

Abstract
Purpose
Effective therapy for malignant melanoma, the leading cause of death from skin cancer, remains an area of significant unmet need in oncology. The elevated expression of PKCε in advanced metastatic melanoma results in the increased phosphorylation of the transcription factor ATF2 on threonine 52, which causes its nuclear localization and confers its oncogenic activities. The nuclear-to-mitochondrial translocation of ATF2 following genotoxic stress promotes apoptosis, a function that is largely lost in melanoma cells, due to its confined nuclear localization. Therefore, promoting the nuclear export of ATF2, which sensitizes melanoma cells to apoptosis, represents a novel therapeutic modality.
Experimental Design
We conducted a pilot high-throughput screen of 3,800 compounds to identify small molecules that promote melanoma cell death by inducing the cytoplasmic localization of ATF2. The imaging-based ATF2 translocation assay was performed using UACC903 melanoma cells that stably express doxycycline-inducible GFP-ATF2.
Results
We identified 2 compounds (SBI-0089410 and SBI-0087702) that promoted the cytoplasmic localization of ATF2, reduced cell viability, inhibited colony formation, cell motility, anchorage-free growth, and increased mitochondrial membrane permeability. SBI-0089410 inhibited the TPA-induced membrane tranlocation of PKC isoforms, whereas both compounds decreased ATF2 phosphorylation by PKCε and ATF2 transcriptional activity. Overexpression of either constitutively active PKCε or phosphomimic mutant ATF2T52E attenuated the cellular effects of the compounds.
Conclusion
The imaging-based high-throughput screen provides a proof-of-concept for the identification of small molecules that block the oncogenic addiction to PKCε signaling by promoting ATF2 nuclear export, resulting in mitochondrial membrane leakage and melanoma cell death.
Keywords: ATF2, PKCε, melanoma, nuclear translocation, high content screen
INTRODUCTION
The incidence of melanoma, the most deadly form of skin cancer, has steadily increased for the past 30 years. Our current understanding of the mechanisms underlying the initiation and progression of melanoma has led to the development of specific inhibitors targeting the major signaling pathways that are known to be deregulated in melanoma, including B-Raf, PI3K, and MEK (1-3). Although clinical trials of B-Raf and MEK inhibitors in melanoma have produced promising results, the development of resistance to such monotherapies represents a barrier that has yet to be overcome (4, 5).
ATF2, a member of the basic helix-loop-helix (HLH) family of transcription factors, is activated via phosphorylation by c-Jun N-terminal kinase or p38 in response to stimuli including stress and cytokines (6-8). ATF2 dimerizes with other members of the AP1 superfamily to activate the transcription of genes implicated in stress and DNA damage responses, growth, differentiation, and apoptosis (6-8). The genetic inactivation of ATF2 in melanocytes has been shown to abolish melanoma formation in the mutant N-Ras/Ink4a−/− genetic mouse model (9), indicating an oncogenic role for ATF2 in melanocyte transformation. Conversely, a tumor suppressor function for ATF2 was suggested by the increased incidence of papillomas (10) and mammary tumors (11) following the genetic inactivation of ATF2 in keratinocytes or mammary tissue, respectively. In our effort to understand the mechanisms underlying the opposing activities of ATF2, we discovered that the subcellular localization dictates the oncogenic or tumor suppressor function of ATF2. Whereas its nuclear localization is required for oncogenic activity, ATF2 must be localized to the cytoplasm to perform its tumor suppressor function. Analysis of tissue microarrays (TMAs) revealed that ATF2 exhibits cytosolic localization in basal cell carcinomas (BCC) or squamous cell carcinomas (SCC) (10) but is primarily nuclear in melanoma tumors, consistent with the constitutive transcriptional activity of ATF2 in these tumors (12). Notably, the nuclear localization of ATF2 is associated with poor prognosis in melanoma patients, suggesting that ATF2 localization might serve as a prognostic marker (12, 13).
We recently found that the nuclear localization of ATF2 is dictated by its phosphorylation on threonine 52 (Thr52) by PKCε (14). Loss of Thr52 phosphorylation, as seen in several non-transformed or non-malignant cell lines following exposure to genotoxic stress, is required to enable the nuclear export and translocation of ATF2 to mitochondria, where it reduces mitochondrial membrane potential and promotes apoptosis. Elevated levels of PKCε, found in the more advanced metastatic melanomas, prevent the nuclear-to-mitochondrial translocation of ATF2 that enable its tumor suppressor function. Notably, the expression of peptides derived from ATF2 (amino acids 50–60 or 50–100) prevents the nuclear localization of ATF2 and sensitizes melanoma cells, but not melanocytes, to apoptosis (15-18). These effects were abolished by the mutation of the peptide at the PKCε phosphorylation site (Thr52) (15), suggesting that the native peptide functions by competitively inhibiting PKCε association with/phosphorylation of endogenous ATF2. Taken together, these findings suggest that small molecule modulators of ATF2 localization could attenuate its oncogenic addiction to PKCε signaling, thereby enhancing its pro-apoptotic functions. Because the nuclear-to-cytoplasmic export of ATF2 also sensitizes mutant B-Raf-expressing melanoma cells to apoptosis, agents that promote the nuclear export of ATF2 are expected to represent a new therapeutic modality for drug-resistant melanomas.
MATERIALS AND METHODS
Cell lines and culture conditions
HEK293T and NIH3T3 cells were obtained from American Type Culture Collection (ATCC, Manassas, VA). Melanoma cell lines were kindly provided by Dr. Meenhard Herlyn (Wistar Institute). The melanoma cell lines UACC903 and 501Melwere kindly provided by Drs. Gavin Robertson (Penn State University) and Ruth Halaban (Yale University), respectively. The cells were maintained at 37°C in a humidified 5% CO2 atmosphere and cultured in DMEM supplemented with 10% fetal bovine serum (FBS), 30 U/ml penicillin, 30 μg/ml streptomycin, and 2 mM L-glutamine (Gibco-Life Technologies, Grand Island, NY). The human melanocytes (Hermes 3A) were maintained in 254 medium supplemented with 10% FBS and human melanocyte growth supplement (Gibco-Life Technologies). The Lenti-X Tet-Off Advanced lentiviral inducible expression system (Clontech Laboratories, Mountain View, CA) was used to generate a stable UACC903 melanoma cell line that could induce the expression of GFP-ATF2. For this purpose, we used the Lenti-X Tet-Off Advanced lentiviral inducible expression system which requires the following 2 lentiviral constructs for tetracycline-controlled expression of ATF2: pLVX-Tet-Off Advanced (which is under G418 selection) and pLVX-Tight into which GFP-ATF2 was cloned (and which is under puromycin selection). The UACC903 melanoma cells were co-transduced with the 2 lentiviruses and selected by growth in G418- and puromycin-containing medium. The expression of GFP-ATF2 was repressed by the addition of the tetracycline analog doxycyline to the growth medium. The transfer of the cells into doxycycline-free medium then enabled the controlled expression of GFP-ATF2. Because melanocytes and melanoma cells are inherently resistant to G418 (19), we used FACS to further enrich for GFP-ATF2-positive cells after withdrawal from doxycycline. The UACC903 GFP-ATF2 Tet-Off (hereafter referred to as GFP-ATF2Tet-Off UACC903) cell line was maintained in DMEM supplemented with 10% FBS, 500 ng/ml doxycycline and 5 μg/ml puromycin, plus 1 mg/ml G418 to maintain the selection of stable lines.
