

Abstract
Microcin C (McC) is heptapeptide-adenylate antibiotic produced by Escherichia coli strains carrying the mccABCDEF gene cluster encoding enzymes, in addition to the heptapeptide structural gene mccA, necessary for McC biosynthesis and self-immunity of the producing cell. The heptapeptide facilitates McC transport into susceptible cells, where it is processed releasing a non-hydrolyzable aminoacyl adenylate that inhibits an essential aminoacyl-tRNA synthetase. The self-immunity gene mccF encodes a specialized serine-peptidase that cleaves an amide bond connecting the peptidyl or aminoacyl moieties of, respectively, intact and processed McC with the nucleotidyl moiety. Most mccF orthologs from organisms other than E. coli are not linked to the McC biosynthesis gene cluster. Here, we show that a protein product of one such gene, MccF from Bacillus anthracis (BaMccF), is able to cleave intact and processed McC and we present a series of structures of this protein. Structural analysis of apo-BaMccF and its AMP-complex reveal specific features of MccF-like peptidases that allow them to interact with substrates containing nucleotidyl moieties. Sequence analyses and phylogenetic reconstructions suggest that several distinct subfamilies form the MccF clade of the large S66 family of bacterial serine peptidases. We show that various representatives of the MccF clade can specifically detoxify non-hydrolyzable aminoacyl adenylates differing in their aminoacyl moieties. We hypothesize that bacterial mccF genes serve as a source of bacterial antibiotic resistance.
Keywords: MccF, serine peptidase, nucleophilic elbow, catalytic triad (Ser-His-Glu), substrate binding loop
INTRODUCTION
Microcins are a small (less than 10 kDa) bacteriocins produced by E. coli and its close relatives1. Some microcins are synthesized as propeptides, which undergo complex posttranslational modifications by dedicated maturation enzymes 2,3. One of these post-translationally modified microcins is microcin C (McC). McC is composed of a heptapeptide covalently attached to adenosine monophosphate (AMP) through the non-hydrolyzable N-acyl-phosphoramidate linkage4,5. An additional aminopropyl moiety is coupled to the phosphate via an ester bond. The McC biosynthetic cluster consists of the mccABCDE operon (Figure 1A). The mccA gene encodes the heptapeptide McC precursor. MccB adenylates the MccA heptapeptide, while MccD and the N-terminal domain of MccE are required for phosphate modification with propylamine. The MccC efflux pump and the C-terminal domain of MccE provide the producing cell with resistance to McC 6.
Figure 1.

A Organization of the mcc gene cluster from E. coli. Arrows indicate mcc genes and the directions of the arrows indicate the direction of transcription (arrows are not drawn to scale). The mccA–E genes form a single operon and are involved in the synthesis of microcin C (chemical structure shown). Genes whose products contribute to McC immunity are highlighted in red. B. The proposed mechanism of McC transport into the cell, intracellular processing, and inhibition of aspartyl-tRNA synthetase. C. Chemical structure of McC analogs aspartyl sulfamoyl adenylate (DSA) and glutamyl sulfamoyl adenylate (ESA).
McC enters the outer membrane of the E. coli cell mostly through the OmpF porin and is transported through the inner membrane by the YejABEF ABC family transporter 7,8. Once McC enters a sensitive cell, the formyl group is removed from the N-terminal methionine by peptide deformylase and subsequently the peptide part of McC is removed by any one of the three aminopeptidases (peptidases A, B, or N)6, 8, 9. The final product of the intracellular processing is a non-hydrolyzable aspartyl-adenylate analogue, which is a potent inhibitor of aspartyl-tRNA synthetase (AspRS)10 (Figure 1A.). Inhibition of this essential enzyme by processed McC leads to the cessation of protein synthesis and cell growth. Intact, unprocessed McC does not affect the aminoacylation reaction, while processed McC has no effect on the growth of McC-sensitive cells at concentrations at which intact McC is highly active 11. McC may thus be viewed as a Trojan horse inhibitor, whose peptide part enables cell entry of the toxic constituent that is subsequently released after intracellular proteolysis 13. There are other inhibitors that also employ the Trojan horse mechanism in targeting aminoacyl-tRNA synthetases. These include albomycin, a nonhydrolyzable seryl pyrimidyl attached to ferritin transport moiety12, and agrocin 84, a nonhydrolyzable leucyl adenylate modified by an opine necessary for transport inside agrobacterial cells13–15. Albomycin targets SerRS, whereas agrocin 84 targets LeuRS.
Earlier studies revealed that mccF, a gene that is adjacent to mccE but transcribed in the direction opposite to the mccABCDE operon transcription direction, contributes to the self-immunity of McC-producing cells 16. A sequence analysis revealed that MccF belongs to the S66 family of serine peptidases 6,17. Recent studies have shown that MccF cleaves a C-N bond adjacent to the N-acyl-phosphoramidate linkage in intact or processed McC thus abolishing the antibacterial activity6. The only other characterized members of the S66 family are LD-carboxypeptidases (LdcA). These enzymes are involved in peptidoglycan recycling and cleave amide bonds between L- and D-amino acids, which occur naturally in bacterial peptidoglycan 18,19. However, unlike MccF, E. coli LdcA is unable to cleave McC6.
Multiple genes encoding proteins homologous to the serine peptidases of the S66 family are present in the genomes of various bacteria. Most of these genes are not associated with other mcc-like genes and their physiological role and biochemical functions are unknown. Here, we show that several MccF homologs are capable of detoxifying various non-hydrolyzable aminoacyl adenylates and provide McC resistance when overepxressed in E. coli. These proteins form a monophyletic group, the MccF clade, of the S66 family that is distinct from the LdcA clade. We determined the first crystal structure of one of the representatives of the clade, MccF from Bacillus anthracis str. Ames (BaMccF) in the apo-form and in complex with AMP. The structures of BaMccF and its mutants, together with bioinformatics analysis and functional data provide insights into the molecular mechanism of substrate binding and catalysis by MccF-related enzymes.
RESULTS
The MccF homolog encoded in the B. anthracis genome is functionally equivalent to MccF encoded by the plasmid-borne microcin C operon of E. coli
The BaMccF (BA_1949) protein is the closest homolog of E. coli MccF (EcMccF) in the B. anthracis genome. It is encoded by a gene of unknown function and is not associated with other mcc genes. Thus, it is an interesting candidate for functional and structural analyses. To determine whether BaMccF is able to hydrolyze McC and McC-like compounds (Figure 1A), the following experiments were performed. A pET-based plasmid containing cloned Ba-mccF was introduced in McC-sensitive E. coli BL21(DE3) cells. Cells transformed with a plasmid expressing EcMccF 10 or an empty pET19 vector were used as a positive and negative control, respectively. Drops of intact McC or DSA, an analog of processed McC6, solutions were deposited on the surface of freshly made lawns of cells containing each plasmid. After an overnight growth at 37 °C, growth inhibition zones around the points where inhibitor solutions were deposited were monitored. The results are presented in Figure 2A. As expected, cells containing only the pET vector were sensitive, while cells overproducing EcMccF were resistant to both McC and DSA at the concentrations tested. Compared to McC, much higher concentrations of aminoacyl adenylates had to be used to observe growth inhibition of sensitive cells due to poor uptake in the absence of the transport peptide. Overproduction of BaMccF also rendered cells resistant to McC and DSA, suggesting that it has an overlapping substrate specificity with EcMccF.
Figure 2.

A. Overproduction of BaMccF renders cell resistant to microcin C and DSA. Cell lawns were made from cultures producing EcMccF, BaMccF and its double active site mutant – S112A, H303A. Cells transformed with the empty pET19 vector for gene overexpression were used as a negative control. The sizes of growth inhibition zones around 2-μl drops of DSA and McC solutions in indicated concentrations deposited on cell lawns are shown. The error bars show standard deviations of measurements obtained in at least three independent experiments. B. BaMccF inactivates microcin C and its analogs in vitro. Purified recombinant EcMccF, BaMccF and S112A, H303A BaMccF were incubated with microcin C and chemical analogs of its processed form – ESA and DSA. Spots of solutions containing reaction products were placed on E.coli BL21 cell lawn. The method is described in “Experimental Procedures”. Sizes of growth inhibition zones recorded after several hours of cell growth are presented. The error bars show standard deviations of measurements obtained in at least three independent experiments. C. In Vivo sensitivity test of B.cereus ATCC4342 and the mutant on a semisolid agar plate. D. mccF− mutant of B.cereus ATCC4342 is hypersensitive to processed microcin C analog. WT and mccF− B.cereus ATCC4342 overnight cultures were diluted in fresh LB broth and grown in the presence of 500 mM DSA. No DSA was added to the control cultures. At the indicated time points the OD600 of the culture was measured. Representatives of several independent experiments are shown.
