

Abstract
The epidemic increase of type 2 diabetes and obesity in developed countries cannot be explained by overnutrition, physical inactivity and/or genetic factors alone. Epidemiologic evidence suggests that an adverse intrauterine environment, in particular a shortage or excess of nutrients is associated with increased risks for many complex diseases later in life. An impressive example for the ‘fetal origins of adult disease’ is gestational diabetes mellitus which usually presents in 1% to >10% of third trimester pregnancies. Intrauterine hyperglycemia is not only associated with increased perinatal morbidity and mortality, but also with increased lifelong risks of the exposed offspring for obesity, metabolic, cardiovascular and malignant diseases. Accumulating evidence suggests that fetal overnutrition (and similarly undernutrition) lead to persistent epigenetic changes in developmentally important genes, influencing neuroendocrine functions, energy homeostasis and metabolism. The concept of fetal programming has important implications for reproductive medicine. Because during early development the epigenome is much more vulnerable to environmental cues than later in life, avoiding adverse environmental factors in the periconceptional and intrauterine period may be much more important for the prevention of adult disease than any (i.e. dietetic) measures in infants and adults. A successful pregnancy should not primarily be defined by the outcome at birth but also by the health status in later life.
Keywords: developmental origins hypothesis, fetal overnutrition, fetal programming, gestational diabetes mellitus, metabolic disease
Introduction
The number of pregnancies complicated by gestational diabetes mellitus (GDM) is increasing worldwide. Depending on the diagnostic criteria and ethnic populations, the incidences range from ∼1 to over 10% (Ben-Haroush et al., 2004). In industrialized countries, approximately every 10th pregnancy is affected and without universal screening programs, many of them may remain unrecognized and untreated (Plagemann, 2011). GDM and type 2 diabetes share a common etiology. The increasing food supply to the fetus during late pregnancy creates a diabetogenic situation (moderate peripheral insulin resistance) for the mother. GDM presents in women without pregestational diabetes, when the maternal β-cell function cannot adapt to the increased insulin demand of late pregnancy; usually it disappears again after delivery (Gilmartin et al., 2008). GDM is diagnosed by an elevated fasting plasma glucose and/or a pathological oral glucose tolerance test (American Diabetes Association, 2006; Coustan et al., 2010). Many pregnant women with GDM can control their glucose levels with a diet, consisting of ∼45% carbohydrate, 30–35% fat and up to 20% protein, although some women require insulin therapy. Common risk factors for GDM are maternal obesity and a family history of diabetes (Kim et al., 2007; Gilmartin et al., 2008; Torloni et al., 2009). Several genetic variants, including genes for β-cell function have been associated with GDM (Robitaille and Grant, 2008; Ridderstråle and Groop, 2009). However, in addition to genetics, overnutrition and physical inactivity, other factors, in particular epigenetic programming of the metabolism early in life appear to play a predominant role (Fernández-Morera et al., 2010; Hanson et al., 2011; Nolan et al., 2011; Pinney and Simmons, 2012).
Children born to mothers with GDM have an increased risk for stillbirth, perinatal complications and notably high birthweight (Schwartz, 1990; Hawdon, 2011). In contrast to the well-known teratogenic effects of periconceptional hyperglycemia in mothers with type 1 or 2 diabetes (Greene, 1999; Corrigan et al., 2009), hyperglycemia in mothers with GDM usually develops during the third trimester when organogenesis is largely completed. Therefore, congenital malformations are not significantly increased. Fetal visceromegaly and macrosomia in neonates exposed to GDM may be largely due to the growth-promoting effects of fetal insulin which is produced in response to the high levels of maternal glucose (overstimulation of fetal β-cells). Since Barker et al. (1993) first noted the association between fetal growth (as a surrogate marker of fetal nutrition) and the occurrence of metabolic disorders later in life, a large number of epidemiologic studies demonstrated that the infants of diabetic mothers are more susceptible to complex diseases, including obesity (Desai et al., 2013), type 2 diabetes (Clausen et al., 2009), metabolic and cardiovascular complications (Wright et al., 2009; Moore, 2010), and even cancer (Wu et al., 2012). Discordant human siblings born before and after the development of maternal diabetes (Dabelea et al., 2000) and embryo transfer experiments in rats (Gill-Randall et al., 2004) provided compelling evidence that these later life phenotypic consequences are caused by the diabetic environment in utero. Maternal obesity (with and without GDM) also leads to fetal overnutrition and appears to be a separate (confounding) risk factor for fetal macrosomia and metabolic disorders in childhood (Boney et al., 2005; Armitage et al., 2008; Drake and Reynolds, 2010).