Reagents
A 3,800 compound subset of the ChemBridge chemical library (ChemBridge, San Diego, CA) was provided by the Conrad Prebys Center for Chemical Genomics at our Institute. Dimethyl sulfoxide (DMSO), formaldehyde solution, crystal violet, Triton X-100 (TX-100), bovine serum albumin (BSA), and sucrose were purchased from Sigma-Aldrich; the PKC inhibitor Gö6850 was purchased from Calbiochem (Billerica, MA). MitoTracker Deep-Red FM was purchased from Invitrogen (Carlsbad, CA). Protease and phosphatase inhibitors cocktails (PhosSTOP and cOmplete, respectively) were purchased from Roche (Indianapolis, IN). The antibodies employed were purchased as follows: ATF2 (#20F1), Stat3 (#9132), Cox IV (3E11), pATF2-T69/71 (#9225), pan-pPKC T514 (#9379), P38 (#9216), pP38 (#9212), pErk1/2 (#9106), Erk1/2 (#9102), PKC isoform sampler kit (#9960), pAkt (#9271), and Akt (#9272) from Cell Signaling Technologies (Danvers, MA); PKCε (C15), ATF2 (N96 & C19), β-tubulin (G8), p53 (FL393), pan-PKC (H300), and GFP (B2) from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA); pS729 PKCε (44977G) from Invitrogen; HSP60 (24) and β-catenin (14) from BD Pharmingen, (San Diego, CA); Actin (ACTN05) from Thermo-Fisher (Pittsburgh, PA); pT52 ATF2 antibodies were produced by PhosphoSolutions Inc. (Aurora, CO). Secondary antibodies were goat anti-rabbit Alexa-680 F(ab’)2, goat anti-rabbit Alexa-568 F(ab’)2, goat anti-mouse Alexa-488 F(ab’)2 (all Molecular Probes, Eugene, OR), and goat anti-mouse IRDye 800 F(ab’)2 (Rockland Immunochemicals, Gilbertsville, PA).
Immunoblotting
Cell lysates were prepared by lysis in RIPA buffer (150 mM NaCl, 1 mM EDTA, 1% NP40, 0.25% sodium deoxycholate, 0.1% SDS, 50 mM Tris HCl pH 7.4, freshly supplemented with phosphatase and protease inhibitors). Lysates were clarified by centrifugation, and proteins were separated by SDS-PAGE and transferred to PVDF membranes (Millipore, Billerica, MA). After blocking with PBS containing 5% non-fat milk, the membranes were incubated with primary antibodies (pT52-ATF2 (1:500); ATF2 (#20F1; 1:1,000)) in PBS containing 5% BSA and 0.1% Tween-20, at room temperature for 1 h or at 4°C overnight. The membranes were washed and incubated for 1 h at room temperature with goat anti-rabbit Alexa Fluor 680 F(ab’)2 or goat anti-mouse IRDye 800 F(ab’)2 diluted at 1:10,000. The blots were subjected to infrared imaging with 2-color detection using an Odyssey Infrared Imaging System (LiCor Biosciences, Lincoln, NE), and quantification of bands was performed according to the manufacturer’s protocols.
Transfections
Plasmid DNAs were transfected using JetPrime reagent (Polypus Transfection Inc., New York, NY) according to the manufacturer’s protocol.
Immunofluorescence
Cells were cultured on glass coverslips, subjected to the indicated treatments, and fixed with 4% formaldehyde in 100 mM phosphate buffer, pH 7.4, for 30 min. After fixation, cells were permeabilized with PBS containing 0.4% Triton X-100 (TX-100) and 0.4% Bovine Serum Albumin (BSA) for 20 min, and incubated for 1-18 h with primary antibodies in staining buffer (0.1% TX-100/0.1% BSA in PBS). Cells were washed 4-5 times with washing buffer (0.2% TX-100/0.2% BSA in PBS), followed by a 3 h incubation with goat anti-rabbit Alexa-568 and/or anti-mouse Alexa-488 F(ab’)2 in staining buffer, followed by two additional washes in washing buffer. The coverslips were then mounted with Vectashield containing DAPI (Vector Laboratories, CA). Fluorescence images were acquired using either an Olympus IX-71 inverted microscope equipped with a digital ORCA-ER camera (Hamamatsu) or with an Olympus FluoView 1000 confocal microscope equipped with a CH350 CCD camera (Hamamatsu). All images were processed with Adobe Photoshop CS5 (Adobe Systems, Mountain View, CA). Fluorescence images were evaluated and quantified using ImageJ software (NIH, Bethesda, MD).
RESULTS
ATF2 Translocation Assay
We developed a cell-based imaging assay to monitor the subcellular localization of ATF2 by expressing GFP-ATF2 in melanoma cells. For this purpose, UACC903 cells were co-transduced with a tetracycline regulator lentivirus (pLVX Tet-Off Advanced) and a response lentivirus (pLVX-Tight puro) into which the GFP-ATF2 insert (GFP-ATF2Tet-Off UACC903 cells; see Methods and Materials) was cloned. We confirmed using fluorescence microscopy that GFP-ATF2 expression was prevented in cultures maintained in the presence of 500 ng/ml doxycycline but was induced and readily detectable in the nuclei within 36-48 h of cell transfer into doxycycline-free medium (Fig. 1A). We also monitored the expression of GFP-ATF2 by immunoblotting, because excessive GFP-ATF2 expression might interfere with the magnitude or kinetics of ATF2 export from the nuclei. As shown in Figure 1, 24-48 h after withdrawal of doxycycline, GFP-ATF2 was expressed at levels comparable to (within 1- to 5-fold as determined by immunoblotting analysis) the endogenous ATF2 levels (Fig. 1B). Taken together, these results demonstrate that in GFP-ATF2Tet-Off UACC903 melanoma cells, GFP-ATF2 was expressed at levels comparable to endogenous ATF2 and was similarly localized within the nucleus.
Figure 1. The induction of GFP-ATF2 expression in the inducible GFP-ATF2Tet-Off UACC903 stable cell line and its application in an imaging-based HTP screen for inducers of ATF2 translocation.
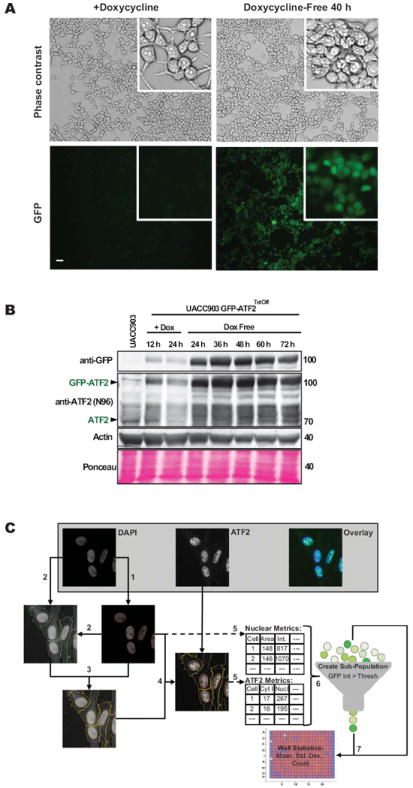
(A) GFP-ATF2 is expressed and readily detectable in GFP-ATF2Tet-Off UACC903 cells following removal of doxycycline from the medium. GFP-ATF2 expression was determined using direct fluorescence microscopy with an Olympus IX71 platform fitted with a 10× air objective lens (NA = 0.31). Scale bar = 20 μm. Insets are further magnified areas of each image. (B) Levels of induced GFP-ATF2 and endogenous ATF2 are comparable (1- to 5-fold) in the GFP-ATF2Tet-Off UACC903 stable cell line. Expression of GFP-ATF2 and endogenous ATF2 was compared in GFP-ATF2Tet-Off UACC903 cells and the parental cell line (negative control). GFP-ATF2Tet-Off UACC903 cells were plated and cultured in the presence or absence of 500 ng/ml doxycycline to inhibit or promote expression of GFP-ATF2, respectively. Cells were cultured for 12-72 h and lysed in RIPA buffer. Lysates were subjected to immunoblot analysis using rabbit anti-ATF2 and mouse anti-GFP antibodies. Actin and Ponceau staining were used for loading control. (C) Imaging-based assay read-out for ATF2 translocation. UACC903 cells stably expressing ATF2-GFP and that were stained with DAPI were imaged with the Opera QEHS system using DAPI and GFP wavelengths (top left and middle respectively, top right shows pseudo-colored overlay). (1) Nuclei detection from DAPI image via thresholding and object splitting (nuclei outlines in red). (2) Cell detection from contrast-stretched DAPI images via thresholding combined with nuclei location information (cell outlines in green). (3) The creation of cytoplasmic regions by subtraction of nuclei regions from the cell regions (cytoplasm outlines in yellow). (4) The overlay of nuclei (red) and cytoplasm (yellow) regions on ATF2 image. (5) Quantify nuclear metrics from nuclei regions of DAPI images and ATF2-based nuclei and cytoplasmic region metrics from ATF2-GFP images on a cell-by-cell level. (6) Create GFP-positive cell sub-population by applying a minimum intensity threshold for whole cell GFP intensity. (7) Calculate well-level assay plate read-outs from cell sub-population statistics of each well. Image analysis was performed using Acapella/Columbus HCS image analysis software.