In the second test of BaMccF function, we incubated purified recombinant BaMccF and EcMccF with McC, aspartyl sulfamoyl adenylate (DSA), and glutamyl sulfamoyl adenylate (ESA). Previously, we showed that EcMccF cleaves these compounds in vitro6. While MccF-catalyzed cleavage inactivates McC, cleavage of DSA and ESA generates sulfamoyl adenosine, which is highly toxic to E. coli cells. The products of McC, DSA, and ESA incubation with various proteins were deposited on lawns of E. coli cells and after several hours of incubation at 37°C growth inhibition zones were monitored. As can be seen from Figure 2B, in this in vitro assay wild-type BaMccF also behaved indistinguishably from EcMccF (caused the disappearance of growth inhibition zones by McC and the appearance of large inhibition zones in reactions containing DSA or ESA due to the generation of sulfamoyl adenosine). Based on this result we conclude that similarly to EcMccF, BaMccF likely cleaves the C-N bond in the carboxamide moiety of in DSA and ESA. Inactivations of McC by BaMccF likely occurs by the same mechanism. We therefore conclude that the MccF proteins from E. coli and B. anthracis are functionally equivalent to one another.
A plasmid expressing a BaMccF mutant substituting Ser112 and His303 for alanines was prepared. Based on a bioinformatics analysis, Ser112 and His303 are part of the catalytic triad and their substitution should abolish the peptidase function. Substitutions in corresponding residues of EcMccF make it unable to detoxify McC, DSA, or ESA6. As expected, E. coli cells expressing mutant BaMccF were sensitive to McC and DSA (Figure 2A), while the purified mutant protein was unable to detoxify McC, DSA, and ESA (Figure 2B).
To determine the physiological role of BaMccF, we deleted the corresponding gene in a surrogate host B. cereus ATCC4342 (the BaMccF and BcMccF sequences are 95% identical and are located in identical genomic positions). The mutant strain was viable and exhibited no defects during growth in rich medium. While both the wild-type and mutant cells were completely resistant to McC at all concentrations tested (most likely due to problems with the drug uptake), we found that DSA did produce growth inhibition zones on lawns of wild-type and mutant B. cereus lawns. To quantitatively monitor the effect of DSA, the wild-type and mutant cells were grown in liquid medium followed by the addition of DSA. As can be seen from Figure 2C, the growth of the wild-type strain in the presence of DSA was considerably faster than that of the mutant strain. We therefore conclude that B. cereus mccF contributes to the resistance of this bacterium to non-hydrolyzable aspartyl adenylates.
Overall structures of BaMccF
Structures of wild-type and mutant (S112A, W180A, F177S, and S238R) BaMccF proteins were determined by synchrotron-based X-ray crystallography methods. Diffraction data were collected at the 19ID beamline of the Structural Biology Center at the Advanced Photon Source, Argonne National Laboratory20. The apo-BaMccF structure was solved by the single-wavelength anomalous dispersive (SAD) method using selenomethionine-substituted protein and refined to 2.1-Å resolution with R/R free =18.1/20.3. The structures of the mutant proteins were determined by the molecular replacement (MR) method. Both the apo- and the S112A-BaMccF proteins crystallized in P43 symmetry. The AMP-bound structure of the S112A-BaMccF mutant was refined to 1.5-Å with R/R free =16.5/18.6. The W180A-BaMccF protein crystallized in the orthorhombic space group P212121 and the structure was determined by MR and refined to 1.75-Å to R/R free =15.9/19.6. The crystals of F177S-BaMccF and S238R-BaMccF were similar to the apo form and the structures were also determined by MR and refined to R/R free =16.9/21.0 and R/R free =16.5/19.0, respectively.
All five structures of BaMccF revealed two molecules in the asymmetric unit related by a 2-fold rotation axis forming an oval-shaped homodimer. These results are consistent with the elution profile obtained during a size exclusion chromatography experiment 21 (as estimated from size-exclusion chromatography, ~ 60 kDa). A calculation of the accessible surface area using the PISA server22 further confirms the dimer is a biologically relevant oligomeric form of BaMccF. An extensive interaction between tightly associated subunits of the dimer buries 3,600 Å2 of the solvent-accessible surface per monomer, which corresponds to 26% of the total solvent-accessible surface area.
The structure fold description is based on the highest resolution structure of the MccF protein (1.5-Å S112A- BaMccF), however all determined structures of BaMccF show an almost identical fold. An overlay between the molecules shows on average, an rmsd of 0.25 Å, indicating nearly identical structures. The overall structure of the BaMccF dimer has an oblong shape and has a large cleft at one face of the dimer. The subunit of the dimer has approximate dimensions of 50 Å × 40 Å × 35 Å and can be divided into two domains: the N-terminal and the C-terminal (Figure 3A,B). The N-terminal domains adopts a Rossmann-like fold, consisting of a central five-stranded parallel open twisted β-sheet, flanked on both sides by a total of five α-helices (S1-H1-H2-S2-H3-S3-H4-H5-S4-H6-S5). The N-terminal domain is connected to the C-terminal domain by a 27-residue loop (L1) containing 2 small β-strands (S7-S8). The C-terminal domain forms an eight-stranded β-barrel (S6-S9-S10-H9-S11-H10-H11-S12-H12-S13-S15-S16) flanked by four α-helices located at one side of the barrel and contributes to the formation of the dimer interface. In the dimer, the N-terminal domain of one subunit interacts with the C-terminal domain of the opposite subunit leading to the formation of a large central cavity, which houses the active sites (Figure 3C).
Figure 3.

Structure of BaMccF. A. Ribbon diagram showing the overall structure of the MccF dimer, with subunits shown in different colors (subunit I in green, magenta, and red; subunit II in teal). Individual domains of the subunit I are labeled and colored in green and red, while a 27 residues long loop (residues 167–194) connecting them is shown in magenta and labeled L1. The catalytic residues are shown as orange sticks. B. Topology diagrams of MccF. The α-helices and β-strands are shown as cylinders and arrows and the color scheme is maintained as in the figure A. The catalytic triad residues are labeled and their position marked with a star. C. Surface rendering as computed by the program PYMOL showing the active site groove in the dimer of BaMccF. The AMP molecules are shown as spheres. Blue and red represent the positive and negative charge potentials at the + 59 kTe−1 and − 59 kTe−1 scales, respectively. Selected secondary elements are labeled.
Similarity of BaMccF and LD-carboxypeptidases
BaMccF is a member of medium-size serine peptidases (family S66), which includes proteins found mainly in bacteria, with very limited representatives distributed in Archaea and Eukaryotes. The amino acid sequence fingerprint of this family is the presence of the catalytic triad constituted by conserved serine, histidine, and glutamic acid residues (in BaMccF: S112, E235, H303). The only member of the S66 family that has been functionally and structurally characterized to date is the LD-carboxypeptidase from Pseudomonas aeruginosa 19. In addition, a structure of putative LD-carboxypeptidase from Novosphingobium aromaticivorans is available in the PDB. Despite low sequence similarity (~19 % identity), superimposition of the BaMccF over LdcA structures using the program Dali (LdcA from P. aeruginosa PDB id 1ZRS, rmsd 2.3 Å, Z-score 33; and rmsd 2.6 Å, Z-score 26 for putative LdcA from N. aromaticivorans PDBid 3G23) reveals nearly identical folds, similar organization of domains, and similar dimerization interfaces (Figure 4A). The greatest similarity is observed in the center of the protein (between residues 108–113 in BaMccF, which correspond to residues 111–116 in 1ZRS LD-carboxypeptidase), around the location of the catalytic serine. This region (a tight turn between a β-strand and α-helix H6) contains a nucleophile elbow motif, which harbors the serine nucleophile and the oxyanion hole (likely formed by the backbone NH group of D113 and G86). This motif is apparent at the sequence level by the presence of a G-X-S-D-X signature sequence motif. The remaining catalytic residues also overlay closely showing analogous orientations (Figure 4B and 5). Some differences between the structures are also observed at the N- and the C-termini of the proteins. However, the major difference between BaMccF and LD-carboxypeptidase structures is the presence of a 27-residue insertion in BaMccF that forms a long loop L1 (Figure 3A,C and 4A), connecting the domains and partially overhanging the central groove (Figure 3C).
