

Abstract
Heightened mammalian target of rapamycin complex 1 (mTORC1) activity by genetic deletion of its direct inhibitor, Tsc1, is associated with aberrant development and dysfunction of the female reproductive tract in mice. Here, we compared the phenotypes of mice with conditional deletion of Tsc1 in the female reproductive tract by either progesterone receptor (PR)-Cre (Tsc1PR(d/d)), which inactivates Tsc1 in all major cell types in the uterus (epithelium, stroma and myometrium), or anti-Mullerian hormone type 2 receptor (Amhr2)-Cre (Tsc1Amhr2(d/d)), which inactivates stromal and myometrial Tsc1. Tsc1PR(d/d) and Tsc1Amhr2(d/d) females are infertile resulting from oviductal hyperplasia, retention of embryos in the oviduct and implantation failure. In contrast to the appropriate embryonic development after fertilization seen in Tsc1Amhr2(d/d) females, embryo development was disrupted in Tsc1PR(d/d) females. In addition, uteri in Tsc1PR(d/d) and Tsc1Amhr2(d/d) females showed epithelial hyperplasia but not endometrial cancer. In conclusion, Tsc1PR(d/d) and Tsc1Amhr2(d/d) have overlapping yet distinct phenotypes in the context of compartment-specific deletion of Tsc1.
Keywords: Tsc1, uterus, ovary, oviduct, mouse
Introduction
There is evidence that heightened mammalian target of rapamycin complex 1 (mTORC1) activity induces dysfunction of the female reproductive tract (Fan et al., 2008; Lu et al., 2008; Reddy et al., 2008; Adhikari et al., 2009, 2010; Contreras et al., 2010; Memarzadeh et al., 2010; Hirota et al., 2011; Tanaka et al., 2012; Tanwar et al., 2012). mTOR is a serine/threonine kinase that plays roles in cell proliferation and survival via multiple signaling pathways, and primarily contributes to two complexes which drive separate signaling pathways: mTORC1 with Raptor, mLST8 and Pras40, and mTORC2 with mSIN1, Rictor, mLST8 and Protor1 (Guertin and Sabatini, 2007; Zoncu et al., 2011; Johnson et al., 2013). The activation of mTORC1 by pAkt is suppressed by tuberous sclerosis complex 1 (Tsc1) and Tsc2 by inhibiting ras homolog enriched in brain (Rheb), which is a direct activator of mTORC1 (Kwiatkowski and Manning, 2005; Zoncu et al., 2011). mTORC1 activation leads to phosphorylation of downstream targets, such as p70 S6 kinase leading to increased protein synthesis (Johnson et al., 2013).
Tsc1 or Tsc2 mutations in humans cause tuberous sclerosis, a rare, autosomal-dominant multisystem tumor syndrome embodied in the development of benign tumors in a variety of organs, including the brain, heart and kidneys (Cheadle et al., 2000; Crino et al., 2006; Curatolo et al., 2008). While mice with homozygous deletion of Tsc1 show embryonic lethality, mice missing one allele of Tsc1 (Tsc1+/−) develop renal cell carcinomas and benign tumors in several organs by 10 months of age. However, only ∼7% of Tsc1+/− mice show leiomyoma by 18 months in the uterus with no evidence of endometrial cancer (Kobayashi et al., 2001). Considering these observations, mice with conditional deletion of Tsc1 were utilized to examine its function in reproductive organs.
It was previously reported that oocyte-specific Tsc1 deletion in mice prematurely activates the entire pool of primordial follicles enhancing follicular depletion in early adult life due to heightened mTORC1 signaling (Adhikari et al., 2010). Furthermore, when Tsc1 was deleted in ovarian granulosa cells (GCs) using anti-Mullerian hormone type 2 receptor (Amhr2)-Cre (Tsc1loxP/loxP/Amhr2cre/+=Tsc1Amhr2(d/d)), significantly fewer primordial follicles were found in deleted mice, suggesting premature ovarian insufficiency (Tanaka et al., 2012). After normal and superovulation, these mice also show compromised oocyte quality. Amhr2-Cre deletes Tsc1 in oviductal stroma and myometrium, inducing oviductal epithelial hyperplasia and blocking oviductal transport of fertilized embryos into the uterus (Tanaka et al., 2012). Collectively, these results indicate that Tsc1 has critical roles in preserving the ovarian reserve of oocytes in addition to maintaining the normal architecture of the oviduct for appropriate transport of preimplantation embryos to the uterus.
mTORC1 activity is also associated with endometrial cancer as a downstream target of Pten (phosphatase and tensin homolog) (Guertin and Sabatini, 2007; Lu et al., 2008; Zoncu et al., 2011). Pten is a tumor suppressor, inactivation of which increases phosphoinositide 3-kinase activity with enhanced Akt activation (Luo et al., 2003). Alteration of Pten is frequently observed in endometrial cancer, particularly in the epithelial compartment (Di Cristofano and Ellenson, 2007; Saal et al., 2007). Increased levels of activated Akt (phosphorylated Akt; pAkt) stimulate mTORC1 activity. In humans, however, mTORC1 activation is more frequently observed in advanced and high-grade endometrial cancer (Darb-Esfahani et al., 2009). This suggests that heightened mTORC1 signaling mainly affects endometrial cancer progression but not initiation.
Conditional deletion of genes in different uterine or oviductal tissue compartments may cause differential phenotypes. For example, conditional deletion of Pten in all major uterine compartments (epithelium, stroma and myometrium) using PR-Cre results in the development of endometrial cancer (Daikoku et al., 2008), whereas the deletion of Pten in the uterine stroma and myometrium by Amhr2-Cre fails to generate endometrial cancer even at the age of 5 months, but rather transforms myometrial cells to adipocytes (Daikoku et al., 2011). Therefore, we generated conditional deletion of Tsc1 in the female reproductive tract with PR-Cre (Tsc1loxP/loxP/PRcre+/− = Tsc1PR(d/d)) which deletes genes in the corpus luteum and oviductal epithelium, in addition to all major uterine tissue compartments. We found that Tsc1PR(d/d) females are totally infertile due to oviductal hyperplasia and retention of embryos similar to the phenotype found in Tsc1Amhr2(d/d) females. However, unlike Tsc1Amhr2(d/d) females, embryo development after fertilization was disrupted in Tsc1PR(d/d) females, but this was not due to poor oocyte quality. We also found that Tsc1PR(d/d) uteri showed epithelial hyperplasia but not cancer even after 300 days of age, but rather exhibited transformation of the uterine stroma and myometrium to benign tumors with heightened mTORC1 signaling in the epithelium, stroma and myometrium. In addition, many of them exhibited low locomotion, hunched-back posturing and vaginal bleeding by this age. These results provide evidence that heightened mTORC1 signaling alone by conditional deletion of Tsc1 is insufficient to initiate endometrial cancer, but allows formation of benign tumors in the uterus, and a similar phenotype was noted when Tsc1 was deleted in the stroma and myometrium by Amhr2-Cre.
