-O-β-D-Lactoside (22)
Compound 22 was prepared from compound 19 as described for the preparation of compound 20 from compound 17. Light-yellow solid, yield 82%, and m.p. >200°C. IR (KBr) ν/cm−1: 3413, 3231, 1618, 1588, 1553, 1518, 1447, 1399, 1260, 1223, 1153, 1122, 1091, 1040, 1018, 958, 895, 863, 829, 780, 675, 618, 541, and 484; 1H NMR (400 MHz, DMSO-d 6): δ 3.38 (2H, s, H-sugar), 3.46 (3H, d, J = 7.1 Hz, H-sugar), 3.49~3.59 (3H, m, H-sugar), 3.61 (2H, s, H-sugar), 3.85~3.92 (9H, s/each, OCH3), 4.23 (1H, d, J = 7.2 Hz, H-sugar), 4.43 (2H, t, J = 5.6 Hz, OH-sugar), 4.52 (1H, d, J = 4.6 Hz, OH-sugar), 4.68 (1H, t, J = 5.1 Hz, OH-sugar), 4.78 (1H, d, J = 5.3 Hz, OH-sugar), 4.81 (1H, d, J = 1.7 Hz, OH-sugar), 5.09 (1H, d, J = 4.3 Hz, OH-sugar), 5.56 (1H, d, J = 6.3 Hz, H-1′′′), 5.67 (1H, d, J = 7.8 Hz, H-1′′), 7.08 (1H, dd, J = 2.4, 8.9 Hz, H-6), 7.13 (1H, d, J = 8.7 Hz, H-8), 7.29 (1H, d, J = 2.3 Hz, H-5′), 7.71 (1H, dd, J = 8.6, 2.0 Hz, H-6′), 7.92 (1H, d, J = 2.1 Hz, H-2′), and 7.99 (1H, d, J = 8.9 Hz, H-5); Anal. Calcd for C30H36O16: C, 55.21; H, 5.56. Found: C, 55.49; H, 5.47.
3.9. In Vitro Cytotoxic Activity Evaluation by SRB Assay
The cytotoxic activity of the chalcones and 5-deoxyflavonoid was evaluated against MDA-MB-231, U251, BGC-823, and B16 tumor cells. MDA-MB-231, U251, BGC-823, and B16 cells were maintained in RPMI-1640 medium supplement with 10% heat inactivated fetal bovine serum (FBS) and incubated at 37°C in a 5% CO2 humidified atmosphere. In order to maintain the cells in log phase cellular suspension, aliquots were refed with fresh RPMI-1640 medium two or three times per week. The stock solutions of the tested compounds were freshly resolved in DMSO and consequently diluted in RPMI-1640. At the final dilutions, the obtained concentration of the solvent never exceeded 0.5%.
The cytotoxic activity was measured in vitro using the SRB colorimetric assay. Cells were inoculated in 96-well microtiter plate (104 cells/well) for 24 h before treatment with the compound(s) to allow attachment of the cell to the wall of the plate. Test compounds were dissolved in DMSO and diluted with saline to the appropriate volume. Different concentrations of the compound under test (0.1, 2.5, 5 and 10 μg/mL) were added to the cell monolayer. Triplicates were prepared for each individual dose. Monolayer cells were incubated with the compound(s) for 48 h, at 37°C, and in atmosphere of 5% CO2. After 48 h, cells were fixed, washed, and stained for 30 min with 0.4% (w/v) SRB dissolved in 1% acetic acid. Unbound dye was removed by four washes with 1% acetic acid, and attached stain was recovered with Tris-EDTA buffer. Color intensity was measured in an ELISA reader. The relation between survival curve for cancer cell lines after the specified time. The concentration required for 50% inhibition of cell viability (IC50) was calculated.
Acknowledgments
This work was supported by the Personal Training Funds in National Basic Science of China (J1103312/J0104) and the Project of Science of Department of Education of Hunan Province (10B012).