Identification of Hit Compounds in a High-Throughput Screen
We performed a pilot study to determine whether the GFP-ATF2Tet-Off UACC903 system was amenable for the high-throughput screening of small molecules that promote ATF2 nuclear export. For this purpose, we selected approximately 3,800 compounds from the ChemBridge compound library, containing representatives of different scaffolds and structures. The cells were plated in twelve 384-well plates. Each plate included 32 wells treated with vehicle (0.2% DMSO) as negative controls and 32 wells treated with the PKC inhibitor Gö6850 (5 μM) as a positive control, whereas the remaining wells were treated with the library compounds (single compound, 10 μM per well). To determine if GFP-ATF2 localization was affected by the compounds, we measured the ratio between the mean nuclear intensity and the mean cytoplasmic intensity of GFP fluorescence (Fig. 1C). Whereas in DMSO-treated cells, GFP-ATF2 was mainly localized to the nucleus, in Gö6850-treated cells, GFP-ATF2 was found both in the nucleus and in the cytosol (Fig. 2A, B). The average Z’-value (21) for this screen based on the positive and negative control wells was 0.47 and ranged between 0.4 and 0.56 between plates. As noted in the Materials and Methods section, the assay was designed to minimize influence of the autofluorescence of the positive control compound Gö6850. It is, however, possible that residual Gö6850 compound fluorescence might have artificially increased the reported Z’-value beyond the true assay window. As expected, most compounds were inactive and GFP-ATF2 localization was mostly nuclear, similar to that observed in the DMSO-treated cells. The automated analysis initially identified 20 wells with activity > 50%, but further examination revealed that 14 of the 20 hits were due to autofluorescence or other artifacts, leaving six potential hits (0.16% hit rate). Two of the 6 compounds failed the PAINS cheminformatics filter (for known Promiscuous and Assay-Interfering Nuisance compounds). The remaining 4 compounds were selected for further analysis using independently ordered powders, which were confirmed in a set of dose-dependent assays (Fig. 2C and data not shown).
Figure 2. A pilot screen of a 3,800 compound ChemBridge library to identify compounds that promote the nuclear export of GFP-ATF2.
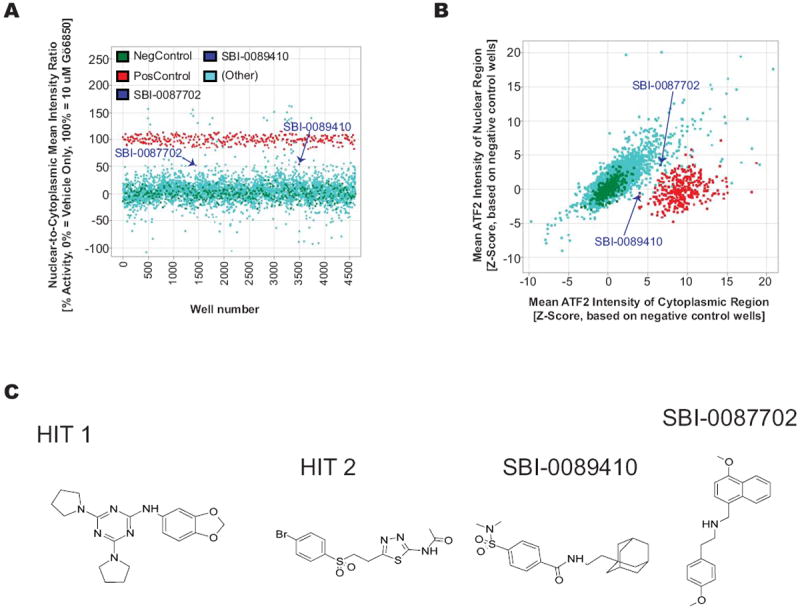
(A) Summary of the results of the complete HTS screen showing the mean ratio of nuclear and cytoplasmic GFP fluorescence intensity for triplicate wells of each compound. (B) Nuclear and cytoplasmic intensities were also evaluated separately for each hit to remove compounds that interfere with the fluorescent protein directly or spectrally as indicated by a significant increase (Z-Score > 5) or decrease (Z-Score < 5) in both regions’ intensities simultaneously. Some autofluorescent compounds are outside of the axes range displayed here. The assay was designed away from the autofluroescent wavelength (Gö6850 is strongly autofluorescent in the red channel) by using green secondary antibodies and emission filters that would minimize the autofluorescence of the Go compound. The average Z’ for this screen was 0.47 and ranged between 0.4 and 0.56 in different plates. Cells were imaged from twelve 384-well plates (4,357 valid total wells 3,669 valid compound wells) using the Opera acquisition system and analyzed with Acapella software to identify GFP-positive cells and delineate nuclear and cytoplasmic regions. DMSO was used as the negative control (blue circles) and the PKC inhibitor Gö6850, which promotes nuclear export of ATF2, was used as the positive control (red circles). As expected, most of the compounds were inactive and exhibited ratios similar to the negative controls. (C) The structure of the 4 selected hits.
Validation of Hit Compounds with Endogenous ATF2
To confirm that the 4 hits identified in the GFP-ATF2Tet-Off UACC903 pilot screen are able to promote the cytoplasmic localization of endogenous ATF2, we ordered new batches of the compounds and tested their effects on endogenous ATF2 translocation in WM793 melanoma cell lines by immunofluorescence microscopy (Fig. 3A). The cells were incubated in the presence of the compounds for either 6 or 24 h. The images were analyzed and quantified using ImageJ software, which confirmed that treatment with hit 3 (SBI-0089410; N-[2-(1-adamantyl)ethyl]-4-[(dimethylamino)sulfonyl]benzamide) or hit 4 (SBI-0087702; N-[(4-methoxy-1-naphthyl)methyl]-2-(4-methoxyphenyl)ethanamine), but not the other 2 hits (hits 1 and 2), promoted cytoplasmic/mitochondrial localization of endogenous ATF2 (Fig. 3B). Partial colocalization between GFP-ATF2 and mitochondria (using HSP60 as surrogate marker of mitochondrial localization) was observed (arrowheads Fig. 3A) following treatment with both hits, thereby confirming their ability to promote ATF2 mitochondrial localization. SBI-0089410 and SBI-0087702 represent low molecular weight compounds that do not appear to be promiscuous based on a SciFinder search of each structure. These compounds do not contain undesirable or reactive functional groups and thus represent possible starting points for a hit-to-lead effort, and also illustrate that synthetically tractable hits can be obtained from a screen. Notably, the nuclear-to-cytoplasmic ratio of endogenous ATF2 was reduced to the same degree seen for the GFP-ATF2 (approximately 30-45% in SBI-0089410- or SBI-0087702-treated cells compared to control, DMSO-treated cells (Fig. 3B). Similar results were also observed in UACC903 melanoma cells (Fig. 3B). Interestingly, the effect of SBI-0087702 on ATF2 localization was detected only after incubation for 24 h, whereas SBI-0089410 induced earlier translocation (after 6 h), which was not sustained at later time points, suggesting that each of these compounds affects ATF2 translocation via a distinct mechanism/target(s). Consistent with the immunostaining data, subcellular biochemical fractionation revealed increased ATF2 in the purified VDAC1-containing mitochondrial fractions, as well as in the β-tubulin-containing cytosolic fraction of WM793 cells after 24 h treatment with SBI-0087702. Similar increase of ATF2 in the mitochondrial fraction was observed in cells treated for 6 h with SBI-0089410 (Supplementary Fig. S1A, B).