Figure 4.
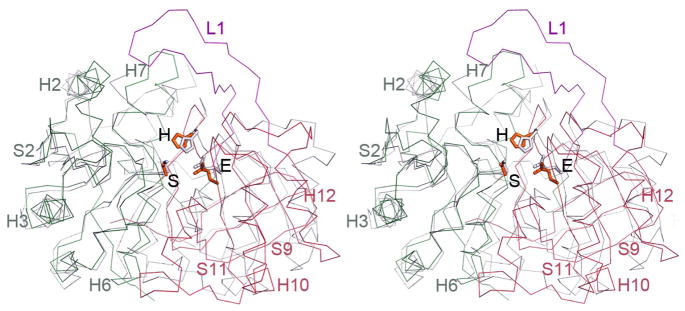
Superimposition of members of the S66 serine protease family. A. Ribbon diagram showing a subunit of BaMccF (green, red and magenta) overlaid over a subunit of the dimer of the LdcA from P. aeruginosa (PDB code: 2AUM) (gray), which has been identified as having the closest matching folds. The catalytic triad in both structures (BaMccF S112, E269, H303 correspond to LdcA residues: S115, E217, H285) are positioned similarly. Selected secondary elements of BaMccF are highlighted.
Figure 5.

Multiple sequence alignment of MccF proteins from B. anthracis and E. coli versus LdcA from P. aeruginosa and putative LdcA from N. aromaticivorans. Sequence identities are highlighted in red and similarities are shown as red letters. Blue letters mark the catalytic triad residues and the letter L describes the active site loops. The corresponding secondary structure of BaMccF is shown on the top (black).
The AMP binding region and the catalytic triad
To gain insight into how substrates might bind to BaMccF we have determined several structures of BaMccF point mutants (S112A, F177S, and S238R) with bound AMP, a product of McC cleavage. The mode of AMP binding is similar in all three structures (Table 1). Clear electron density features corresponding to AMP molecules were observed at each end of the central groove (Figure 2C and 5). The AMP binding pocket is formed by the loop (L1) of the C-terminal domain (top) and the helix H6 of the N-terminal domain (bottom of the pocket), which houses conserved S112 of the catalytic triad. The sides of the AMP pocket are flanked on one side by the loop with highly conserved G85 and G86 from the N-terminal domain (G86, positioned at loop L2 is also contributes to the oxyanion hole), while the opposite side is formed by three loops (L3, L4, L5). Loops L3 and L4 are supplying the remaining residues of the catalytic triad (H303, E235), while loop L5 supplies E269. The interaction of the AMP molecule with the protein residues is summarized in Figure 5. The adenine ring of the AMP binds to the hydrophobic sub-cavity formed by the L1 loop turn, and is flanked by W180 at one side and F177 at the opposite site. The ribose moiety of AMP interacts with the protein through the hydroxyl groups of the ribose. The 2′ and 3′ hydroxyl groups form hydrogen bonds with the oxygen atoms of the side chain of E269. The phosphate moiety forms hydrogen bonds with the peptide bond amide of G85 and N2 nitrogen of the imidazole ring of H303. S112 is axially positioned underneath the phosphate atom of AMP within 3.5 Å. The importance of the catalytic triad residues was validated by in vivo and in vitro experiments. As already mentioned, the double S112A and H303A mutant (Figure 1A and B) and the triple mutant of S112A, H303, E325 (data not shown) completely abolished the activity of BaMccF.
TABLE 1.
| Data collection statistics | BaMccF | MccF-W180A | MccF-F177S | MccF-S238R | MccF-S112A |
|---|---|---|---|---|---|
| Space group | P43 | P212121 | P43 | P43 | P43 |
| Unit cell (Å) | a = 119 b = 119 c = 55.5 |
a = 51.3 b = 97.1 c = 125.6 |
a = 118 b = 118 c = 55.0 |
a = 119 b = 119 c = 55.5 |
a = 119 b = 119 c = 55.5 |
| Wavelength (Å) | 0.9794 | 0.9794 | 0.9794 | 0.9794 | 0.9794 |
| Resolution (Å) | 40-2.10 | 40-1.50 | 40-2.04 | 34-1.60 | 40-1.75 |
| Number of observed reflections | 174606 | 391668 | 184837 | 456168 | 286294 |
| Number of unique reflections | 45124 | 93524 | 47231 | 101972 | 77550 |
| Rmerge (%)a | 9.3(56.0)b | 8.2 (56.0)b | 8.8(55.8) b | 7.5(73.1) b | 6.9 (48.5)b |
| Completeness (%) | 99.3(97.8) | 91.7 (68.7) | 98.1(88)b | 99.8(100) b | 99.3(100)b |
| I/σ I | 15.2(2.1)b | 20.2 (2.3)b | 18.0(2.1) b | 20.8 (1.8)b | 22.3 (2.9)b |
| Phasing method | SAD | MR | MR | MR | MR |
| Phasing resolution range (Å) | 40-3.8 | 40-3.0 | 40-3.0 | 34-3.0 | 40-3.0 |
| Number of Se-Met | 7 | - | - | - | - |
| FOM Molphere DM |
0.31 0.72 |
- | - | - | - |
| Rcryst (%) | 18.9 | 15.0 | 16.9 | 16.5 | 18.5 |
| Rfree (%) | 23.2 | 17.8 | 21.0 | 19.0 | 20.2 |
| Number of protein residues per chain | 334/333 | 333/271 | 334/330 | 335/333 | 334/333 |
| AMP/GOL | −/− | −/− | 2/− | 2/− | 2/1 |
| Solvent molecules | 230 | 644 | 260 | 509 | 628 |
| Bond lengths (Å) | 0.020 | 0.014 | 0.016 | 0.015 | 0.004 |
| Bond angles (deg) | 1.69 | 1.55 | 1.88 | 1.61 | 0.97 |
| B-factors (Å2) | |||||
| Protein main chain | 42.8 | 23.3 | 44.5 | 30.1 | 32.6 |
| Protein side chain | 47.8 | 28.3 | 47.6 | 34.9 | 38.0 |
| ADP/GOL | - | - | 58.5/− | 35.5/− | 66.9/22.4 |
| Ramachandran (%)c | |||||
| Most favored regions | 96.2 | 97.5 | 96.7 | 97.3 | 97.5 |
| Additionally allowed regions | 3.8 | 2.5 | 3.3 | 2.7 | 2.5 |
| PDB ID | 3GJZ | 3SR3 | 3TYX | 3U1B | 3T5M |
Rmerge = ΣhklΣi|Ii − <I>|/ΣhklΣi|<I>|, where Ii is the intensity for the ith measurement of an equivalent reflection with indices h, k, and l.
Numbers in parentheses are values for the highest-resolution bin.
As defined by MOLPROBITY.
Phylogenetic analysis of S66 family serine peptidases and the role of L1 loop as a structural and sequence determinant of substrate specificity
Homologs of the S66 family of serine peptidases were identified using a PSI-BLAST search against complete microbial genomes stored in the Refseq database23 with a profile for COG1619 as a query. This search revealed 587 sequences, which were further clustered in order to reduce sequence redundancy. The representative set of 174 sequences was used to build multiple sequence alignments (Supporting material Fig. S1). Confidently aligned blocks were used further for phylogenetic tree reconstruction. The resulting phylogenetic tree is shown in Figure 7 (also see Supporting material Fig. S2 to see details on bootstrap support values). As can be seen, the closest MccF and LdcA homologs belong to two distinct branches separated by several other major and minor branches (Figure 7).