Materials and Methods
Mice. Tsc1loxP/loxP mice (stock number 005680) were obtained from the Jackson Laboratory. PR-Cre mice were obtained from Francesco DeMayo and John Lydon (Baylor College of Medicine), and Amhr2-Cre mice were obtained from Richard Behringer (MD Anderson Cancer Center). Tsc1loxP/loxP/PRcre/+ (Tsc1PR(d/d)) mice and Tsc1loxP/loxP/Amhr2cre/+ (Tsc1Amhr2(d/d)) were created by breeding Tsc1loxP/loxP mice with PR-Cre mice and Amhr2-Cre mice, respectively. For experiments, littermate floxed and gene-deleted mice were utilized within the same set of experiments to minimize the influence of genetic background variability and to ensure the validity of our results. All mice used in this investigation were housed in barrier facilities in the Cincinnati Children's Hospital Medical Center's Animal Care Facility according to National Institutes of Health and institutional guidelines for the use of laboratory animals. All protocols for the present study were reviewed and approved by the Cincinnati Children's Research Foundation's Institutional Animal Care and Use Committee. Mice were provided with autoclaved rodent Lab Diet 5010 (Purina) and UV light-sterilized RO/DI constant circulation water ad libitum.
LacZ staining
LacZ staining was performed as previously described (Daikoku et al., 2011). In brief, tissues were embedded in OCT after fixation in 0.2% paraformaldehyde and infusion in 30% sucrose at 4°C. Frozen sections were stained with 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside overnight at 37°C. The sections were counterstained with eosin.
Immunohistochemistry
Immunohistochemistry was performed as previously described (Daikoku et al., 2006). Tissues were fixed in PROTOCOL Safefix II (Thermo Fisher) and embedded in paraffin. Uterine sections (6 μm) were subjected to immunostaining using antibodies to phosphorylated Rps6 (pS6, Cell signaling), cytokeratin 8 (CK8; Developmental Studies Hybridoma Bank), α-smooth muscle actin (αSMA; Abcam) or Ki67 (Thermo Fisher). After deparaffinization and hydration, the sections were subjected to antigen retrieval by autoclaving in 10 mM sodium citrate solution (pH = 6) for 10 min. A diaminobenzidine kit (Invitrogen) was used to visualize antigens. The sections were counterstained with hematoxylin or eosin.
Western blot analysis
Tissue samples were prepared as previously described (Daikoku et al., 2006). After measuring protein concentrations, supernatants were boiled with SDS–PAGE sample buffer for 5 min. Samples were run on 10% SDS–PAGE gels and transferred onto PVDF membranes. Membranes were blocked with 10% milk or 5% bovine serum albumin in TBST and probed with antibodies to pS6 (Cell Signaling), Rps6 (S6, Cell Signaling), or actin (Santa Cruz) overnight at 4°C. After washing, blots were incubated in peroxidase-conjugated donkey anti-goat or donkey anti-rabbit IgG (Jackson Immuno Research Laboratories, Inc.). All signals were detected using chemiluminescent reagents (GE Healthcare). S6 and actin served as loading controls.
RNA extraction and RT–PCR
Tsc1PR(f/f) (Tsc1loxP/loxP/PR+/+) or Tsc1PR(d/d) uterine and oviductal RNA samples were prepared and analyzed as previously described (Daikoku et al., 2006). In brief, total RNA was extracted with Trizol (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's protocol. After DNase treatment (Ambion, Austin, TX), 1 μg of total RNA was reverse transcribed with Superscript II (Invitrogen). A PCR was performed using primers 5′-GGAGTGAGCTTTGGAAGTGG-3′ and 5′-CTTCTGAGAGACCTGGCTGAG-3′ for Tsc1; 5′-CATTTGGGCCAGCTAAACAT-3′ and 5′-CCCGGCAAAACAGGTAGTTA-3′ for Cre; 5′-GTGGGCCGCCCTAGGCACCAG-3′ and 5′-CTCTTTGATGTCACGCACGATTTC-3′ for Act β.
In situ hybridization
Paraformaldehyde-fixed frozen sections were hybridized with 35S-labeled cRNA probes as previously described (Daikoku et al., 2006).
Analysis of ovulation, fertilization, embryo development, implantation and term pregnancy
Six to eight weeks old littermate Tsc1PR(f/f), Tsc1PR(d/d), Tsc1Amhr2(f/f) (Tsc1loxP/loxP/Amhr2+/+) or Tsc1Amhr2(d/d) females were mated with wild-type fertile males to induce pregnant mice. The day of vaginal plug formation was considered Day 1 of pregnancy. To examine fertilization, mice were sacrificed on Day 2 of pregnancy and oviducts were flushed with saline to recover embryos. To examine implantation, pregnant dams were assessed on Day 5 morning. Implantation sites were visualized by intravenous injection of a Chicago blue dye solution, and the number of implantation sites — demarcated by distinct blue bands—was recorded. For litter size analysis, pregnant females were monitored from Day 17 through Day 21 by observing mice daily for parturition timing and litter size.
In vitro fertilization
In vitro fertilization (IVF) was performed as previously described (Sun et al., 2009). Wild-type or mutant females were superovulated by intraperitoneal injections of 5 IU of eCG (Sigma), followed by injections of 5 IU of hCG (Sigma) 48 h later. Cumulus–oocyte complexes from each female were collected from the oviduct ampulla 13–14 h after hCG injection and placed in 50-μl droplets of human tubal fluid (HTF) medium (Chemicon). Sperms were collected from the cauda of the epididymis from mature WT male mice and placed into 0.2 ml of HTF medium for counting. After 2 h incubation in a humidified 5% CO2 incubator at 37°C to allow capacitation, sperms (~1.2–1.5 × 106 sperm/ml) were then co-incubated with eggs to allow fertilization. After 6 h, eggs were washed and placed in 20-μl droplets of KSOM medium (Chemicon) for incubation. The cleavage rate (two-cell stage) after 24 h was used as an index of fertilization. Formation of blastocysts at 120 h indicated developmental potential of fertilized embryos.
Measurement of serum estradiol and progesterone levels
Mouse blood samples were collected on Day 4 of pregnancy, and serum levels of estradiol-17β (E2) and progesterone (P4) were measured using EIA kits (Cayman).
Statistical analysis
Student's t-test was performed to determine statistical significance between groups. P < 0.05 was considered significant.