Figure 3. SBI-0089410 and SBI-0087702 promote the translocation of endogenous ATF2 to the cytoplasm/mitochondria.
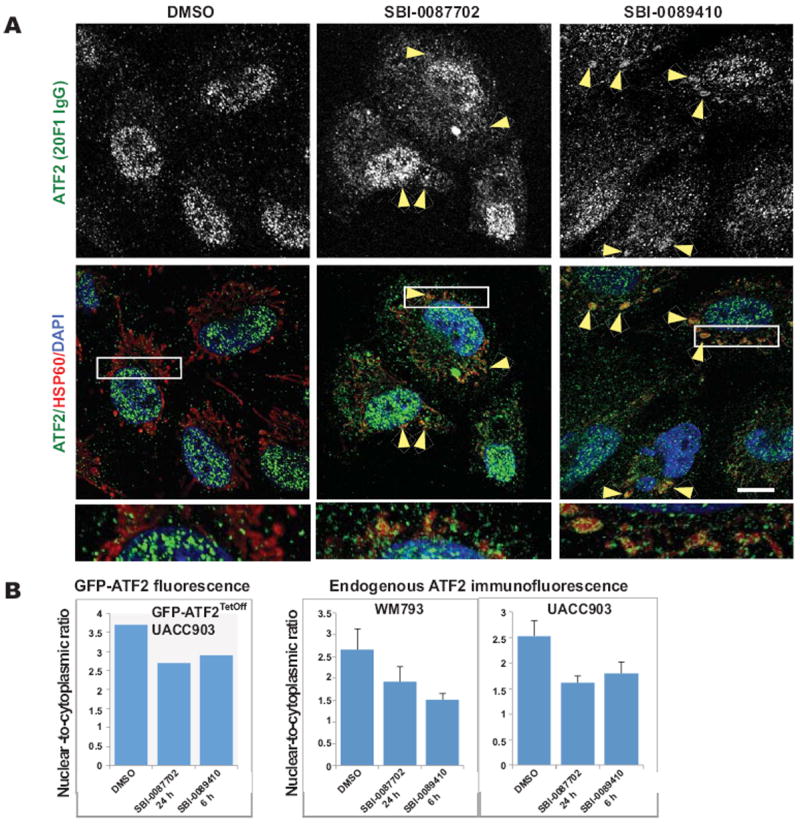
(A) Representative images of WM793 melanoma cells following treatment with DMSO, SBI-0089410, or SBI-0087702, showing increased localization of ATF2 to the cytoplasm and mitochondria (arrowheads). WM793 cells were cultured on glass coverslips and incubated with DMSO (0.2%, 24 h), SBI-0089410 (10 μM, 6 h), or SBI-0087702 (10 μM, 24 h). The cells were then fixed and stained with specific anti-ATF2 antibodies (green channel), anti-HSP60 antibodies to label mitochondria (red channel), and DAPI to label nuclei (blue channel). The cells were imaged using a FluoView1000 confocal microscope with an 100x oil immersion objective. Scale bar = 10 μm. Similar results were obtained with UACC903 cells (data not shown). (B) To measure ATF2 translocation, either the GFP fluorescence intensity of GFP-ATF2-expressing UACC903 (graph on left) or the ATF2 immunofluorescence intensity in WM793 and UACC903 cells (graphs on right) in the nucleus and cytoplasm were quantified using ImageJ software. Between 6 and 17 cells were analyzed for each condition. Quantification reveals a reduction of approximately 40-50% in the nuclear-to-cytoplasmic ATF2 ratio.
Notably, the treatment of UACC903 cells with DMSO, SBI-0089410, SBI-0087702, or Gö6850 did not affect the nuclear compartmentalization of p53, (Supplementary Fig. S1C), a transcription factor that partially localizes at the mitochondria following oxidative stress (22). Furthermore, neither SBI-0089410 nor SBI-0087702 affected the localization of Stat3 and β-catenin, under conditions that induced ATF2 translocation (Supplementary Fig. S2), suggesting that these compounds do not have pleiotropic effect on other transcripiton factors that can be also localized at the mitochondria.
SBI-0089410 and SBI-0087702 Induce Apoptosis and Inhibit Melanoma Cell Growth in an ATF2-Dependent Manner
To determine whether SBI-0089410 or SBI-0087702 affected melanoma cell growth, we measured the colony formation of 501Mel cells. Notably, although both compounds reduced colony formation, SBI-0087702 exhibited the greatest effect, reducing the number of colonies by ~90% (Fig. 4A). The inhibition of colony formation is likely an outcome of the cytotoxicity of these compounds. To verify that the cell death observed following treatment with SBI-0089410 and SBI-0087702 was mediated by ATF2 translocation, we examined whether overexpression of a constitutively nuclear ATF2 mutant (ATFT52E) could rescue cell death. Indeed, ATFT52E-stably transduced UACC903 cells exhibited reduced degrees (up to 50%) of cell death upon treatment by SBI-0089410 or SBI-0087702, compared with control empty vector (EV)-transduced cells (Fig. 4B and 4C). Furthermore, similar to their effect on UACC903 cells (which harbor mutated B-Raf and wild-type N-Ras), SBI-0089410 and SBI-0087702 induced apoptosis in WM1346 melanoma cells (which harbor wild-type B-Raf and mutated N-Ras), which was dependent on ATF2 (Fig. 4B, 4C and Supplementary Fig. S3A-C). Notably, the combined treatment of PLX4032-resistant melanoma cell lines (501Mel & UACC903) with low dose (2 μM) PLX4032 and 10 μM of either of the 2 compounds suppressed their viability and colony-forming ability compared to PLX4032 alone (see data below). Importantly, the two compounds did not induce apoptosis in melanocytes, even when used at a higher concentration (20 μM; Supplementary Fig. S3D), although in human foreskin (BJ) fibroblasts, they induced moderate levels of apoptosis (Supplementary Fig. S3D) relative to the melanoma cell lines. Treatment with SBI-0089410 (6 h) resulted in an increased G2/M phase population, whereas SBI-0087702 (24 h) reduced the G2/M population and increased sub-G1 (dead cell) content (Supplementary Fig. S3E), indicating that both compounds affect melanoma cells by distinct, albeit ATF2-dependent, mechanisms. Interestingly, the ability of ATF2T52E to attenuate the effect of SBI-0087702 was also accompanied by changes in cell morphology; ATF2T52E-expressing cells appeared more rounded and compact than EV-expressing cells (Supplementary Fig. S4A).
Figure 4. SBI-0089410 and SBI-0087702 inhibit colony formation, cell motility, anchorage-independent growth, and promote the apoptosis of melanoma cells in an ATF2-dependent manner.