Figure 7.

Phylogenetic tree of S66 family of serine peptidases. The tree was constructed using confidently aligned blocks corresponding to conserved regions of multiple alignments of the S66 family of serine peptidases (174 sequences in total, 188 aligned positions; see Supporting material Fig. S1 for details). Three branches of the MccF clade and LcdA branch are shaded. The bootstrap value for the MccF clade is indicated. The EcMccF(black star) and BaMccF (black dot) sequences are indicated by magenta; sequences corresponding to proteins with determined substrate specificity are indicated by orange. Each terminal node of the tree is labeled by the Genbank Identifier (GI) number, five-letter taxonomy code of an organism and full systematic name of an organism. The taxonomy code is as follows: Gamma – Gammaproteobacteria; Beta – Betaproteobacteria; Alpha - Alphaproteobacteria; delta - Deltaproteobacteria; Bacil – Bacilli; Clost – Clostridia; Deino - Deinococcus-Thermus group; Syner - Synergistetes; Therm - Thermotogae; Acido - Acidobacteria group; Gemma - Gemmatimonadetes; Chroo – Chroococcales; Oscil – Oscillatoriales; Proch - Prochlorales; Nosto -Nostocales; Molli – Mollicutes; Spiro - Spirochaetes; Halob – Halobacteria; Actin – Actinobacteria; Fusob – Fusobacteria; Fibro – Fibrobacteres; Bacte - Bacteroidetes; Negat - Negativicutes; Thepl – Thermoplasmatales; Thepr – Thermoproteales.
In order to gain insights to which branches specificity towards aminoacyl adenylates as substrates could be attributed, we compared multiple alignments of major branches in the region of the L1 loop, which was shown above to play a major role in the stabilization of the substrate via the interaction with the adenine moiety of the AMP. A structural analysis suggests that two aromatic residues from L1 loop, BaMccF, F177 and W180, stack with the adenine moiety of the AMP and thus may be particularly important for the recognition of aminoacyl adenylates. A structural analysis of F177S-BaMccF shows that the presence of only W180 is sufficient to preserve proper stacking and AMP binding, since superimposition of the F177S-BaMccF AMP complex over the AMP bound structure of S112A-BaMccF shows identical positions of AMP molecules in both structures. In contrast, the structure of W180A-BaMccF revealed the active site loop in an open conformation, indicating that W180 may play a dual role: in substrate binding and loop stabilization (Figure 8A). The, F177S substitution appeared to have no effect on BaMccF activity in our in vivo or in vitro tests (data not shown). In contrast, W180A significantly decreased MccF activity in vivo (Figure 8B, compare growth inhibition zones in the presence of 10 μM McC) and in vitro (Figure 8C, compare the size of growth inhibition zones obtained after cleavage of ESA, recall that smaller zones mean less MccF activity due to decreased formation of sulfamoyl adenylate). Based on these results we conclude that W180 contributes to the hydrolyzing activity of BaMccF, most likely by stabilizing the substrate through stacking interactions with the adenine base.
Figure 8.
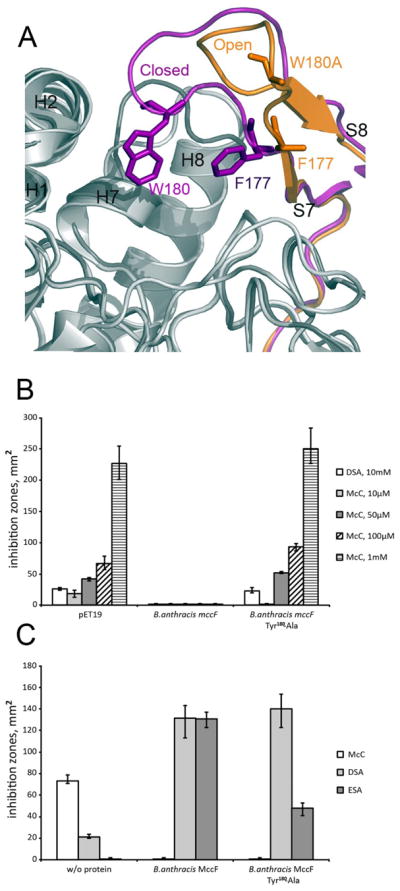
A.The active site loop L1. Two conformation of the active site loop L1 observed in the apo form (magenta) and in the W180A mutant (orange) of BaMccF. B. The active site loop L1 W180A mutant shows decreased activity in vivo. Cell lawns were made from cultures producing BaMccF, and the adenosine interacting residue mutant –W180A. Cells transformed with the empty pET19 vector for gene overexpression were used as a negative control. Sizes of growth inhibition zones with around 2-μl drops of DSA and McC solutions in indicated concentrations deposited on cell lawns are shown. The error bars show standard deviations of measurements obtained in at least three independent experiments. C. The active site loop L1 W180A substitution compromise BaMccF activity in vitro. Purified recombinant BaMccF, and W180A BaMccF were incubated with microcin C and chemical analogs of its processed form – ESA and DSA. Spots of solutions containing reaction products were placed on E.coli BL21 cell lawn. The method is described in “Experimental Procedures”. Sizes of growth inhibition zones recorded after several hours of cell growth are presented. The error bars show standard deviations of measurements obtained in at least three independent experiments.
In addition to the MccF branch, two additional subfamilies typified by YocD from B. subtilis and BA_3275 from B. anthracis show conservation of L1 loop aromatic residues in the position corresponding to W180 in BaMccF (Supporting material Fig. S1) and may thus utilize aminoacyl adenylates as substrates. The position of F177 in contrast is not conserved. Importantly, in addition to the conservation of aromatic amino acids in the L1 loop region in YocD and BA_3275 subfamilies they group together with MccF in the phylogenetic tree to form a distinct clade with statistically significant bootstrap support, suggesting that the substrate specificity is likely monophyletic (Figure 7). S66 family proteins other than MccF, YocD, and BA_3275 subfamilies proteins contain short L1 loops without conserved aromatic residues (Supporting material Fig. S1).
Functional analysis of BaMccF and other MccF clade representatives
Since the results of the phylogenetic analysis suggested that additional proteins from B. anthracis and other pathogenic bacteria may possess an MccF-like activity, the corresponding genes from B. anthracis, B. cereus, Vibrio cholerae, Listeria monocytogenes, Agrobacterium tumefaciens, and Streptococcus pneumoniae were cloned in E. coli expression plasmids and the ability of E. coli cells to grow in the presence of McC and various non-hydrolazable aminoacyl sulfamoyl adenylates was tested. Growth inhibition zones around the points where inhibitor solutions were deposited on freshly made lawns of cells containing each plasmid were monitored after an overnight growth. The results are presented in Table 2. As expected, cells containing only the empty vector were sensitive to both McC and each of the aminoacyl sulfamoyl adenylates (DSA, GSA, PSA, FSA, and ASA) used. Overproduction of EcMccF rendered cells completely resistant to McC and DSA, but had no effect on their sensitivity to other inhibitors, an expected result since previously it was shown that EcMccF has a strong preference for aminoacyl adenylate substrates containing acidic amino acid side chains6. The patterns of sensitivity of cells overproducing BaMccF and V. cholerae homolog from MccF subfamily were very similar, though cells expressing V. cholerae MccF were partially sensitive to the highest concentration of McC used (1 mM), an effect that may either reveal poorer affinity of this protein to McC or, alternatively, be caused by differences in expression levels.
Table 2.