References
- 1.Kumar SK, Hager E, Pettit C, Gurulingappa H, Davidson NE, Khan SR. Design, synthesis, and evaluation of novel boronic-chalcone derivatives as antitumor agents. Journal of Medicinal Chemistry. 2003;46(14):2813–2815. doi: 10.1021/jm030213+. [DOI] [PubMed] [Google Scholar]
- 2.Lopez A, Ming DS, Neil Towers GH. Antifungal activity of benzoic acid derivatives from piper lanceaefolium. Journal of Natural Products. 2002;65(1):62–64. doi: 10.1021/np010410g. [DOI] [PubMed] [Google Scholar]
- 3.Salem MM, Werbovetz KA. Antiprotozoal compounds from Psorothamnus polydenius. Journal of Natural Products. 2005;68(1):108–111. doi: 10.1021/np049682k. [DOI] [PubMed] [Google Scholar]
- 4.Edwards ML, Stemerick DM, Sunkara PS. Chalcones: a new class of antimitotic agents. Journal of Medicinal Chemistry. 1990;33(7):1948–1954. doi: 10.1021/jm00169a021. [DOI] [PubMed] [Google Scholar]
- 5.Wu JH, Wang XH, Yi YH, Lee KH. Anti-AIDS agents 54. A potent anti-HIV chalcone and flavonoids from genus Desmos . Bioorganic & Medicinal Chemistry Letters. 2003;13(10):1813–1815. doi: 10.1016/s0960-894x(03)00197-5. [DOI] [PubMed] [Google Scholar]
- 6.Lin L-C, Chiou C-T, Cheng JJ. 5-Deoxyflavones with cytotoxic activity from Mimosa diplotricha . Journal of Natural Products. 2001;74(9):2001–2004. doi: 10.1021/np200307r. [DOI] [PubMed] [Google Scholar]
- 7.Nguyen PH, Dao TT, Kim J, Phong DT, Ndinteh JT, Mbafaor WKO. New 5-deoxyflavonoids and their inhibitory effects on protein tyrosine phosphatase 1B (PTP1B) activity. Bioorganic & Medicinal Chemistry. 2011;19(11):3378–3383. doi: 10.1016/j.bmc.2011.04.037. [DOI] [PubMed] [Google Scholar]
- 8.Deng J, Sanchez T, Al-Mawsawi LQ, et al. Discovery of structurally diverse HIV-1 integrase inhibitors based on a chalcone pharmacophore. Bioorganic & Medicinal Chemistry. 2007;15(14):4985–5002. doi: 10.1016/j.bmc.2007.04.041. [DOI] [PubMed] [Google Scholar]
- 9.Avila HP, Smania EDFA, Monache FD, Smânia A., Jr. Structure–activity relationship of antibacterial chalcones. Bioorganic & Medicinal Chemistry. 2008;16(22):9790–9794. doi: 10.1016/j.bmc.2008.09.064. [DOI] [PubMed] [Google Scholar]
- 10.Chiruta C, Schubert D, Dargusch R, Maher P. Chemical modification of the multitarget neuroprotective compound fisetin. Journal of Medicinal Chemistry. 2012;55(1):378–389. doi: 10.1021/jm2012563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wells L, Vosseller K, Hart GW. Glycosylation of nucleocytoplasmic proteins: signal transduction and O-GlcNAc. Science. 2001;291(5512):2376–2378. doi: 10.1126/science.1058714. [DOI] [PubMed] [Google Scholar]
- 12.Zhang X, Simmons CG, Coreg DR. Liver cell specific targeting of peptide nucleic acid oligomers. Bioorganic & Medicinal Chemistry Letters. 2001;11(10):1269–1272. doi: 10.1016/s0960-894x(01)00198-6. [DOI] [PubMed] [Google Scholar]
- 13.Wu Z, Liang ZY, Li W, Ren YM, Wang QA. Synthesis of naturally occurring neolignans demethylnitidanin, herpetol and salvinal as well as their glycosyl derivatives. Chemical Research in Chinese Universities. 2011;27(6):949–954. [Google Scholar]
- 14.Furniss BS, Hannaford AJ, Smith PWGAR. Textbook of Practical Organic Chemistry. 5th edition. New York, NY, USA: John Wiley & Sons; 1989. [Google Scholar]
- 15.Zhao J, Zhang ZP, Chen HS, Zhang XQ, Chen XH. Synthesis of baicalin derivatives and evaluation of their anti-human immunodeficiency virus(HIV-1) activity. Acta Pharmacologica Sinica. 1998;33:p. 22. [PubMed] [Google Scholar]
- 16.Guan LP, Yin XM, Quan HM, Quan ZS. Synthesis of hydroxylated chalcones and related derivatives. Chinese Journal of Organic Chemistry. 2004;24(10):1274–1277. [Google Scholar]
- 17.Anthoni U, Rosalba ED, Nielsen PH, Christophersen G. Huazhongilexone is not 3′,5,5′,7-tetrahydroxyflavanone. Preparation of 3′,5′-dimethoxy-5,7-dihydroxyflavanone. Acta Chemica Scandinavica. 1998;52(10):1243–1246. [Google Scholar]
- 18.Neves MP, Cravo S, Lima RT, et al. Solid-phase synthesis of 2′-hydroxychalcones. Effects on cell growth inhibition, cell cycle and apoptosis of human tumor cell lines. Bioorganic & Medicinal Chemistry. 2012;20(1):25–33. doi: 10.1016/j.bmc.2011.11.042. [DOI] [PubMed] [Google Scholar]
- 19.Simpson TH, Garden L. 905. Chelate systems. Part I. Journal of the Chemical Society. 1952:4638–4644. [Google Scholar]
- 20.Oyamada T. A new general method for the synthesis of flavonol derivatives. The Chemical Society of Japan. 1934;55:1256–1261. [Google Scholar]