Results
Differential recombination of loxP sites in the female reproductive organs by PR-Cre and Amhr2-Cre
Our major objective was to examine whether heightened mTORC1 by deletion of Tsc1 by PR-Cre (Tsc1PR(d/d)) shows differential phenotype compared with Tsc1-deleted mice by Amhr2-Cre (Tsc1Amhr2(d/d)). As previously shown (Daikoku et al., 2011), both PR-Cre and Amhr2-Cre recombined loxP sites in female reproductive organs, however the recombination by each Cre is different depending on the cell type (Fig. 1). Using a Rosa26-LacZ reporter mouse line, we have shown that PR-Cre recombines loxP sites in all major uterine tissue compartments (epithelium, stroma and myometrium) of the uterus, while Amhr2-Cre did so only in the stroma and myometrium. In the ovary, PR-Cre recombined loxP sites primarily in the corpora lutea (CL), while Amhr2-Cre showed recombination in follicular GCs and CL of the ovary. PR-Cre mainly recombined the loxP sites in the epithelium of the oviductal isthmus with some recombination also occurring in the stroma and muscle layer; signals were undetectable in the ampullary epithelium. On the other hand, Amhr2-Cre primarily recombined loxP sites in the stroma and muscle layer, although modest expression of LacZ was noted in epithelia of both the ampulla and isthmus. Taken together, we speculated that a set of distinct and overlapping phenotypes will be observed in Tsc1PR(d/d) and Tsc1Amhr2(d/d) females.
Figure 1.
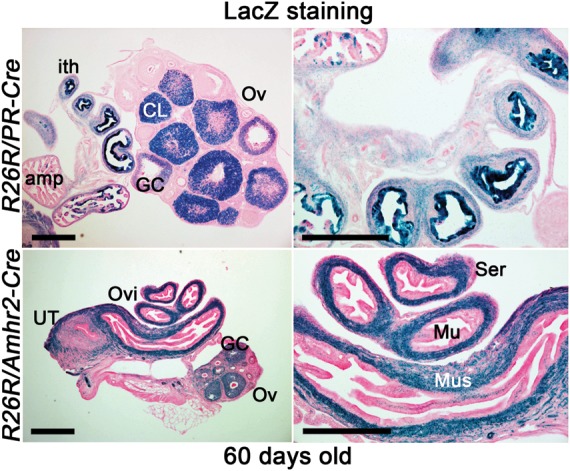
Localization of recombination in PR-Cre and Amhr2-Cre oviducts and ovaries. The conditional gene recombination by Cre recombinase induced by the PR promoter (upper panel) or Amhr2 promoter (lower panel) was visualized by LacZ staining. R26R/PR-Cre females show recombination primarily in corpus luteum (CL), GCs and the isthmus of the oviduct (ith), while R26R/Amhr2-Cre females show recombination in the oviductal muscularis layer (Mus) and ovarian follicular GC. Tissues were harvested at the age of 60 days. Left panels are of lower magnification: left panel bars, 400 µm. Right panels are of higher magnification: bars, 100 µm. Ovi, oviduct; Ov, ovary; amp, oviductal ampulla; Mu, mucosa; Ser, serosa; UT, uterus.
Pregnancy fails in Tsc1PR(d/d) females due to oviductal retention of embryos
To evaluate fertility, we first examined pregnancy outcome in Tsc1PR(d/d) females. Deletion of Tsc1 in Tsc1PR(d/d) oviducts was first confirmed by RT–PCR (Fig. 2A). These dams did not produce any pups (n = 6) compared with Tsc1PR(f/f) littermates (7.3 ± 0.8 pups/litter, n = 14). We then examined the status of implantation on Day 5 of pregnancy in 6- to 8-week-old Tsc1PR(f/f) and Tsc1PR(d/d) littermate females. Tsc1PR(d/d) uteri did not show any implantation sites as examined by the blue dye method and no unfertilized eggs or embryos were recovered from the uterus. Despite the recovery of comparable numbers of fertilized eggs between control and experimental groups on Day 2, morulae and blastocysts along with several degenerating eggs were recovered from the oviducts on Day 5 of pregnancy (Tables I and II). These results indicate that pregnancy failure was due to retention of embryos in Tsc1PR(d/d) oviducts similar to the phenotype seen in Tsc1Amhr2(d/d) females (Tanaka et al., 2012).
Figure 2.
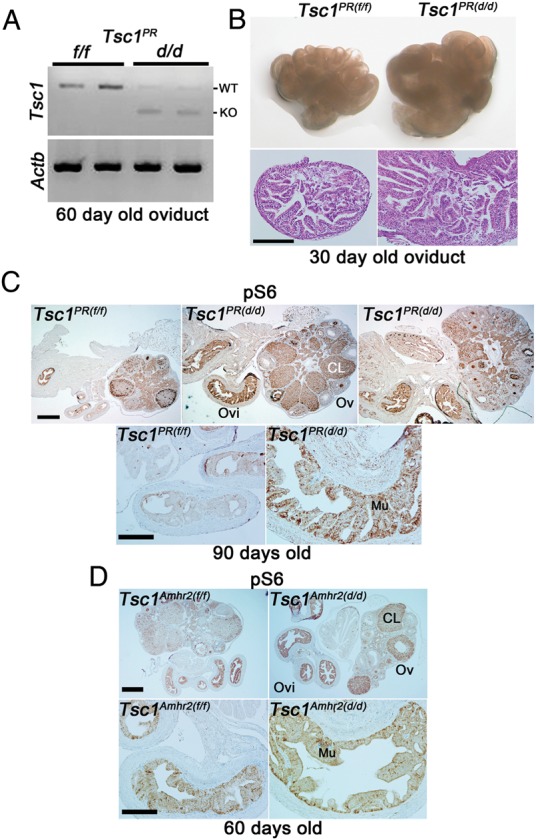
Tsc1PR(d/d) oviducts exhibit higher epithelial hyperplasia and increased mTORC1 signaling. (A) RT–PCR showing inactivation of Tsc1 in Tsc1PR(d/d) oviducts compared with Tsc1PR(f/f) oviducts from mice at 60 days of age. Actb (encoding β-actin) served as a loading control. (B) Morphology and histology of oviduct and ovary of Tsc1PR(f/f) and Tsc1PR(d/d) from mice 6–8 weeks of age showing an increased oviductal size and epithelial hyperplasia in Tsc1PR(d/d) oviducts. Seven Tsc1PR(f/f) and nine Tsc1PR(d/d) females were assessed for oviductal and ovarian morphology, while one Tsc1PR(f/f) and three Tsc1PR(d/d) oviducts were assessed for histology. Bar, 100 µm. (C and D) Immunostaining for pS6 in oviducts and ovaries of Tsc1PR(f/f)(n = 3), Tsc1PR(d/d) (n = 3), Tsc1Amhr2(f/f) (n = 4), or Tsc1Amhr2(d/d) (n = 4) females indicated increased mTORC1 signaling primarily in Tsc1PR(d/d) oviductal mucosa (Mu). Ovaries and oviducts from Tsc1Amhr2(f/f) or Tsc1Amhr2(d/d) females were collected at 60 days (n = 2) or 90 days of age (n = 2). The intensity of staining was similar between these two groups. Top panels show lower magnification: bars, 400 µm; bottom panels show higher magnification: bars, 100 µm. WT; wild-type, KO; knockout, Ovi, oviduct; Ov, ovary; CL, corpus luteum.