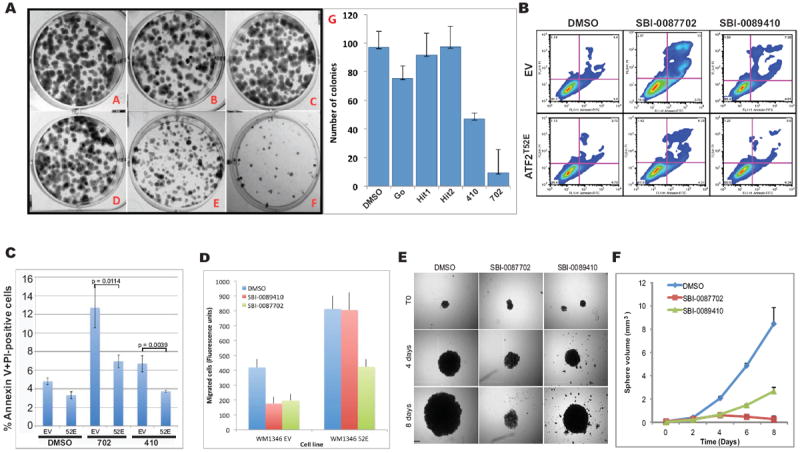
(A) Panels A-F in right image show representative wells containing colonies of 501Mel melanoma cells cultured in the presence of screen hits and controls as follows: (A) DMSO, (B) Gö6850, (C) hit 1, (D) hit 2, (E) SBI-0089410, and (F) SBI-0087702. 501Mel cells were plated at low density (500 cells/well in 6-well plates) and were grown in medium containing the indicated compounds. The number of colonies formed after 7 days in culture was determined by crystal violet staining. SBI-0089410 (410; E) and SBI-0087702 (702; F) were the most potent inhibitors of colony formation compared with Gö6850 (G). Similar results were obtained with the UACC903 cell line (data not shown). (B) Representative FACS profiles for pBabe empty vector (EV) or pBabe ATF2T52E (ATF2T52E)-stably transduced UACC903 cells that were treated with DMSO, 10 μM SBI-0089410 (410) or 10 μM SBI-0087702 (702) for 24 h. The cells were harvested, stained with Annexin V and propidium iodide (PI), and subjected to FACS analysis. N = 10,000 cells per replicate sample. (C) The histogram represents data from 3 independent experiments in (B) (ATF2T52E = 52E). (D) Quantification of the effect of hit compounds on the motility of WM1346 cell lines stably expressing phospho-mimic ATF2 mutant (ATF2 52E) or empty vector (EV). Expression of ATF2 enhanced cell motility by approximately 2-fold (right columns). Both hit compounds inhibited migration of EV cells but expression of ATF2 52E rescued the inhibitory effect. Cell motility was determined in a modified Boyden chamber assay using the Calcein-AM fluorecent staining as described in material and methods. The experiments were performed in triplicate wells and results show averages and standard deviations. (E) SBI-0089410 and SBI-0087702 inhibited the spheroid growth of SW1 melanoma cells. Representative images demonstrating the effect of hit compounds on SW1 spheroids growth. Spheroids were generated by the hanging drop method and subsequently transferred to separate wells and treated with the indicated compounds (10 μM) or vehicle (DMSO 0.08%) every other day for 8 days, in triplicates. Scale bar = 200 μm. (F) Quantification of spheroid growth showing average and standard deviation from 3 independent wells. Phase-contrast images of spheroids were obtained using an Olympus light microscope and spheroid diameter was measure using the SlideBook software. Similar results were obtained with UACC903 cells.
We next examined the effect of these two compounds on migration, a major phenotype of melanoma cells. Both SBI-0089410 and SBI-0087702 inhibited the motility of WM1346 cells across a porous membrane in a Boyden chamber-type assay. Notably, the expression of ATF2T52E in the WM1346 cells effectively reversed the inhibitory effect of SBI-0089410 and to lesser extent the effect of SBI-0087702 (Fig. 4D and Supplementary Fig. S4A-C). These results demonstrate that both compounds promote the apoptosis of melanoma cells and inhibit their migration in an ATF2-dependent manner.
SBI-0089410 and SBI-0087702 Perturb Mitochondrial Membrane Integrity and Promote Leakage
We previously showed that cytoplasmic ATF2 promotes cell death by binding to protein complexes at the mitochondrial membrane, resulting in reduced mitochondrial membrane potential, increased mitochondrial leakage, and subsequent apoptosis (14). Consistent with its effects on ATF2 localization, we found that SBI-0087702 compromised mitochondrial membrane potential and promoted mitochondrial leakage in WM793 and UACC903 cells (Fig. 5A, B and Supplementary Fig. S5, respectively). SBI-0089410 induced a transient decrease in mitochondrial membrane potential at 6 h, which is consistent with its transient effect on ATF2 cytoplasmic localization.
Figure 5. SBI-0089410 and SBI-0087702 promote mitochondrial leakage and reduced viability, which is block by expression of constitutively active PKCε.
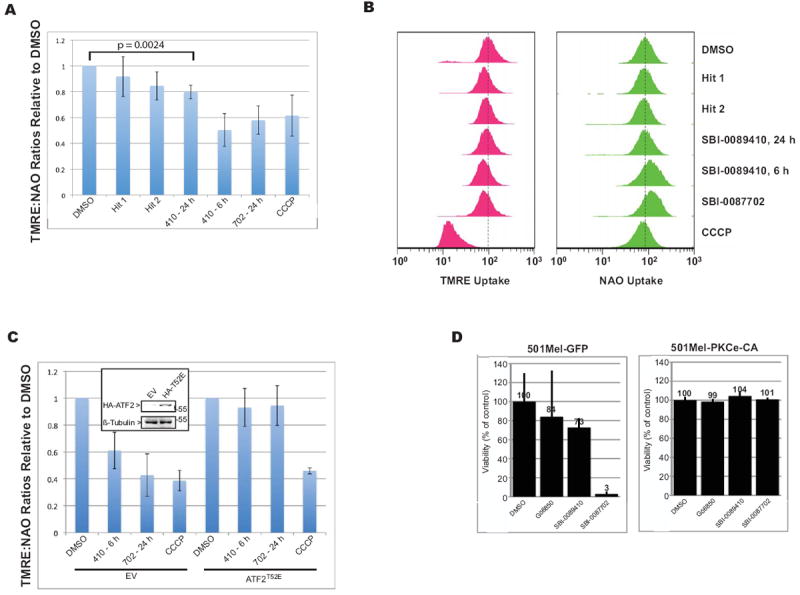
(A) SBI-0089410 (410) and SBI-0087702 (702) induce ATF2-dependent mitochondrial membrane leakage. WM793 cells were incubated DMSO alone, 10 μM hits 1 or 2 for 24 h, 10 μM SBI-0089410 for 6 and 24 h, 10 μM SBI-0087702 for 24 h, or with 50 μM carbonyl cyanide 3-chlorophenylhydrazone (CCCP) for 45 min. Cell were then labeled with TMRE (250 μM) or NAO (10 nM) and analyzed by FACS. Samples were gated on whole cells by forward and side scatter and 10,000 gated cells were analyzed per sample. (B) Red FACS profiles represent TMRE uptake (reflective of mitochondrial membrane potential), whereas the green profiles represent NAO uptake (mitochondrial mass). Leftward peak shifts indicate decreased TMRE or NAO uptake, whereas rightward peak shifts indicate increased TMRE or NaO uptake. The dashed line indicates the median uptake values for the DMSO-treated samples. (C) WM793 cells were transiently transfected with control empty vector (EV) or ATF2T52E (pEF-HA-ATF2T52E) for 48 h. Cells were then treated with DMSO alone, 10 μM SBI-0089410 (410) for 6 h, 10 μM SBI-0087702 (702) for 24 h, or 50 μM CCCP for 45 min. The cells were labeled and analyzed as described for (A). Inset in (C): Western blot analysis showing HA-ATF2 expression. Histograms for (A) and (C) show the mean TMRE/NAO ratio values ± S.D. of 3 independent experiments. (D) Transient expression of a constitutively active form of PKCε (PKCε-CA) confers resistance to SBI-0087702-induced cell death. 501Mel melanoma cells were transiently transfected with either control GFP or PKCε-CA and incubated with the indicated compounds (10 μM final concentration) for 3 days before cell viability was measured using the CellTiter-Blue fluorescence assay (Promega). SBI-0087702-induced cell death was prevented by the expression of PKCε-CA.
To verify that the decrease in mitochondrial membrane potential observed following treatment with SBI-0089410 and SBI-0087702 was mediated by ATF2 translocation, we examined whether the overexpression of ATFT52E (the constitutively nuclear ATF2 mutant) would prevent mitochondrial leakage (Fig. 5C). Indeed, the ectopic expression of ATF2T52E effectively attenuated the loss of mitochondrial membrane potential induced by both SBI-0089410 and SBI-0087702, but not by the mitochondrial uncoupler CCCP, which promotes mitochondrial leakage in an ATF2-independent fashion.