Sensitivity of E.coli BL21(DE3) and E.coli AG1 cultures producing proteins from MccF, BS_YocD, BA_3275 and LdcA branches of the phylogenetic tree of MccF homologs to McC, DSA (aspartyl-sulfamoyl-adenosine), GSA (glycyl-sulfamoyl-adenosine), PSA (prolyl-sulfamoyl-adenosine), FSA (phenylalanyl-sulfamoyl-adenosine) and ASA (alanyl-sulfamoyl adanylates). Sensitivity is determined using the in vivo sensitivity test described in “experimental procedures”. If growth inhibition zone of used microcin analog is absent on the cell lawn of cultures overproducing protein, this culture is designated as resistant (“R”). If the growth inhibition zones is of the same size as zone on the negative control culture, tested culture is designated as sensitive (“S”). If zone is present but smaller than in case of the negative control culture, the tested culture is designated as partially resistant (“PR”). For E.coli BL21(DE3) cultures cells carrying the pET19 vector served as the negative control. For E.coli AG1 + E.coli LcdAcells cultures carrying the pCA24N vector served as the control.
| Clade | Source organism | McC | DSA | GSA | ASA | FSA | PSA |
|---|---|---|---|---|---|---|---|
| MccF | Escherichia coli | R | R | S | S | S | S |
| Bacillus anthracis | R | R | S | S | S | S | |
| Vibrio cholerae | PR | R | S | S | S | S | |
| BS_YocD | Bacillus anthracis | S | S | PR | R | R | R |
| Listeria monocytogenes | S | PR | R | R | R | R | |
| Agrobacterium radiobacter | S | S | S | S | S | S | |
| BA_3275 | Streptococcus pneumoniae | PR | PR | PR | PR | PR | S |
| Bacillus cereus | PR | PR | PR | R | PR | PR | |
| LdcA | Escherichia coli | S | S | S | S | S | S |
Cells expressing YocD from B. anthracis and L. monocytogenes were fully sensitive to McC and DSA. However, these cells were fully or partially resistant to other aminoacyl sulfamoyl adenylates at the concentrations used. Cells expressing Ba_3275 subfamily member from S. pneumoniae, were partially resistant to McC and DSA, FSA, GSA, and ASA, and sensitive to PSA. Cells expressing another Ba_3275 subfamily member from B. cereus showed a similar pattern of sensitivity to McC, DSA, FSA and GSA, while they are resistant to ASA and partially resistant to PSA. Since the B. cereus MccF homolog from the Ba_3275 branch detoxifies DSA in E. coli, it may be responsible for the residual resistance level of B. cereus with deleted BcMccF gene (above).
Cells expressing AgnC5 and LdcA were as sensitive to all compounds used as cells carrying the corresponding empty vector plasmid.
Overall, consistent with bioinformatics predictions the data indicate that many MccF and YocD homologs are able to render cells resistant to toxic aminoacyl adenylates, presumably through their peptidase activity, but specificity towards the aminoacyl moiety of the hydrolyzed substrate appears to be quite variable. In contrast, LdcA homologs are unable to detoxify aminoacyl adenylates.
CONCLUSION
Our finding that BaMccF is functionally equivalent to its ortholog from E. coli was unexpected, as B. anthracis has not been linked to microcin production and is naturally resistant to McC. To date, the mccF gene products have only been associated with Enterobacteriaceae in which the mccF gene is located immediately downstream of the mccABCDE operon. Analysis of the genetic neighborhood of the mccF gene as well as the whole genome of B. anthracis confirmed the absence of other mcc operon members. Thus, stand-alone genes homologous to E. coli mccF and coding for products with identical biochemical activities exist. In fact, functional analysis revealed that MccF orthologs from other pathogenic species (V. cholerae, L. monocytogenes, A. tumefaciens and S. pneumoniae) also provide resistance to McC and various non-hydrolyzable aminoacyl adenylates. The presence of the mccF-like genes in many representatives of pathogenic bacteria suggests that their products could be used for detoxification of various aminoacyl adenylates that are either produced inside the cell or are exogenous.
In order to understand the mechanism of MccF resistance to McC at the molecular level, we have structurally characterized BaMccF. A series of structures of the apo- and AMP-bound mutants of BaMccF unveiled the enzyme’s architecture, its active site cavity, and provided insights into the enzyme function and substrate recognition. A mutagenesis analysis revealed that BaMccF acts as a serine protease, and uses a conserved triad to process the carboxamide bond of McC and it derivatives. The catalytic triad (S112, E235 and H303 residues) located at the bottom of the active site cleft hydrolyzes the peptide bond, likely according to a model established in other well-characterized peptidases24–26. In this mechanism, the S112-OH group acts as a nucleophile, attacking the carbonyl carbon of the last peptide bond of the heptapeptide substrate. The H303 side chain nitrogen has the ability to accept the hydrogen from the S112-OH group, thus coordinating the attack of the peptide bond. The carboxyl group on the E235 forms a hydrogen bound with H303 making it more electronegative. Structures of BaMccF in complex with AMP provide evidence that such a mechanism is highly possible, since S112 is in a good position for a nucleophilic attack.
A comparison of the BaMccF structure with the only other characterized member of the serine peptidases S66 family LdcA shows that both enzymes share a highly similar core albeit with low sequence similarity (~19%). The central and the most conserved, part of the core is established by the “nucleophilic elbow” motif. The motif is characterized by an α-helix that is connected with a β-strand via a sharp β-II type turn containing the nucleophile (S112 in BaMccF). While the elbow motifs are common to α/β hydrolases19, 27,28, the consensus signature sequence of the motif (G-X-S-D-X, with X- describing any residue) appears to be unique to the S66 family. Preservation of the elbow motif between the S66 peptidases confirms their functional significance and provides a strong argument confirming a universal mechanism. Another common feature of the S66 peptidases, which were characterized to date, is their oligomeric state. Both BaMccF and LdcA enzymes were observed to form dimers. Also, a recently determined structure of the putative LdcA from N. aromaticivorans (PDB id 3G23) revealed a dimer as the biological unit, suggesting that S66 proteases likely favor this state.
The key difference between the LdcA and MccF enzymes is the presence of the ~ 27 residue insertion region forming the L1 loop. Structures of BaMccF revealed that the loop is located over the active site groove and binds to the adenine moiety of the AMP. Based on the structural and mutagenic evidences we propose that the loop is responsible for differing specificities of MccF and LdcA, however due to the lack of the structural (A structure of LdcA complexed with the substrate has not been determined yet) and biochemical data it remains speculative. Looking from an evolutionary point of view modification of the L1 loop seems to be the simplest and most attractive avenue to evolve new specificity in an already established mechanistically scaffold of the S66 peptidase.
MATERIALS AND METHODS
Bioinformatics analysis
The PSI-BLAST program (PMID: 9254694) was used to retrieve homologs of the S66 family of serine peptidases from the NCBI Refseq database (PMID: 17130148; March 2010 release) of complete microbial genomes. The position-specific scoring matrices (PSSM) built from the sequences assigned to this family in the COG database for COG1619 (PMID: 12969510) was used as a query with an E-value threshold 0.01. The BLASTCLUST program (PMID:17993672) with a length coverage cutoff of 0.9 and a score coverage threshold of 1.5 (bit score divided by alignment length) was used to reduce sequence redundancy. A few fragmented sequences were manually removed and the sequence for MccF from E.coli (gi: 165975420) was added to form a representative set of 174 homologs of the S66 family serine peptidases. Multiple alignments for this set were constructed using the Muscle program29 (PMID: 15318951) with default parameters, followed by a minimal manual correction on the basis of local alignments obtained using PSI-BLAST30 (PMID: 9254694) in the region of the L1 loop. The confidently aligned blocks (Supporting material Fig. S1) with 188 informative positions were used for maximum likelihood tree reconstruction by the FastTree program31 (PMID: 20224823) with default parameters (default parameters : JTT evolutionary model, discrete gamma model with 20 rate categories). The same program was used for the calculation of bootstrap values.
Gene cloning and protein expression
The coding genes of full length BaMccF and BaYocD were amplified by PCR from Bacillus anthracis str. Ames genomic DNA (ATCC) with KOD DNA polymerase using conditions and reagents according to a standard protocol described previously32. An identical procedure was used to produce expression clones of MccF homologs from V. cholerae O1 biovar El Tor str N16961, Listeria monocytogenes EGD-e, and Streptococcus pneumoniae str TIGR4. This process generated expression clones of fusion proteins, which have the N-terminal His6-tag followed by a TEV protease recognition site (ENLYFQ↓S). The fusion protein was expressed in an E. coli BL21 (DE3)-derivative that harbored the pMAGIC plasmid encoding one of the rare E. coli tRNAs, Ile (ATA). The transformed BL21 (DE3) cells were grown at 37°C in LB medium and protein expression was induced with 1mM isopropyl-β-d-thiogalactoside (IPTG). The cells were then incubated at 18°C overnight. The harvested cells were re-suspended in lysis buffer (500mM NaCl, 5% (v/v) glycerol, 50mM HEPES, pH 8.0, 10mM imidazole, 10mM 2-mercaptoethanol) and stored at −20°C. The B. cereus mccF homolog from the BA_3275 clade was cloned into the pET19b vector. The preparation of expression clones of AgnC5 from Agrobacterium radiobacter str. K84, and LdcA from E.coli was described previously6.