Table I.
Normal fertilization in Tsc1-deleted mice. Day 2.
| Genotype | No. of mice examined | Mice with fertilized eggs (%) | Total No. of recovered eggs | No. of degenerated eggsa | Total No. of fertilized eggs (%b) |
|---|---|---|---|---|---|
| Tsc1PR(f/f) | 7 | 7 (100%) | 60 | 13 | 31 (65 ± 16) |
| Tsc1PR(d/d) | 9 | 6 (67%) | 176 | 86 | 51 (49 ± 14) |
| Tsc1Amhr2(f/f) | 8 | 7 (87.5%) | 73 | 0 | 60 (95 ± 3) |
| Tsc1 Amhr2(d/d) | 3 | 3 (100%) | 59 | 27 | 28 (82 ± 12) |
aDegenerated eggs from the last cycle.
b% is shown by mean ± SEM and is not statistically different between the control and experimental groups (P > 0.05).
Table II.
Retention of eggs in the oviduct in Tsc1-deleted mice. Day 5.
| Genotype | No. of mice examined | No. of mice with IS | No. of IS (mean ± SEM) | % of mice with oviductal eggs |
Total No. of eggs from the oviduct |
||
|---|---|---|---|---|---|---|---|
| Eggs | Embryo | Embryo | Degenerated eggs | ||||
| Tsc1PR(f/f) | 6 | 6 (100%) | 8.0 ± 1.5 | 17%a | 17% | 1 | 6 |
| Tsc1PR(d/d) | 7 | 0 (0%) | 0 | 100% | 85.7% | 26b | 71 |
| Tsc1Amhr2(f/f) | 3 | 3 (100%) | 9.0 ± 0.6 | 0% | 0% | 0 | 0 |
| Tsc1 Amhr2(d/d) | 4 | 0 (0%) | 0 | 100% | 100% | 9c | 28 |
IS, implantation sites.
aOne mouse had one IS and seven eggs (six degenerated and one blastocyst) retained in the oviduct.
bThese include 7 blastocysts, 16 morula, one 8-cell and two 2-cell.
cThese include eight blastocysts and one morula.
We next compared the histology of oviducts between Tsc1PR(f/f)and Tsc1PR(d/d) females. Tsc1PR(d/d) oviducts were morphologically larger than those of Tsc1PR(f/f) females (Fig. 2B). Consistent with this observation, we found epithelial hyperplasia in Tsc1PR(d/d) oviducts as early as 30 days of age, similar to the phenotype seen in Tsc1Amhr2(d/d) oviducts (Tanaka et al., 2012). Most wild-type oviducts showed normal histology with no epithelial hyperplasia and timely passage of embryos through the oviduct into the uterus. These results suggest that this hyperplasia is a potential cause of oviductal retention of embryos in both Tsc1PR(d/d) and Tsc1Amhr2(d/d) females.
Heightened mTORC1 signaling after Tsc1 deletion was examined by immunohistochemistry for the phosphorylated form of ribosomal protein Rps6 (pS6), a downstream target of mTORC1 (Fig. 2C). Indeed, intensity of pS6 signals was higher primarily in the mucosa in Tsc1PR(d/d) oviducts; the signal was only slightly higher in the muscular layer. In comparison, deletion of Tsc1 by Amhr2-Cre resulted in a modest increase of pS6 in the oviductal mucosa (Fig. 2D).
The above results led us to examine the ovulation and fertilization status in Tsc1PR(d/d)and Tsc1Amhr2(d/d) females. Cre is primarily expressed in oocytes and surrounding GCs in ovaries of Amhr2-Cre females, whereas it is primarily present in CL in PR-Cre ovaries (Fig. 1). The deletion of Tsc1 in different compartments of the ovary did not affect ovulation in either model. The average number of two-cell embryos was comparable in Tsc1Amhr2(d/d)and Tsc1PR(d/d) females, suggesting that the process of ovulation is not adversely affected in both of these deleted females. In addition, the fertilization rates in these two models were comparable with those of their corresponding wild-type counterparts (Tables I and II). These results suggest that sperms can still travel into oviducts despite oviductal epithelial hyperplasia, and ovulated eggs are capable of fertilization in Tsc1Amhr2(d/d)and Tsc1PR(d/d) oviducts.
On Day 5 of pregnancy, implantation sites can be visualized by distinct blue bands after an intravenous blue dye injection, and the implantation sites are evenly distributed in wild-type uteri (Paria et al., 1993). Interestingly, we could not detect any implantation sites in uteri of Tsc1Amhr2(d/d)and Tsc1PR(d/d) females. Instead, embryos and eggs were recovered from oviducts, indicating persistent oviductal retention of embryos and eggs (Tables I and II). These results suggest that the hyperplasia resulting from deletion of Tsc1 in epithelial cells, along with epithelial shedding into the oviductal lumen, causes defective oviductal embryo transport. Many of the recovered eggs were degenerate, presumably resulting from the previous ovulation; this may provide one possible explanation for recovery of degenerated eggs and embryos from deleted oviducts on Day 2 of pregnancy.
On Day 5 of pregnancy, examination of embryos recovered from oviducts showed compromised embryonic development in both Tsc1Amhr2(d/d) and Tsc1PR(d/d) females. Embryos ranging from two-cell to blastocyst stages were recovered from deleted oviducts with a large number of embryos undergoing degeneration (Tables I and II). The results suggest that compromised embryo development is due to sub-optimal hyperplastic oviductal environment, inherent defects in oocyte competency or both. As shown in Table III, 50% of eggs from Tsc1PR(d/d) females fertilized in vitro developed into blastocysts, comparable with the rate of fertilization in Tsc1PR(f/f) females. These results suggest that the quality and fertilization competency of ovulated eggs from Tsc1PR(d/d) females were apparently normal and that the Tsc1PR(d/d) oviductal environment is not conducive to embryo development. In contrast, most IVF-derived embryos from Tsc1Amhr2(d/d) females did not develop into blastocysts, indicating inherent defects in oocyte quality in these mice.
Table III.
Differential development of IVF-derived embryos from Tsc1PR(d/d) and Tsc1Amhr2(d/d).
| Genotype | No. of donor mice | No. of eggs used for IVF | IVF rate | Development |
|---|---|---|---|---|
| No. of two-cell embryo (%) | No. of blastocysts/total No. of two-cell embryos (%) | |||
| Tsc1PR(f/f) | 7 | 194 | 137 (70.6) | 47/137 (34.3)* |
| Tsc1PR(d/d) | 5 | 159 | 138 (86.8) | 69/138 (50.0)*,*** |
| Tsc1Amhr2(f/f) | 7 | 108 | 73 (67.6) | 39/73 (53.4)** |
| Tsc1 Amhr2(d/d) | 7 | 45 | 35 (77.8) | 2/35 (5.7)**,*** |
*not statistically different (P > 0.05).