To determine the potential of SBI-0089410 and SBI-0087702 to inhibit tumor growth, we assessed their ability to inhibit 3D, anchorage-free growth using the spheroid growth assay (Fig. 4E, F). We found that after 8 days, SBI-0089410 and SBI-0087702 inhibited spheroid growth by >70% and >90%, respectively. These results suggest that these compounds are stable enough to affect long-term growth and are able to penetrate into 3D structures, characteristics required for effective therapy.
SBI-0089410 and SBI-0087702 Inhibit Melanoma Cell Growth and Thr52 phosphorylation of ATF2 in a PKCε-Dependent Manner
In cell viability assays, treatment with 10 μM SBI-0087702 was sufficient to inhibit the growth of 501Mel cells by >90% (Fig. 5D, left). Significantly, the ectopic expression of a constitutively active form of PKCε (PKCε-CA) prevented the cell death induced by SBI-0087702 treatment, suggesting that this compound mediates its effect on ATF2 translocation and cell viability via inhibition of PKCε (Fig. 5D, right). Because PKCε-mediated phosphorylation of ATF2 on T52 controls its subcellular localization, we next investigated whether SBI-0089410 and SBI-0087702 affect the phosphorylation of endogenous ATF2 at Thr52. Lysates from DMSO-, SBI-0089410-, or SBI-0087702-treated NIH3T3 cells were subjected to immunoblot analysis for altered phosphorylation on T52, as well as on T69/71, which are the sites for JNK/p38 phosphorylation. SBI-0089410, and to a lesser degree SBI-0087702, reduced the level of ATF2 phosphorylation on T52 (SBI-0089410 exhibited comparable effects to that of the PKC inhibitor Gö6850; Fig. 6A). Interestingly, both Gö6850 and SBI-0087702 also attenuated the degree of ATF2 phosphorylation on T69/71, indicative of the effect of these compounds on JNK/p38-mediated phosphorylation of ATF2. Unlike the broad PKC inhibitor Gö6850, which also reduced the total ATF2 expression level, the selected hits were more selective and did not exhibit such effect (Fig. 6A). We further tested whether SBI-0089410 might elicit similar effects on ATF2 phosphorylation in UACC903 cells (Fig. 6B). Indeed, SBI-0089410 inhibited ATF2 phosphorylation on Thr52 but to a lesser degree on Thr 69/71. Additionally, SBI-0089410 inhibited p38 phosphorylation. The inhibitory effect on phosphorylation was transient, peaking at 6 h after incubation with SBI-0089410. This transient effect on ATF2 phosphorylation is consistent with the effect on ATF2 translocation (Fig. 3) and on mitochondrial leakage (Fig. 5 and Supplementary Fig. S5). Notably, SBI-0089410 and SBI-0087702 did not inhibit other key signaling molecules including Erk1/2, Akt, pan-PKC (Supplementary Fig. S6A), suggesting that these compounds do not affect signal transduction pathways that are central in melanomagenesis. Since SBI-0089410 and SBI-0087702 affected ATF2 phosphorylation by PKCε but not the phosphorylation of PKC itself, we set to assess their possible effect on the recruitment of PKC to cellular membranes. Interestingly, SBI-0089410, but not SBI-0087702, inhibited the recruitment of PKCε and other PKC isoforms to the membrane within 3 min of stimulation with TPA (Fig. 6C, D and Supplementary Fig. S6B, C, D, and E). At 10 μM, SBI-0089410 inhibited PKC translocation, although this effect was stronger with increasing dose (10-50 μM) or at lower TPA concentration (Fig. 6C and S6E). Taken together, these results suggest that SBI-0089410 and SBI-0087702 inhibit ATF2 phosphorylation on Thr52, thus preventing ATF2 nuclear translocation, and that the effect of SBI-0089410 is mediated by impairing PKC-to-membrane translocation.
Figure 6. SBI-0089410 and SBI-0087702 inhibit melanoma cell growth, ATF2 transcriptional activity, and affect the expression of ATF2 target genes in a PKCε-dependent manner.
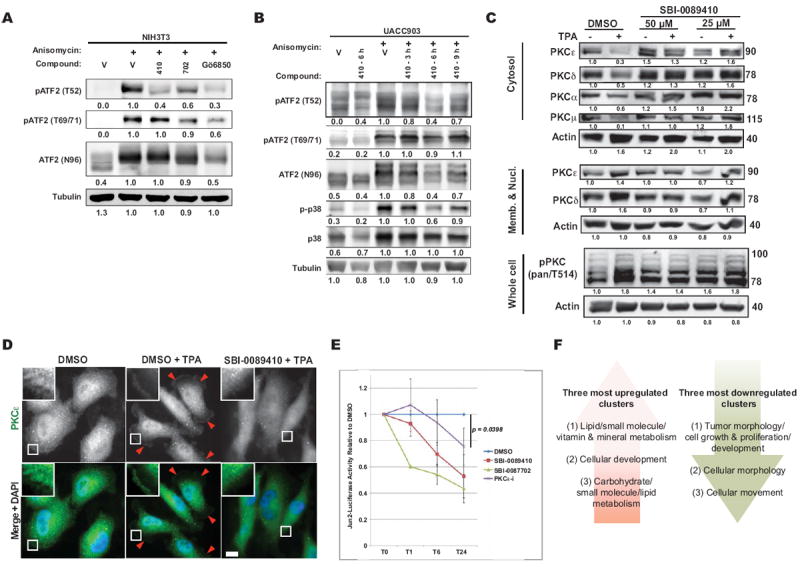
(A) NIH3T3 cells were incubated with DMSO, 10 μM SBI-0089410 (410) for 6 h, 10 μM SBI-0087702 (702) for 24 h, or with 10 μM of the PKCε inhibitor Gö6850. Anisomycin was then added for 30 min to prevent de novo protein synthesis and to activate cellular stress responses. Cells were lysed in RIPA buffer and analyzed by western blotting with the indicated antibodies. Protein bands were quantified using ImageJ software (numbers below). (B) SBI-0089410 (410) transiently inhibited ATF2 and p38 phosphorylation in UACC903 melanoma cells. Cells were pretreated with SBI-0089410 for 3-9 h, before stimulation with anisomycin. SBI-0089410 inhibited ATF2 phosphorylation at Thr52 and p38 phosphorylation with maximum activity after 6 h incubation. (C) SBI-0089410 inhibited the TPA-induced membrane translocation of PKC. WM793 cells were incubated in the presence of SBI-008410 or vehicle (DMSO) for 6 h, then stimulated with 100 nM TPA for 3 min, washed in cold PBS, lysed in lysis buffer, and the membrane and cytosol fractions were isolated according to the protocol detailed in the Material and Methods section. The different fractions were analyzed by Western blot using the antibodies indicated. (D) WM793 cells were treated with compounds and TPA as in (C) and subsequently fixed with 4% formaldehyde prior to processing for immunofluorescence using anti-PKCε primary and FITC-conjugated goat anti-rabbit-IgG secondary antibodies. Imaging was conducted using an Olympus IX-71 fluorescent microscope. Arrowheads point to PKCε staining at the plasma membrane and the insets show enlarged region near the plasma membrane. Scale-bar = 10 μm. (E) Luciferase assay evaluation of the effects of SBI-0089410 and SBI-0087702 on ATF2 transcriptional activity. UACC903 cells were stably transfected with a secreted 3X Jun2-(Gaussia) Luciferase construct. The cells were then transiently transfected with pCMV-Cypridina luciferase (normalization control), followed by treatment with DMSO, 10 μM SBI-0089410, 10 μM SBI-0087702, or with 10 μM PKCε translocation peptide inhibitor (PKCε-i). At the indicated time-points (0, 1, 6, 24 h = T0, T1, T6, T24), Gaussia and Cypridina luciferase activities were measured, and Gaussia luciferase activity was normalized to Cypridina luciferase activity. The graph represents the mean Jun2-luciferase activities ± S.D. relative to DMSO of 3 replicate experiments. (F) IPA analysis of gene expression changes observed upon treatment with SBI-0087702 identifies main clusters that are up- or downregulated. The lists of top 20 genes for each cluster are provided in Supplementary Tables 1 and 2. The original data were deposited into GEO (GSE43135).