In Vivo Sensitivity test
E. coli BL21(DE3) cells carrying pET19 control3, pET19 + E. coli mccF, pET19+ B. cereus mccF, pMCSG7 + B. anthracis mccF, pMCSG7 with several homologs of B. anthracis mccF, pMCSG7 with several mutants of B. anthracis mccF, pET21+A.radiobacter mccF homolog, and E. coli AG1 cells carrying the pCA24N, pCA24N + E. coli ldcA were grown in 2 ml of LB broth at 37 °C to OD600 of ~1 in the presence of ampicillin (100 μg/ml) or kanamycin (50 μg/ml) in case of E. coli AG1 cultures. 150 μl of the cell culture was added to 5ml of melted top agar (0.65 g/l of agar in LB broth) cooled to ~50°C. The mixture was poured on the surface of an LB agar plate supplemented with ampicillin (100 μg/ml) or kanamycin (50 μg/ml). After the agar solidified, 2 μl drops of the solutions of Microcin C and aminoacyl-sulfamoyl-adenylates derivatives (see section Microcin C analogs) were placed on the surface and then dried out. Plates were incubated for 4–6 hours at 37 °C and documented using a scanner. Cultures of B. cereus ATCC4342 and B. cereus ATCC4342 mccF− were grown in 2 ml of LB at 37°C overnight. The mccF− cells were cultured in the presense of erythromycin (5 μg/ml). The overnight cultures were diluted to OD600=0.001 in fresh LB broth (supplemented with 5μg/ml of erythromycin in the case of the mccF-strain) and DSA was added to the mixture to a final concentration of 500μM. The cultures were incubated at 37 °C with vigorous shaking. The cell growth was monitored hourly.
Gene Deletion in B.cereus
The deletion mutant of mccF in B. cereus ATCC4342 was constructed according to the following procedure. A segment of the B. cereus ATCC4342 gene (from 345 to 764 nt) was amplified and inserted into the polylinker of the pHY304 plasmid (designated as pHY304+mccF). The B. cereus ATCC4342 cells were transformed with the pHY304+mccF plasmid by electroporation as described below. Cells were plated on an LB agar plate supplemented with erythromycin (10 μg/ml) and grown at 28 °C until colonies appeared (24–48 hours). Single colonies were replated on a fresh LB agar plate with the same concentration of erythromycin and were grown at 37 °C overnight. Since the pHY304 vector is not maintained in B.cereus at 37 °C, therefore erythromycin resistant clones grown at this temperature insert the plasmid into the chromosome. Colonies were picked and grown in LB in the presence of erythromycin (10 μg/ml) at 37 °C until the culture reached the stationary phase. The deletion of the mccF gene was confirmed by PCR.
Electroporation of B. cereus
3 ml of B. cereus ATCC4342 overnight culture grown on Brain-Heart Infusion (BHI) media (Difco) were added to 150 ml of the fresh media in a 750 ml flask. The culture was grown for 1.5 hours at 28 °C with vigorous shaking. All subsequent manipulations were performed at 4°C. The cells were harvested by centrifugation at 4000 × g for 20 minutes, then washed in 80 ml of buffer (10% glycerin, 1mM HEPES pH 7.0) and resuspended in 2.5 ml of the same buffer. Immediately after the preparation a 400μl aliquot of the cell suspension was mixed with 0.5–5 μg of plasmid DNA and transferred into an ice-cold electroporation cuvette (0.2 mm). A single pulse of 7kVwas delivered (time constant=3.8–4.5 msec). The cells were mixed with 3ml of pre-warmed BHI media and grown at 28 °C for 1.5 hours prior to plating.
Purification and Crystallization
The wild-type and mutants of BaMccF proteins were purified according to a standard Ni-NTA affinity chromatography protocol 32; 33. The harvested cells were resuspended in lysis buffer and lysozyme was added at a final concentration of 1 mg/mL and protease inhibitor cocktail (at concentration of 50 μl/g of wet cells, Roche, Indianapolis, IN). The cell mixtures were incubated on ice for 20 min and then sonicated. The lysates were clarified by centrifugation at 36,000×g for 1 h. The clarified lysates were applied to 5-mL HiTrap Ni-NTA columns (GE Health Systems) connected to an ÄKTAexpress system (GE Health Systems). The His6-tagged proteins were eluted with elution buffer (500 mM NaCl, 5% glycerol, 50 mM HEPES, pH 8.0, 250 mM imidazole, and 10 mM 2-mercaptoethanol), and the fusion tag was removed by treatment with recombinant His6-tagged TEV protease (a gift from Dr. D. Waugh, NCI). Ni-NTA affinity chromatography steps were performed to remove the His6-tag, uncut protein, and His6-tagged TEV protease. The proteins were concentrated using Amicon Ultra concentrators with 10000-MW cutoff (EMD Millipore). Subsequently the proteins were further purified by size-exclusion chromatography on a Superdex-200 column (GE Healthcare) using a standard crystallization buffer containing 200 mM NaCl, 20 mM HEPES pH 8.0, and 2 mM DTT and concentrated to 40 mg/mL. Crystallizations were performed using the sitting-drop vapor-diffusion method at 16 °C using a Mosquito liquid handling robot and the commercial screens in 96 well-plates (Index HT and PEG/Ion HT from Hampton Research and Wizard ™ from Emerald Biosystems). Crystals of apo-BaMccF were grown at 16 °C in sitting drops containing 0.4 μl of precipitant solution (56 μM Dihydogen phosphate, 1.35M di-potassium hydrogen phosphate, pH 8.2) and 0.4μl of protein. In order to obtain BaMccF in the complex with AMP (adenine monophosphate, A1752 Sigma), a batch of the apo-protein (including mutants) was incubated with 20mM AMP for 30 min on ice, prior to crystallization. Crystals of AMP-bound S112A, F177S and S238R BaMccF mutants grew from the conditions containing (0.2 M Potassium Chloride and 20% Peg 3350). The crystals of the apo- and AMP-bound S112A, F177, S238R BaMccF mutants grew within two weeks and belonged to the primitive tetragonal space group P43 with the unit cell parameters a=119 Å, b=119 Å, c=55.5 Å. The orthorhombic crystals (P212121) of the W180A BaMccF mutant grew in 10 days from a solution containing (0.2 M Ammonium fluoride and 20% PEG 3350, pH 6.5, and 20mM ATP). For data collection the mother-liquid containing 20% glycerol was used as a cryoprotectant. Each crystal was picked up with a nylon loop, dragged through the cryoprotectant solution and flash-frozen in liquid nitrogen.
MccF peptidase assay
Purified recombinant EcMccF6, BaMccF32 and its mutant derivatives were incubated with Microcin C, DSA and ESA for 2 hours at 37°C. In the case of Microcin C 50 μl of reaction contained 1μg of protein, 200 pmol of Microcin C, 20mM Tris-HCl pH8.0 and 50mM NaCl. 20 nm of DSA or ESA were incubated in the total volume of 200 μl with 4 μg of protein in the same reaction buffer. After incubation the reactions were frozen, lyophilized and dissolved in 5 μl of deionized water. Aliquots of redissolved solutions were placed on the surface of LB agar plates then overlaid with 5 ml of top agar, which was premixed with 150 μl of the E. coli BL21(DE3) culture grown to a density of OD600 of ~1. After the drops of solutions dried out, the plates were incubated and documented as described above.
Preparation of mutants
The site-directed mutagenesis was performed using the QuikChange™ site-directed mutagenesis protocol (Agilent). The selected amino acids (S112, F177, W180, S238) were mutated to alanines. The pMCSG7 vector34 containing the wild type BaMccF DNA was used as a template. The standard PCR mixture was prepared as follows: 10 ng of the template DNA and 65 ng of each mutagenizing primers were mixed in a 25ul reaction volume.