**P < 0.02.
***P < 0.02.
Deletion of Tsc1 increases mTORC1 signaling and alters uterine receptivity
Our next objective was to examine whether uterine deletion of Tsc1 alters mTORC1 signaling and whether this alteration corresponds to changes in the expression of genes known to be critical for uterine receptivity since these mice show implantation failure. We first confirmed Cre expression and level of Tsc1 deletion in non-pregnant Tsc1PR(d/d) uteri from 30-day old mice by RT–PCR analysis. As shown in Fig. 3A, uterine Tsc1 mRNA levels are drastically reduced in these mice with low to undetectable levels by 60 days of age. We next examined whether a loss of Tsc1 led to increased pS6 levels by western blotting and immunohistochemistry (Fig. 3B and C). pS6 was indeed upregulated in Tsc1PR(d/d) uteri with an increased intensity of pS6 immunostaining observed in the epithelium and circular muscle with much less signals in the stroma. Unexpectedly, pS6 levels were not uniform in Tsc1PR(d/d) epithelia; a positive signal was primarily restricted to the luminal epithelium with low to undetectable levels in the glandular epithelium.
Figure 3.
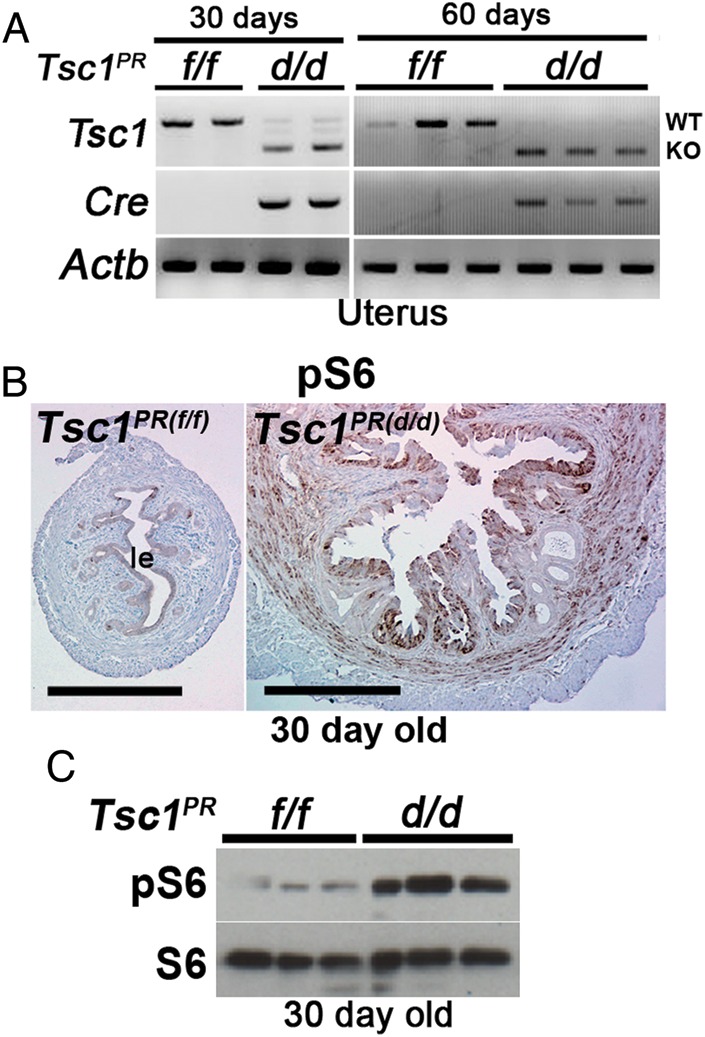
Heightened mTORC1 signaling in Tsc1-deleted uteri. (A) RT–PCR showing inactivation of Tsc1 in Tsc1PR(d/d) uteri compared with Tsc1PR(f/f) uteri from mice at 30 or 60 days of age. Actb served as a loading control. (B) Immunohistochemistry of pS6 shows increased mTORC1 signaling in Tsc1PR(d/d) uteri (n = 4 females) compared with control Tsc1PR(f/f) uteri (n = 4 females) from mice at 30 days of age. Bars, 400 µm. le, luminal epithelium. (C) Western blotting of pS6 indicates increased mTORC1 signaling in Tsc1PR(d/d) uteri from mice at 30 days of age. S6 served as a loading control.
We next examined Tsc1 expression in the pregnant uterus during the peri-implantation period (Fig. 4A and B). Tsc1 was localized in the epithelium and myometrium on Day 1, glandular epithelium on Day 4 and in the stroma on Days 5 and 8 of pregnancy. However, the deletion of uterine Tsc1 in Tsc1PR(d/d) females did not affect estrogen-responsive leukemia inhibitor factor (Lif) expression or progesterone-responsive Hoxa10 and Indian hedgehog (Ihh) expressions on Day 4 of pregnancy (Fig. 4C). We also examined uterine cell proliferation and differentiation status by Ki67 immunostaining on Day 4 when epithelial cells normally become differentiated and stromal cells undergo extensive proliferation in the preparation for implantation and decidualization in wild-type mice. Epithelial cells on Day 4 of pregnancy were still Ki67-positive in some Tsc1PR(d/d) uteri with sparse Ki67-positive cells in the stroma, indicating impaired epithelial differentiation and stromal cell proliferation in some females but not in others (Fig. 4D). Notably, serum progesterone and estradiol-17β levels were not significantly altered between control and deleted females on Day 4 of pregnancy (Fig. 4E and F). These results indicate that Tsc1 deficiency compromised epithelial–mesenchymal interactions required for uterine receptivity for implantation. In fact, a previous report showed failure of implantation after embryo transfer experiments using Tsc1Amhr2(d/d) mice (Tanaka et al., 2012).
Figure 4.

Uterine receptivity is compromised in Tsc1PR(d/d) uteri. (A and B) RT–PCR and in situ hybridization of Tsc1 in wild-type peri-implantation uteri. Tsc1 expression is primarily seen in the epithelium and myometrium (myo) on Day 1, glandular epithelium (ge) on Day 4 and in the stroma (s) on Days 5 and 8 of pregnancy (n = 1 per day of pregnancy). Magnification of Days 1, 4, and 5 of pregnancy: bar, 500 µm; Magnification of Day 8 of pregnancy: bar, 1 mm. Arrowhead denotes the site of blastocyst. Actb served as a loading control. le, luminal epithelium; Dec, decidua. (C) In situ hybridization of Lif, Ihh and Hoxa10 expression Tsc1PR(f/f) (n = 3 females) and Tsc1PR(d/d) (n = 2 females) uteri on Day 4 of pregnancy show comparable expression between the two groups. Upper bar, 250 µm; lower bar, 500 µm. (D) Normal Ki67 staining is seen in Tsc1PR(f/f) females Day 4 (n = 3, left panel), while uteri of two of three Tsc1PR(d/d) females showed compromised epithelial differentiation and stromal proliferation (middle panel); uteri from one Tsc1PR(d/d)mouse showed a relatively normal expression pattern (right panel). Le, luminal epithelium; s, stroma. Bar, 400 µm. (E and F) Serum levels of E2 and P4 on Day 4 of pregnancy (mean ± SEM). Three independent samples were examined in each group.