Mapping transcriptional programs that are affected by SBI-0087702
To measure the effects of the 2 compounds on ATF2 transcriptional activities, we performed transcription-based reporter assays and a qPCR analysis of known ATF2 transcriptional target genes. Using an ATF2-targeted (Jun2) promoter-luciferase assay system, we evaluated how SBI-0089410 and SBI-0087702 affect the transcriptional activity of ATF2 (Fig. 6E). Consistent with the cytoplasmic/mitochondrial translocation effects of SBI-0089410 and SBI-0087702 on ATF2, these compounds reduced the transcriptional activity of ATF2 in UACC903 cells at 24 h by approximately 47% and 56%, respectively. Notably, the effect of the compound was more pronounced than a PKCε peptide inhibitor, which reduced ATF2 transcriptional activity by approximately 24%. Assessment of the Jun2-luciferase activity in another melanoma cell line, WM793, revealed that SBI-0089410 and SBI-0087702 similarly reduced ATF2 transcriptional activity at 24 h by approximately 30% and 58%, respectively, whereas the PKCε peptide inhibitor reduced ATF2 transcriptional activity by approximately 37% (Supplementary Fig. S6F). Evaluation of the effects of these compounds on the activity of the c-Jun-targeted TRE promoter did not reveal inhibition, but rather slightly increased c-Jun activity (Supplementary Fig. S6G). To further define the specificity of these compounds, we examined the effects of SBI-0089410 and SBI-0087702 on the transcriptional activities of the heat shock response element (HSRE) and androgen receptor (AR), which are representative of independent transcriptional regulatory elements. As shown Supplementary Figure S6H and I, neither of the 2 compounds caused notable changes in the transcriptional activity from these unrelated promoters.
In comparing the effect of the 2 compounds on ATF2 translocation, phosphorylation and sensitization of melanoma cells to apoptosis, SBI-0087702 appeared to be superior. Therefore, we further characterized the transcriptional changes elicited by this compound. To this end, we subjected WM793 cells treated with either DMSO or 10 μM SBI-0087702 for 24 h to microarray-based expression analysis (see details in the Materials and Methods section; data deposited in GEO accession # GSE43135). The top 3 functional networks upregulated in response to SBI-0087702 (Fig. 6F) were as follows: (A) lipid/small molecule/vitamin and mineral metabolism, (B) cellular development, and (C) carbohydrate/small molecule/lipid metabolism. Notably, 2 of the 3 major networks that were upregulated are associated with lipid metabolism, consistent with the increase seen in mitochondrial mass (Supplementary Fig. 5B). The top 3 downregulated clusters were: (A) tumor morphology/cell growth and proliferation/development, (B) cell morphology, and (C) cellular movement. Here, all of the 3 major networks affected by SBI-0087702 are associated with cell morphology and growth, consistent with the effects elicited by this compound on the migratory behavior of the melanoma cells (Fig. 4 and Supplementary Fig. S4). The corresponding top 20 up- and downregulated genes are shown in Supplementary Tables S1 and S2.
DISCUSSION
Oncogene addiction is the phenomenon whereby the survival of cancer cells depends on constitutive oncogenic cues, often mediated by mutated, or otherwise activated oncogenes (23). Our recent studies have pointed to the oncogenic addiction of melanoma to ATF2, which is controlled by PKCε (14). Constitutively active PKCε-mediated phosphorylation of ATF2 impairs its nuclear export following cellular stress and damage, which under normal circumstances would promote the association of ATF2 with the mitochondrial membrane proteins voltage-dependent anion channel 1 and hexokinase 1 (VDAC1 and HK1), resulting in the loss of mitochondrial membrane integrity and the induction of apoptosis (14).
In melanoma, ATF2 is largely confined to the nucleus, predominantly due to its constitutive phosphorylation by PKCε, which confers its oncogenic addiction and attenuates its role in genotoxic stress-induced cell death (14). We therefore sought to establish an imaging-based high-throughput screen to identify small molecules capable of inducing the nuclear export of ATF2 in melanoma cells. We report here on the establishment and the results of such proof-of-concept screen. The GFP-ATF2Tet-Off UACC903 cell line was used to perform the initial screen of 3,800 compounds, from which we identified 2 small molecules that promote cytoplasmic/mitochondrial localization of ATF2 in melanoma cells. The compounds were confirmed to reduce mitochondrial membrane potential and to have concomitant effects on melanoma growth and colony formation. Importantly, the effects of the compounds could be partially blocked by the overexpression of a constitutively active form of PKCε or a phosphomimic ATF2 mutant that is constitutively active transcriptionally. These observations strongly suggest that the compounds mediate their inhibitory effects through PKCε and ATF2.
Interestingly, SBI-0089410 reduced ATF2 phosphorylation on Thr52 by a similar degree as the PKCε inhibitor Gö6850, suggesting that this compound directly affects PKCε or its phosphorylation of ATF2. SBI-0087702 was effective in inhibiting ATF2 Thr52 phosphorylation, albeit to lesser degree compared with SBI-0089410, suggesting that its mechanism of action might be at least partially independent of the PKCε-ATF2 axis. Consistent with this possibility, the duration of the effects of these 2 compounds were different. SBI-0087702 elicited sustained ATF2 translocation, whereas the effect of SBI-0089410 was more rapid but transient, perhaps due to its reduced stability or alternate cellular target. Likely, SBI-0089410, and SBI-087702 elicit their effect on ATF2 and melanoma cells through different pathways. Consistent with the gene expression profiling, SBI-087702 reduces the expression of genes associated with growth and proliferation while increasing mitochondrial lipid biosynthesis, corresponding to the inhibition of melanoma growth and tumorigenicity seen in cultured cells. Arguably, each of the compounds identified in this screen exhibit a partial profile seen for Gö6850, pointing to a more selective effect. Among the 2 selected compounds that were characterized here, SBI-0087702 exhibits the more desired profile with respect to ATF2 translocation, phosphorylation and melanoma sensitization to apopotosis.
Although we do not yet understand the mechanism by which the small molecules might induce ATF2 nuclear export and function at the mitochondria, several possibilities exist. The compounds might inhibit PKCε activity (as appears to be the case for SBI-0089410), or they might activate a protein phosphatase, which would increase T52 dephosphorylation, thereby promoting ATF2 nuclear export. Alternatively, the compounds might modify ATF2 to facilitate its interaction with nuclear export factors. We cannot exclude the possibility that the compounds might elicit more indirect and global effects; for example, by influencing the function of import/export proteins in general. However, we note that the hit validation testing funnel was designed to exclude hits that affect ATF2 localization indirectly (by monitoring the subcellular partitioning of other transcription factors and by the ability to attenuate ATF2 translocation upon expression of ATF2T52E).
Although we validated our observations on GFP-ATF2 by examining endogenous ATF2, it is possible that GFP-tagged ATF2 might not completely phenocopy the dynamics or the action of endogenous ATF2 co-expressed in the same cells. This issue could be circumvented by inhibiting endogenous ATF2 expression and by titrating doxycycline to limit the expression of exogenous GFP-ATF2. In this study, however, our secondary analysis confirmed that the hits indeed affected ATF2 translocation and sensitized the melanoma cells to cell death, which was the ultimate purpose of the screen.
Can small molecules that promote ATF2 nuclear export offer a novel therapeutic modality? Evidence from our previous work suggests an affirmative answer to this important question. We showed that ATF2-derived peptides that affected ATF2 subcellular localization and transcriptional activity also effectively sensitized the melanoma cells to death induced by chemotherapeutic drugs, and inhibited melanoma development and metastasis in human and mouse models (9, 15, 17). Supporting this notion, the treatment of melanoma cells that are resistant to PLX4032 with either SBI-0089410 or SBI-0087702 and low-dose PLX4032 reduced their viability and colony-forming ability relative to PLX4032 alone (Supplementary Fig. S6J and S6K). However, given our current understanding of the melanoma therapeutic landscape, one might consider this as a complementary approach to existing therapies that target different components along the MAPK signaling pathways. Genetic evidence supports the importance of ATF2 in N-Ras melanoma development (9), pointing to the possibility that small molecules identified in these screens could be used to treat N-Ras melanomas, for which there is currently no effective therapy.