Multi-site mutagenesis was introduced in a stepwise fashion. The methylated template plasmid was digested with Dpn I, and 2 μl of each reaction was used to transform competent E. coli BL21(DE3) pMAGIC cells. The plasmids were purified from ampicillin-resistant colonies using the QIAprep Spin mini prep kit as described by the manufacturer (Qiagen). DNA sequencing at the University of Chicago Cancer Research DNA Sequencing Facility verified the presences of the desired alanine substitutions. Mutated proteins were purified according to the same protocol as for the wild type protein.
Microcin C analogs
Various chemical analogs of processed microcin C were used in the studies. Aspartyl-sulfamoyl-adenylate (DSA) contains an aspartyl residue with an alpha-carboxy group replaced by a carboxamide moiety. Carboxamide nitrogen forms a sulfamoyl bond with the sulfate group attached to the adenosine 5′-OH. The overall structure of DSA mimics aspartyl-adenylate and processed microcin C. A glutamyl-sulfamoyl-adenylate (ESA), which is identical to DSA except it has a glutamyl side chain instead of aspartyl in the aminoacid residue. Glycyl- (GSA), prolyl- (PSA), phenylalanyl- (FSA) and alanyl- (ASA) sulfamoyl adanylates have structures similar to DSA, the difference is that the glutamyl side chain is replaced by the corresponding amino acid side chains.
Data Collection, Structures Solution and Refinement
Data collections were carried out on the 19-ID beamline of the Structural Biology Center 20 at the Advanced Photon Source. Data to 2.1-Å (apo-BaMccF), 1.75-Å (W180A-BaMccF), 1.5-Å (S112A-BaMccF), 2.04-Å (F177S- BaMccF) and 1.60-Å (S238R-BaMccF) were collected at a wavelength of 0.9794 Å from the single crystals and were processed using the program HKL300035 (Table 1). The structures of the apo-BaMccF were determined by SAD phasing, density modification, and initial protein model building as implemented in the HKL3000software package. The initial models were further processed with the ARP/wARP40 software, and the final models were built with the COOT41 program and refined with the PHENIX42 and REFMAC43 programs. The data collection and refinement statistics are shown in Table 1. The structures of the BaMccF mutants have been determined by molecular replacement using the subunit of BaMccF as a search model. Molecular replacement searches were completed with the MOLREP44 program of the CCP4 suite45. The initial models were subsequently rebuilt manually using the program COOT41 and refined using the program PHENIX42. For all models the final round of refinement was carried out by using translation/libration/screw (TLS) refinement with 15 TLS groups46. Analysis and validation of structures were performed with the aid of MOLPROBITY47 and COOT41 validation tools. The figures were prepared using the PYMOL program.
Protein Data Bank accession code
The atomic coordinates and structure factor files for the structures of the catalytic domain of apo-BaMccF and W180A-BaMccF, and S112A- BaMccF, F177S-BaMccF, and S238R-BaMccF have been deposited in the RCSB Protein Bank with accession code 3GJZ, 3SR3, 3T5M, 3TYX, 3U1B, respectively.
Supplementary Material
Figure 6.

BaMccF active site with AMP. 2Fo–Fc omit map (light blue) contoured at 1s covering the molecule of AMP. Residues involved in catalysis and binding of the AMP molecule are shown as sticks. Distances in Angstroms are shown as dashed lines. All structural figures were prepared using the program PYMOL.
Highlights.
The first crystal structure of MccF protein.
MccF is S66 family serine peptidase, which process McC and it derivatives.
MccF and LdcA folds are very similar despite of only 19% sequence similarity.
MccF clade of bacterial serine peptidases in S66 family identified.
Acknowledgments
We wish to thank all members of the Structural Biology Center at Argonne National Laboratory for their help in conducting these experiments. This project has been funded in whole or in part with Federal funds from the National Institute of Allergy and Infectious Diseases, National Institute of Health, Department of Health and Human Services, under Contract No. HHSN272200700058C. and by the U. S. Department of Energy, Office of Biological and Environmental Research, under contract DE-AC02-06CH11357. AT was partially supported by a fellowship from the Dynasty Foundation. Work in the KS laboratory was supported by a Charles and Johanna Busch Memorial Fund research grant and by a Russian Academy of Sciences Presidium project grant in Bionanotechnology.
NOTE
While this manuscript was being revised a manuscript on MccF from E. coli was published. Agarwal V, Tikhonov A, Metlitskaya A, Severinov K, Nair SK. Structure and function of a serine carboxypeptidase adapted for degradation of the protein synthesis antibiotic microcin C7. Proc Natl Acad Sci U S A. 2012 Mar 20;109(12):4425-30. Epub 2012 Mar 2.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Baquero F, Bouanchaud D, Martinez-Perez MC, Fernandez C. Microcin plasmids: a group of extrachromosomal elements coding for low-molecular-weight antibiotics in Escherichia coli. J Bacteriol. 1978;135:342–7. doi: 10.1128/jb.135.2.342-347.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Severinov K, Semenova E, Kazakov A, Kazakov T, Gelfand MS. Low-molecular-weight post-translationally modified microcins. Mol Microbiol. 2007;65:1380–94. doi: 10.1111/j.1365-2958.2007.05874.x. [DOI] [PubMed] [Google Scholar]
- 3.Destoumieux-Garzon D, Peduzzi J, Rebuffat S. Focus on modified microcins: structural features and mechanisms of action. Biochimie. 2002;84:511–9. doi: 10.1016/s0300-9084(02)01411-6. [DOI] [PubMed] [Google Scholar]
- 4.Guijarro JI, Gonzalez-Pastor JE, Baleux F, San Millan JL, Castilla MA, Rico M, Moreno F, Delepierre M. Chemical structure and translation inhibition studies of the antibiotic microcin C7. J Biol Chem. 1995;270:23520–32. doi: 10.1074/jbc.270.40.23520. [DOI] [PubMed] [Google Scholar]
- 5.Metlitskaya AZ, Katrukha GS, Shashkov AS, Zaitsev DA, Egorov TA, Khmel IA. Structure of microcin C51, a new antibiotic with a broad spectrum of activity. FEBS Lett. 1995;357:235–8. doi: 10.1016/0014-5793(94)01345-2. [DOI] [PubMed] [Google Scholar]
- 6.Tikhonov A, Kazakov T, Semenova E, Serebryakova M, Vondenhoff G, Van Aerschot A, Reader JS, Govorun VM, Severinov K. The mechanism of microcin C resistance provided by the MccF peptidase. J Biol Chem. 2010;285:37944–52. doi: 10.1074/jbc.M110.179135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vondenhoff GH, Blanchaert B, Geboers S, Kazakov T, Datsenko KA, Wanner BL, Rozenski J, Severinov K, Van Aerschot A. Characterization of peptide chain length and constituency requirements for YejABEF-mediated uptake of microcin C analogues. J Bacteriol. 2011;193:3618–23. doi: 10.1128/JB.00172-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nocek B, Mulligan R, Bargassa M, Collart F, Joachimiak A. Crystal structure of aminopeptidase N from human pathogen Neisseria meningitidis. Proteins. 2008;70:273–9. doi: 10.1002/prot.21276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kazakov T, Vondenhoff GH, Datsenko KA, Novikova M, Metlitskaya A, Wanner BL, Severinov K. Escherichia coli peptidase A, B, or N can process translation inhibitor microcin C. J Bacteriol. 2008;190:2607–10. doi: 10.1128/JB.01956-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Van de Vijver P, Vondenhoff GH, Kazakov TS, Semenova E, Kuznedelov K, Metlitskaya A, Van Aerschot A, Severinov K. Synthetic microcin C analogs targeting different aminoacyl-tRNA synthetases. J Bacteriol. 2009;191:6273–80. doi: 10.1128/JB.00829-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Novikova M, Metlitskaya A, Datsenko K, Kazakov T, Kazakov A, Wanner B, Severinov K. The Escherichia coli Yej transporter is required for the uptake of translation inhibitor microcin C. J Bacteriol. 2007;189:8361–5. doi: 10.1128/JB.01028-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Benz G, Schroder T, Kurz J, Wunsche C, Karl W, Steffens G, Pfitzner J, Schmidt D. Constitution of the Deferriform of the Albomycins Delta-1, Delta-2, and Epsilon. Angewandte Chemie-International Edition in English. 1982;21:527–528. [Google Scholar]