Uterine deletion of Tsc1 induces epithelial hyperplasia but not cancer
Our next objective was to examine whether uterine deletion of Tsc1 in mice results in endometrial cancer. To address this question, we measured uterine wet weight as an index of uterine growth and compared uterine histology between Tsc1PR(f/f) and Tsc1PR(d/d) females. At 30 days of age, Tsc1PR(d/d) uteri became rigid and weighed considerably more (103.4 ± 26.0 mg, n = 8) compared with Tsc1PR(f/f) uteri (24.8 ± 5.3 mg, n = 8). At this age, Tsc1PR(d/d) uteri showed epithelial hyperplasia (Fig. 5A and Table IV). Although complex hyperplasia was noted in a few cases (2 of 8 females), most uteri showed simple hyperplasia relative to normal epithelial cells. Furthermore, the circular muscle layer appeared thicker with a disrupted architecture. At 90 days of age, the weight of Tsc1PR(d/d) uteri continued to increase (290.3 ± 28.4 mg, n = 8) compared with those of Tsc1PR(f/f) uteri (78.3 ± 6.1 mg, n = 7). At this time, the circular muscle appeared to be more disrupted and the boundary between the stromal and circular muscle compartments was disturbed in Tsc1PR(d/d) uteri (Fig. 5B). Tsc1PR(d/d) glandular epithelia showed simple hyperplasia but no signs of cancer (Fig. 5B and Table IV). Furthermore, the luminal epithelium seemed to be intact. We also observed hyperplasia in Tsc1Amhr2(d/d) uterine glandular epithelium (data not shown).
Figure 5.

Tsc1PR(d/d) uteri show epithelial hyperplasia but not cancer. (A–C) Histology (H&E) and immunostaining of Tsc1PR(f/f) and Tsc1PR(d/d) uteri from mice at 30 days (A), 90 days (B) and more than 270 days of age (C). Bars, 400 µm. CK8 and αSMA are markers for epithelia and myometrium, respectively. Le, luminal epithelium; ge, glandular epithelium; s, stroma; myo, myometrium. The number of females examined per genotype is indicated in Table IV.
Table IV.
Uterine histology of Tsc1-deleted mice.
| Age (days) | Genotype | No. of mice examined | Uterine histology | Mice with indicated histology (%) |
|---|---|---|---|---|
| 30 | Tsc1PR(f/f) | 8 | Normal | 8 (100%) |
| Tsc1PR(d/d) | 8 | Simple hyperplasiaa | 5 (62.5%) | |
| Complex hyperplasia with luminal hyperplasia | 2 (25%) | |||
| Complex hyperplasia | 1 (12.5%) | |||
| 60 | Tsc1PR(f/f) | 7 | Normal | 7 (100%) |
| Tsc1PR(d/d) | 9 | Moderate to marked simple hyperplasia | 9 (100%) | |
| 90 | Tsc1PR(f/f) | 7 | Normal | 6 (85.8%) |
| Mild simple hyperplasia | 1 (14.2%) | |||
| Tsc1PR(d/d) | 8 | Simple hyperplasia | 8 (100%) | |
| Survivalb | Tsc1PR(f/f) | 8 | Normal | 6 (75%) |
| Simple hyperplasia | 2 (25%) | |||
| Tsc1PR(d/d) | 6 | Simple hyperplasia, stromal polyp with stromal overgrowth | 4 (66.6%) | |
| Simple hyperplasia with focal atypia, stromal polyp with stromal overgrowth | 1 (16.7%) | |||
| Marked simple hyperplasia Leiomyosarcoma | 1 (16.7%)c |
aRelative to normal epithelium.
bTsc1PR(f/f), 323–383 days old; Tsc1PR(d/d), 271–308 days old.
cThis uterus possibly has a malignant spindle cell tumor.
Long-term survival studies predicted shorter lifespan of Tsc1 deleted mice. While control mice (Tsc1f/f) survived for longer than 320 days of age, general malaise and vaginal bleeding were apparent in Tsc1PR(d/d) females as early as 270 days and thus, experiments were terminated (Table IV). Tsc1PR(d/d)uteri were grossly enlarged (Tsc1f/f : 0.13 ± 0.02 g; Tsc1PR(d/d) : 12.39 ± 0.49 g (including ovaries due to extensive adhesions), n = 6), showing excessive stromal overgrowth and polyp formation. However, the epithelium still exhibited simple hyperplasia and did not transform into cancer although the degree of hyperplasia increased with age in Tsc1PR(d/d) uteri (Fig. 5C). Notably, only one Tsc1PR(d/d) uterus exhibited leiomyosarcoma, while the others showed only benign tumors (Table IV). These results indicate that Tsc1 deletion is not sufficient to initiate endometrial cancer.
Discussion
There is considerable evidence that mTORC1 signaling plays distinct roles in the female reproductive tract ranging from fertility to cancer. We have previously shown that conditional deletion of uterine Pten initiates endometrial cancer in mice with 100% penetrance (Daikoku et al., 2008). In this study, we have shown that Tsc1 deletion in oviducts by PR-Cre causes retention of embryos in the oviduct and interferes with their development after fertilization. The occurrence of oviductal embryo retention in Tsc1 deleted oviduct by Amhr2-Cre has previously been reported and is confirmed here (Tanaka et al., 2012). Intriguingly, we find that the deletion of oviductal Tsc1 compromises embryo development when trapped in the oviduct, suggesting that the etiology of early pregnancy failure in Tsc1-deleted females is 2-fold: compromised embryo passage through the oviduct and abnormal development within the oviduct. Although the exact nature of the causative factors that influence the quality of the oviductal environment and retention of embryos is not clear, hyperplasia of the epithelium and shedding of these epithelial cells into the lumen could be a potential cause. Further studies using these mouse models are warranted to understand the mechanisms underlying oviductal embryo retention. Phenotypical differences in embryo development in vitro between mice with deletion of Tsc1 by PR-Cre and Amhr2-Cre are not clear at this time, but may result from in vitro conditions that perpetuated some inherent defects in oocytes from Tsc1Amhr2(d/d) mice.
Compromised Tsc activity and increased mTORC1 signaling are associated with endometrial cancer in humans (Mak and Yeung, 2004; Lu et al., 2008; McCampbell et al., 2010). Although the importance of the mTORC1 pathway in endometrial cancer was previously shown in mice using conditional deletion of uterine epithelial Lkb1 which indirectly inhibits the pathway mediated by Tsc1 and Tsc2 complexes, it is still unclear whether heightened mTORC1 in the uterine epithelium itself is sufficient to initiate endometrial cancer since Lkb1 also regulates alternate pathways in addition to the mTORC1 pathway (Katajisto et al., 2007; Contreras et al., 2010). In the present study, we demonstrate that heightened mTORC1 signaling, resulting from deletion of Tsc1, is not sufficient to initiate cancer but can give rise to epithelial hyperplasia, suggesting that heightened mTORC1 activation alone is not sufficient to initiate endometrial cancer but promotes its progression. In fact, mTORC1 activation is more frequently observed in advanced-stage and high-grade endometrial cancer in humans (Darb-Esfahani et al., 2009).