Overall, this study provides proof-of-concept for the high-content screening of small molecules to promote the nuclear export of ATF2 in PKCε-addicted melanomas. This approach can be readily adapted to evaluate other transcription factors that elicit opposing functions dependent on their subcellular localization.
Supplementary Material
Translational Relevance.
The identification of new therapeutic modalities is among the most important priorities for improving cancer treatment. Among those is the need to halt oncogene addiction, which underlies tumor development, progression, and the development of resistance. We have recently demonstrated the oncogenic addiction of melanoma to ATF2 through PKCε, which mediates its confined nuclear localization. Here, we have developed a platform to identify small molecules that impair the oncogenic function of ATF2 by permitting its translocation from the nucleus to the mitochondria, thereby enabling its tumor suppressor activities to facilitate melanoma cell death. Our initial screen identified and characterized 2 compounds that sensitize melanoma cells to death, offering a proof-of-concept for the therapeutic paradigm whereby changing the subcellular localization of a protein can limit its oncogenic contributions and promote its tumor suppressor activities.
Acknowledgments
We thank Drs. Meenhard Herlyn, Ruth Halaban, and Gavin Robertson for the melanoma cell lines used in this study. We also thank Dr. Edward Mosonov and the Cell-Imaging Facility for help with confocal microscopy, and Amy Cortez and the Flow Cytometry Facility for help with cell sorting.
Financial Support: Support by NIH grant CA099961 (to ZR) and a Melanoma Research Alliance grant (to ZR). EL supported in part by NIH T32 grant CA121949.
Abbreviations used
- ATF2
activating transcription factor 2
- PKCε
protein kinase C epsilon
- FACS
fluorescence-activated cell sorting
- DMSO
dimethyl sulfoxide
Footnotes
Conflict of interest: There are no conflicts to disclose.
Additional Methods can be found online at CCR web page.
References
- 1.Ko JM, Fisher DE. A new era: melanoma genetics and therapeutics. The Journal of pathology. 2011;223:241–50. doi: 10.1002/path.2804. [DOI] [PubMed] [Google Scholar]
- 2.Shepherd C, Puzanov I, Sosman JA. B-RAF inhibitors: an evolving role in the therapy of malignant melanoma. Current oncology reports. 2010;12:146–52. doi: 10.1007/s11912-010-0095-2. [DOI] [PubMed] [Google Scholar]
- 3.Smalley KS. Understanding melanoma signaling networks as the basis for molecular targeted therapy. The Journal of investigative dermatology. 2010;130:28–37. doi: 10.1038/jid.2009.177. [DOI] [PubMed] [Google Scholar]
- 4.Villanueva J, Vultur A, Lee JT, Somasundaram R, Fukunaga-Kalabis M, Cipolla AK, et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer cell. 2010;18:683–95. doi: 10.1016/j.ccr.2010.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bollag G, Hirth P, Tsai J, Zhang J, Ibrahim PN, Cho H, et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature. 2010;467:596–9. doi: 10.1038/nature09454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lopez-Bergami P, Lau E, Ronai Z. Emerging roles of ATF2 and the dynamic AP1 network in cancer. Nat Rev Cancer. 2010;10:65–76. doi: 10.1038/nrc2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bhoumik A, Lopez-Bergami P, Ronai Z. ATF2 on the double - activating transcription factor and DNA damage response protein. Pigment Cell Res. 2007;20:498–506. doi: 10.1111/j.1600-0749.2007.00414.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lau E, Ronai ZA. ATF2 - at the crossroad of nuclear and cytosolic functions. Journal of cell science. 2012;125:2815–24. doi: 10.1242/jcs.095000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shah M, Bhoumik A, Goel V, Dewing A, Breitwieser W, Kluger H, et al. A role for ATF2 in regulating MITF and melanoma development. PLoS Genet. 2010;6:e1001258. doi: 10.1371/journal.pgen.1001258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bhoumik A, Fichtman B, Derossi C, Breitwieser W, Kluger HM, Davis S, et al. Suppressor role of activating transcription factor 2 (ATF2) in skin cancer. Proc Natl Acad Sci U S A. 2008;105:1674–9. doi: 10.1073/pnas.0706057105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maekawa T, Shinagawa T, Sano Y, Sakuma T, Nomura S, Nagasaki K, et al. Reduced levels of ATF-2 predispose mice to mammary tumors. Mol Cell Biol. 2007;27:1730–44. doi: 10.1128/MCB.01579-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Berger AJ, Kluger HM, Li N, Kielhorn E, Halaban R, Ronai Z, et al. Subcellular localization of activating transcription factor 2 in melanoma specimens predicts patient survival. Cancer research. 2003;63:8103–7. [PubMed] [Google Scholar]
- 13.Gould Rothberg BE, Berger AJ, Molinaro AM, Subtil A, Krauthammer MO, Camp RL, et al. Melanoma prognostic model using tissue microarrays and genetic algorithms. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2009;27:5772–80. doi: 10.1200/JCO.2009.22.8239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lau E, Kluger H, Varsano T, Lee K, Scheffler I, Rimm DL, et al. PKCepsilon promotes oncogenic functions of ATF2 in the nucleus while blocking its apoptotic function at mitochondria. Cell. 2012;148:543–55. doi: 10.1016/j.cell.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bhoumik A, Jones N, Ronai Z. Transcriptional switch by activating transcription factor 2-derived peptide sensitizes melanoma cells to apoptosis and inhibits their tumorigenicity. Proc Natl Acad Sci U S A. 2004;101:4222–7. doi: 10.1073/pnas.0400195101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bhoumik A, Ivanov V, Ronai Z. Activating transcription factor 2-derived peptides alter resistance of human tumor cell lines to ultraviolet irradiation and chemical treatment. Clinical cancer research : an official journal of the American Association for Cancer Research. 2001;7:331–42. [PubMed] [Google Scholar]
- 17.Bhoumik A, Huang TG, Ivanov V, Gangi L, Qiao RF, Woo SL, et al. An ATF2-derived peptide sensitizes melanomas to apoptosis and inhibits their growth and metastasis. J Clin Invest. 2002;110:643–50. doi: 10.1172/JCI16081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bhoumik A, Gangi L, Ronai Z. Inhibition of melanoma growth and metastasis by ATF2-derived peptides. Cancer research. 2004;64:8222–30. doi: 10.1158/0008-5472.CAN-04-0714. [DOI] [PubMed] [Google Scholar]
- 19.Halaban R, Alfano FD. Selective elimination of fibroblasts from cultures of normal human melanocytes. In vitro. 1984;20:447–50. doi: 10.1007/BF02619590. [DOI] [PubMed] [Google Scholar]
- 20.Kelm JM, Timmins NE, Brown CJ, Fussenegger M, Nielsen LK. Method for generation of homogeneous multicellular tumor spheroids applicable to a wide variety of cell types. Biotechnology and bioengineering. 2003;83:173–80. doi: 10.1002/bit.10655. [DOI] [PubMed] [Google Scholar]
- 21.Zhang JH, Chung TD, Oldenburg KR. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. Journal of biomolecular screening. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 22.Vaseva AV, Marchenko ND, Ji K, Tsirka SE, Holzmann S, Moll UM. p53 Opens the Mitochondrial Permeability Transition Pore to Trigger Necrosis. Cell. 2012;149:1536–48. doi: 10.1016/j.cell.2012.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weinstein IB, Joe A. Oncogene addiction. Cancer research. 2008;68:3077–80. doi: 10.1158/0008-5472.CAN-07-3293. discussion 80. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.