- 13.Roberts WP, Tate ME, Kerr A. Agrocin 84 Is a 6-N-Phosphoramidate of an Adenine-Nucleotide Analog. Nature. 1977;265:379–381. doi: 10.1038/265379a0. [DOI] [PubMed] [Google Scholar]
- 14.Tate ME, Murphy PJ, Roberts WP, Kerr A. Adenine N6-Substituent of Agrocin 84 Determines Its Bacteriocin-Like Specificity. Nature. 1979;280:697–699. doi: 10.1038/280697a0. [DOI] [PubMed] [Google Scholar]
- 15.Murphy PJ, Tate ME, Kerr A. Substituents at N6 and C-5′ Control Selective Uptake and Toxicity of the Adenine-Nucleotide Bacteriocin, Agrocin 84, in Agrobacteria. European Journal of Biochemistry. 1981;115:539–543. doi: 10.1111/j.1432-1033.1981.tb06236.x. [DOI] [PubMed] [Google Scholar]
- 16.Gonzalez-Pastor JE, San Millan JL, Castilla MA, Moreno F. Structure and organization of plasmid genes required to produce the translation inhibitor microcin C7. J Bacteriol. 1995;177:7131–40. doi: 10.1128/jb.177.24.7131-7140.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rawlings ND, Barrett AJ, Bateman A. Merops: The Peptidase Database. Nucleic Acids Research. 2010;38:D227–D233. doi: 10.1093/nar/gkp971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Metz R, Henning S, Hammes WP. Ld-Carboxypeptidase Activity in Escherichia-Coli .2. Isolation, Purification and Characterization of the Enzyme from Escherichia-Coli K-12. Archives of Microbiology. 1986;144:181–186. doi: 10.1007/BF00414732. [DOI] [PubMed] [Google Scholar]
- 19.Korza HJ, Bochtler M. Pseudomonas aeruginosa LD-carboxypeptidase, a serine peptidase with a Ser-His-Glu triad and a nucleophilic elbow. J Biol Chem. 2005;280:40802–12. doi: 10.1074/jbc.M506328200. [DOI] [PubMed] [Google Scholar]
- 20.Rosenbaum G, Alkire RW, Evans G, Rotella FJ, Lazarski K, Zhang RG, Ginell SL, Duke N, Naday I, Lazarz J, Molitsky MJ, Keefe L, Gonczy J, Rock L, Sanishvili R, Walsh MA, Westbrook E, Joachimiak A. The Structural Biology Center 19ID undulator beamline: facility specifications and protein crystallographic results. J Synchrotron Radiat. 2006;13:30–45. doi: 10.1107/S0909049505036721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nocek B, Chang C, Li H, Lezondra L, Holzle D, Collart F, Joachimiak A. Crystal structures of delta1-pyrroline-5-carboxylate reductase from human pathogens Neisseria meningitides and Streptococcus pyogenes. J Mol Biol. 2005;354:91–106. doi: 10.1016/j.jmb.2005.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–97. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 23.Pruitt KD, Tatusova T, Klimke W, Maglott DR. NCBI Reference Sequences: current status, policy and new initiatives. Nucleic Acids Res. 2009;37:D32–6. doi: 10.1093/nar/gkn721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Page MJ, Di Cera E. Serine peptidases: classification, structure and function. Cell Mol Life Sci. 2008;65:1220–36. doi: 10.1007/s00018-008-7565-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carter P, Wells JA. Dissecting the catalytic triad of a serine protease. Nature. 1988;332:564–8. doi: 10.1038/332564a0. [DOI] [PubMed] [Google Scholar]
- 26.Kraut J. Serine proteases: structure and mechanism of catalysis. Annu Rev Biochem. 1977;46:331–58. doi: 10.1146/annurev.bi.46.070177.001555. [DOI] [PubMed] [Google Scholar]
- 27.Schrag JD, Li YG, Wu S, Cygler M. Ser-His-Glu triad forms the catalytic site of the lipase from Geotrichum candidum. Nature. 1991;351:761–4. doi: 10.1038/351761a0. [DOI] [PubMed] [Google Scholar]
- 28.Ekici OD, Paetzel M, Dalbey RE. Unconventional serine proteases: variations on the catalytic Ser/His/Asp triad configuration. Protein Sci. 2008;17:2023–37. doi: 10.1110/ps.035436.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–7. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Price MN, Dehal PS, Arkin AP. FastTree 2--approximately maximum-likelihood trees for large alignments. PLoS One. 5:e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim Y, Babnigg G, Jedrzejczak R, Eschenfeldt WH, Li H, Maltseva N, Hatzos-Skintges C, Gu M, Makowska-Grzyska M, Wu R, An H, Chhor G, Joachimiak A. High-throughput protein purification and quality assessment for crystallization. Methods. doi: 10.1016/j.ymeth.2011.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nocek B, Evdokimova E, Proudfoot M, Kudritska M, Grochowski LL, White RH, Savchenko A, Yakunin AF, Edwards A, Joachimiak A. Structure of an amide bond forming F(420):gamma-glutamyl ligase from Archaeoglobus fulgidus -- a member of a new family of non-ribosomal peptide synthases. J Mol Biol. 2007;372:456–69. doi: 10.1016/j.jmb.2007.06.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stols L, Gu M, Dieckman L, Raffen R, Collart FR, Donnelly MI. A new vector for high-throughput, ligation-independent cloning encoding a tobacco etch virus protease cleavage site. Protein Expr Purif. 2002;25:8–15. doi: 10.1006/prep.2001.1603. [DOI] [PubMed] [Google Scholar]
- 35.Minor W, Cymborowski M, Otwinowski Z, Chruszcz M. HKL-3000: the integration of data reduction and structure solution--from diffraction images to an initial model in minutes. Acta Crystallogr D Biol Crystallogr. 2006;62:859–66. doi: 10.1107/S0907444906019949. [DOI] [PubMed] [Google Scholar]
- 36.Sheldrick GM. A short history of SHELX. Acta Crystallogr A. 2008;64:112–22. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 37.Otwinowski Z. Maximum likelyhood refinement of heavy atom parameters. In: Wolf W, Evans PR, Leslie AGW, editors. Isomorphous replacement and Anomalous scattering. Proceedings of the Study Weekend held at Daresbury laboratory. 1991. pp. 80–86. [Google Scholar]
- 38.Terwilliger T. SOLVE and RESOLVE: automated structure solution, density modification and model building. J Synchrotron Radiat. 2004;11:49–52. doi: 10.1107/s0909049503023938. [DOI] [PubMed] [Google Scholar]
- 39.Cowtan K. DM: an automated procedure for phase improvement by density modification. Joint CCP4 and ESFEACBM Newsletter on Protein Crystallogr. 1994:31. [Google Scholar]
- 40.Perrakis A, Morris R, Lamzin VS. Automated protein model building combined with iterative structure refinement. Nat Struct Biol. 1999;6:458–63. doi: 10.1038/8263. [DOI] [PubMed] [Google Scholar]
- 41.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–32. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 42.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Echols N, Headd JJ, Hung LW, Jain S, Kapral GJ, Grosse Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner RD, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. The Phenix software for automated determination of macromolecular structures. Methods. doi: 10.1016/j.ymeth.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–55. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 44.Vagin A, Teplyakov A. Molecular replacement with MOLREP. Acta Crystallogr D Biol Crystallogr. 66:22–5. doi: 10.1107/S0907444909042589. [DOI] [PubMed] [Google Scholar]
- 45.The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–3. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 46.Painter J, Merritt EA. Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr D Biol Crystallogr. 2006;62:439–50. doi: 10.1107/S0907444906005270. [DOI] [PubMed] [Google Scholar]
- 47.Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, 3rd, Snoeyink J, Richardson JS, Richardson DC. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35:W375–83. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.