Based on our present findings that mTORC1 activation did not uniformly occur in all uterine epithelia and that activation was not sustained past 90 days of age in Tsc1PR(d/d) females, we believe that the deletion of Tsc1 cannot initiate uterine cancer due to compensatory regulatory mechanisms limiting mTORC1 signaling. Another possibility is that an additional insult must be superimposed upon mTORC1 activation to initiate endometrial cancer. Accordingly, it has been reported that ∼25% of patients with endometrial cancer previously had benign endometrial biopsies (Torres et al., 2012). It would be interesting to examine whether deletion of a second tumor suppressor superimposed on Tsc1 deletion can more readily initiate endometrial cancer. Alternatively, since the uterine epithelium is considered the site of origin for endometrial cancer, it is not yet known whether heightened mTORC1 signaling after uterine epithelial deletion of Tsc1 contributes to the initiation and/or progression of this cancer.
Uterine deletion of Tsc1 also increased mTORC1 signaling in the myometrium and stroma and transformed these tissue compartments into large yet benign tumors. Another interesting observation is that mesenchymal Tsc1 deletion with Amhr2-Cre caused uterine epithelial hyperplasia, although at present, we do not understand the epithelial–mesenchymal interactions impacting this epithelial transformation. These results suggest that the effects of Tsc1 deletion on mTORC1 activation vary depending on the cell type. Although the mechanisms governing these interactions remain unknown, further studies which add to our understanding of these disease processes using models with cell-type specific deletions of critical tumor suppressors may help to establish better strategies to treat endometrial cancer and other gynecologic diseases in which mTORC1 signaling is implicated.
Authors' roles
T.D. and S.K.D. designed research; T.D., M.Y., H.X., X.S., J.C. and S.K.D. performed research; L.H.E. analyzed histology; T.D., M.Y., H.X., X.S., J.C., L.H.E. and S.K.D. analyzed data; and T.D., X.S., J.C., and S.K.D. wrote the paper.
Funding
This study was supported in parts by the Ohio Cancer Research Associates (T. Daikoku), and NIH grants RO1HD068524 and PO1CA77839 (S.K.D.). X.S. is supported by a Lalor Foundation Postdoctoral Fellowship, and J.C. is supported by an NIH NRSA Fellowship (F30AG040858) and the University of Cincinnati Medical Scientist Training Program (T32 GM063483).
Conflict of interest
None declared.
Acknowledgements
We thank Serenity Curtis for editing the manuscript, Amanda Bartos for immunohistochemistry and Dustin Sams for genotyping. We are grateful to Richard M. Behringer (MD Anderson Cancer Center, Houston, TX, USA), and Francesco DeMayo and John B. Lydon (Baylor College of Medicine, Houston, TX, USA) for providing the Amhr2-Cre and PR-Cre mice, respectively.
References
- Adhikari D, Flohr G, Gorre N, Shen Y, Yang H, Lundin E, Lan Z, Gambello MJ, Liu K. Disruption of Tsc2 in oocytes leads to overactivation of the entire pool of primordial follicles. Mol Hum Reprod. 2009;15:765–770. doi: 10.1093/molehr/gap092. doi:10.1093/molehr/gap092. [DOI] [PubMed] [Google Scholar]
- Adhikari D, Zheng W, Shen Y, Gorre N, Hamalainen T, Cooney AJ, Huhtaniemi I, Lan ZJ, Liu K. Tsc/mTORC1 signaling in oocytes governs the quiescence and activation of primordial follicles. Hum Mol Genet. 2010;19:397–410. doi: 10.1093/hmg/ddp483. doi:10.1093/hmg/ddp483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheadle JP, Reeve MP, Sampson JR, Kwiatkowski DJ. Molecular genetic advances in tuberous sclerosis. Hum Genet. 2000;107:97–114. doi: 10.1007/s004390000348. doi:10.1007/s004390000348. [DOI] [PubMed] [Google Scholar]
- Contreras CM, Akbay EA, Gallardo TD, Haynie JM, Sharma S, Tagao O, Bardeesy N, Takahashi M, Settleman J, Wong KK, et al. Lkb1 inactivation is sufficient to drive endometrial cancers that are aggressive yet highly responsive to mTOR inhibitor monotherapy. Dis Model Mech. 2010;3:181–193. doi: 10.1242/dmm.004440. doi:10.1242/dmm.004440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crino PB, Nathanson KL, Henske EP. The tuberous sclerosis complex. N Engl J Med. 2006;355:1345–1356. doi: 10.1056/NEJMra055323. doi:10.1056/NEJMra055323. [DOI] [PubMed] [Google Scholar]
- Curatolo P, Bombardieri R, Jozwiak S. Tuberous sclerosis. Lancet. 2008;372:657–668. doi: 10.1016/S0140-6736(08)61279-9. doi:10.1016/S0140-6736(08)61279-9. [DOI] [PubMed] [Google Scholar]
- Daikoku T, Tranguch S, Trofimova IN, Dinulescu DM, Jacks T, Nikitin AY, Connolly DC, Dey SK. Cyclooxygenase-1 is overexpressed in multiple genetically engineered mouse models of epithelial ovarian cancer. Cancer Res. 2006;66:2527–2531. doi: 10.1158/0008-5472.CAN-05-4063. doi:10.1158/0008-5472.CAN-05-4063. [DOI] [PubMed] [Google Scholar]
- Daikoku T, Hirota Y, Tranguch S, Joshi AR, DeMayo FJ, Lydon JP, Ellenson LH, Dey SK. Conditional loss of uterine Pten unfailingly and rapidly induces endometrial cancer in mice. Cancer Res. 2008;68:5619–5627. doi: 10.1158/0008-5472.CAN-08-1274. doi:10.1158/0008-5472.CAN-08-1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daikoku T, Jackson L, Besnard V, Whitsett J, Ellenson LH, Dey SK. Cell-specific conditional deletion of Pten in the uterus results in differential phenotypes. Gynecol Oncol. 2011;122:424–429. doi: 10.1016/j.ygyno.2011.04.022. doi:10.1016/j.ygyno.2011.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darb-Esfahani S, Faggad A, Noske A, Weichert W, Buckendahl AC, Muller B, Budczies J, Roske A, Dietel M, Denkert C. Phospho-mTOR and phospho-4EBP1 in endometrial adenocarcinoma: association with stage and grade in vivo and link with response to rapamycin treatment in vitro. J Cancer Res Clin Oncol. 2009;135:933–941. doi: 10.1007/s00432-008-0529-5. doi:10.1007/s00432-008-0529-5. [DOI] [PubMed] [Google Scholar]
- Di Cristofano A, Ellenson LH. Endometrial carcinoma. Annu Rev Pathol. 2007;2:57–85. doi: 10.1146/annurev.pathol.2.010506.091905. doi:10.1146/annurev.pathol.2.010506.091905. [DOI] [PubMed] [Google Scholar]
- Fan HY, Liu Z, Cahill N, Richards JS. Targeted disruption of Pten in ovarian granulosa cells enhances ovulation and extends the life span of luteal cells. J Mol Endocrinol. 2008;22:2128–2140. doi: 10.1210/me.2008-0095. doi:10.1210/me.2008-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer cell. 2007;12:9–22. doi: 10.1016/j.ccr.2007.05.008. doi:10.1016/j.ccr.2007.05.008. [DOI] [PubMed] [Google Scholar]
- Hirota Y, Cha J, Yoshie M, Daikoku T, Dey SK. Heightened uterine mammalian target of rapamycin complex 1 (mTORC1) signaling provokes preterm birth in mice. Proc Natl Acad Sci U S A. 2011;108:18073–8. doi: 10.1073/pnas.1108180108. doi:10.1073/pnas.1108180108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SC, Rabinovitch PS, Kaeberlein M. mTOR is a key modulator of ageing and age-related disease. Nature. 2013;493:338–345. doi: 10.1038/nature11861. doi:10.1038/nature11861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katajisto P, Vallenius T, Vaahtomeri K, Ekman N, Udd L, Tiainen M, Makela TP. The LKB1 tumor suppressor kinase in human disease. Biochim Biophys Acta. 2007;1775:63–75. doi: 10.1016/j.bbcan.2006.08.003. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Minowa O, Sugitani Y, Takai S, Mitani H, Kobayashi E, Noda T, Hino O. A germ-line Tsc1 mutation causes tumor development and embryonic lethality that are similar, but not identical to, those caused by Tsc2 mutation in mice. Proc Natl Acad Sci USA. 2001;98:8762–8767. doi: 10.1073/pnas.151033798. doi:10.1073/pnas.151033798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski DJ, Manning BD. Tuberous sclerosis: a GAP at the crossroads of multiple signaling pathways. Hum Mol Genet. 2005;14(Spec No. 2):R251–R258. doi: 10.1093/hmg/ddi260. doi:10.1093/hmg/ddi260. [DOI] [PubMed] [Google Scholar]
- Lu KH, Wu W, Dave B, Slomovitz BM, Burke TW, Munsell MF, Broaddus RR, Walker CL. Loss of tuberous sclerosis complex-2 function and activation of mammalian target of rapamycin signaling in endometrial carcinoma. Clin Cancer Res. 2008;14:2543–2550. doi: 10.1158/1078-0432.CCR-07-0321. doi:10.1158/1078-0432.CCR-07-0321. [DOI] [PubMed] [Google Scholar]
- Luo J, Manning BD, Cantley LC. Targeting the PI3K-Akt pathway in human cancer: rationale and promise. Cancer Cell. 2003;4:257–262. doi: 10.1016/s1535-6108(03)00248-4. doi:10.1016/S1535-6108(03)00248-4. [DOI] [PubMed] [Google Scholar]
- Mak BC, Yeung RS. The tuberous sclerosis complex genes in tumor development. Cancer Invest. 2004;22:588–603. doi: 10.1081/cnv-200027144. doi:10.1081/CNV-200027144. [DOI] [PubMed] [Google Scholar]
- McCampbell AS, Broaddus RR, Walker CL. Loss of inhibitory insulin receptor substrate-1 phosphorylation: an early event in endometrial hyperplasia and progression to carcinoma. Cell Cycle. 2010;9:2698–2699. doi: 10.4161/cc.9.14.12618. doi:10.4161/cc.9.14.12618. [DOI] [PubMed] [Google Scholar]
- Memarzadeh S, Zong Y, Janzen DM, Goldstein AS, Cheng D, Kurita T, Schafenacker AM, Huang J, Witte ON. Cell-autonomous activation of the PI3-kinase pathway initiates endometrial cancer from adult uterine epithelium. Proc Natl Acad Sci USA. 2010;107:17298–17303. doi: 10.1073/pnas.1012548107. doi:10.1073/pnas.1012548107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paria BC, Huet-Hudson YM, Dey SK. Blastocyst's state of activity determines the ‘window’ of implantation in the receptive mouse uterus. Proc Natl Acad Sci USA. 1993;90:10159–10162. doi: 10.1073/pnas.90.21.10159. doi:10.1073/pnas.90.21.10159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy P, Liu L, Adhikari D, Jagarlamudi K, Rajareddy S, Shen Y, Du C, Tang W, Hamalainen T, Peng SL, et al. Oocyte-specific deletion of Pten causes premature activation of the primordial follicle pool. Science. 2008;319:611–613. doi: 10.1126/science.1152257. doi:10.1126/science.1152257. [DOI] [PubMed] [Google Scholar]
- Saal LH, Johansson P, Holm K, Gruvberger-Saal SK, She QB, Maurer M, Koujak S, Ferrando AA, Malmstrom P, Memeo L, et al. Poor prognosis in carcinoma is associated with a gene expression signature of aberrant PTEN tumor suppressor pathway activity. Proc Natl Acad Sci USA. 2007;104:7564–7569. doi: 10.1073/pnas.0702507104. doi:10.1073/pnas.0702507104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Wang H, Okabe M, Mackie K, Kingsley PJ, Marnett LJ, Cravatt BF, Dey SK. Genetic loss of Faah compromises male fertility in mice. Biol Reprod. 2009;80:235–242. doi: 10.1095/biolreprod.108.072736. doi:10.1095/biolreprod.108.072736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y, Park JH, Tanwar PS, Kaneko-Tarui T, Mittal S, Lee HJ, Teixeira JM. Deletion of tuberous sclerosis 1 in somatic cells of the murine reproductive tract causes female infertility. Endocrinology. 2012;153:404–416. doi: 10.1210/en.2011-1191. doi:10.1210/en.2011-1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanwar PS, Kaneko-Tarui T, Zhang L, Tanaka Y, Crum CP, Teixeira JM. Stromal liver kinase B1 [STK11] signaling loss induces oviductal adenomas and endometrial cancer by activating mammalian target of rapamycin Complex 1. PLoS Genet. 2012;8:e1002906. doi: 10.1371/journal.pgen.1002906. doi:10.1371/journal.pgen.1002906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres ML, Weaver AL, Kumar S, Uccella S, Famuyide AO, Cliby WA, Dowdy SC, Gostout BS, Mariani A. Risk factors for developing endometrial cancer after benign endometrial sampling. Obstet Gynecol. 2012;120:998–1004. doi: 10.1097/aog.0b013e31826b9fef. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12:21–35. doi: 10.1038/nrm3025. doi:10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]