The phenomenon that adverse environmental exposures during early life, especially with regard to nutrition, increase the lifelong risk of many chronic diseases is now referred to as developmental programming or the ‘Barker hypothesis’ (Barker et al., 2002; Gillman, 2005; Gluckman et al., 2009). Interestingly, there is a U-shaped relationship between birthweight, which reflects the uteroplacental supply of nutrients and hormones to the fetus, and disease risk (Harder et al., 2007; Baker et al., 2008; Caughey and Michels, 2009). Transient overnutrition and undernutrition in early stages of life appear to have the same negative and long-lasting impact on setting (and fine-tuning) of the neuroendocrine control systems of the metabolism, leading to a lifelong increased morbidity. The mechanisms underlying this developmental programming of metabolic and cardiovascular diseases are still largely unclear. Epigenetic modifications are generally assumed to mediate gene–environment interactions, leading to persistent changes of gene regulation and metabolic pathways (Fernández-Morera et al., 2010; Hanson et al., 2011). Tissue culture experiments demonstrated that elevated insulin and glucose levels can interfere with the epigenetic programming machinery (Chiang et al., 2009; Wellen et al., 2009).
Epigenetics studies the inheritance of information beyond the DNA sequence. Biochemical changes, i.e. in forms of DNA methylation and histone modifications, control the spatial, temporal and parent-specific highly coordinated gene expression patterns. Each of the >200 cell types of the body is endowed with a specific combination of silenced and expressed genes, which is established during development and differentiation and then stably inherited during cell divisions. The epigenome is the sum of the epigenetic modifications which bring a cellular phenotype into being. The most thoroughly studied epigenetic modification is the methylation of cytosine-phosphatidyl-guanine (CpG) sites. In contrast to non-coding regions of the genome where most CpGs are methylated to prevent retrotransposition activity (Yoder et al., 1997), CpG islands in 5′ cis-regulatory regions of genes are usually unmethylated. Methylation of these CpG islands during development or disease processes is associated with post-translational histone modifications that lead to a locally condensed inactive chromatin structure and gene silencing (Jaenisch and Bird, 2003; Weber et al., 2007). One important hallmark of the epigenome is its enormous plasticity in response to internal (i.e. during differentiation) and environmental factors (Faulk and Dolinoy, 2011; Feil and Fraga, 2012). In this light, epigenetics is by far the most likely mechanism by which the intrauterine environment affects health and disease of the offspring.
Epigenetic effects of maternal nutritional environment on the offspring: evidence from animal models
A number of elegant animal studies have shown that nutritional factors can modify the epigenome of the developing offspring. One of the most impressive examples is the Agouti viable yellow (Avy) mouse model, in which insertion of a transposable IAP element has created a metastable epiallele that can be turned on or off during early development. Depending on the degree of methylation at this IAP element, which is increased and decreased by methyl donor supplements (Waterland and Jirtle, 2003) and endocrine disruptors (Anderson et al., 2012) in the maternal diet, respectively, Avy/a offspring show a pseudoagouti (comparable to wild-type), a mottled (mosaic) or a viable yellow phenotype. The latter is characterized by the yellow coat color and susceptibility to metabolic diseases and cancer.
Neonatal overfeeding in rats was associated with epigenetic changes in the hypothalamic proopiomelanocortin (Pomc) and insulin receptor (Insr) genes that are important for the neurovegetative control of body weight and metabolism. The degree of hypermethylation was directly dependent on the glucose level (Plagemann et al., 2009, 2010). Feeding pregnant rats a protein-restricted diet altered the DNA methylation and expression patterns of the glucocorticoid receptor (Nr3c1) and peroxisome proliferator-activated receptor alpha (Ppara) genes in the liver and heart of the offspring (Lillycrop et al., 2005, 2007; Slater-Jefferies et al., 2011). In a rat model for fetal programming of hypertension, exposure to a low-protein diet and/or maternal glucocorticoid in early pregnancy led to reduced methylation and increased expression of the adrenal angiotensin receptor (At1b) gene (Bogdarina et al., 2010). Treatment of pregnant guinea pigs with glucocorticoids in late gestation induced changes in global DNA methylation and expression of several genes involved in epigenetic regulation in the offspring and subsequent generations (Crudo et al., 2012). However, in general, even when statistical significance was reached, the absolute methylation differences in the above-mentioned studies were low. The offspring of mouse dams experiencing dietary protein restriction showed robust expression changes in several imprinted (i.e. Gnas and Grb10) and non-imprinted (Ppara) genes in the liver, whereas the DNA methylation patterns remained largely unaltered, suggesting that epigenetic mechanisms other than DNA methylation contribute to developmental programming (Ivanova et al., 2012).
Indeed, in a genome-wide chromatin immunoprecipitation survey, it was shown that streptozotocin-induced maternal diabetes and, independently, maternal consumption of a high-fat diet affected the histone H3 and H4 acetylation in chromatin of the mouse embryo. Increased H3K27 acetylation marks were enriched near neural tube defect genes, suggesting that epigenetic changes contribute to the teratogenic effects of maternal diabetes (Salbaum and Kappen, 2012). In cultured cells, hyperglycemia increased histone acetylation via the citrate lyase pathway (Wellen et al., 2009). A conceptually related study in primates (Aagaard-Tillery et al., 2008) also revealed that in utero exposure to a caloric-dense high-fat maternal diet alters fetal chromatin structure (increased hepatic H3 acetylation).
Effects of maternal malnutrition in humans
In humans strong evidence for the long-lasting effects of an adverse early nutritional environment on health and disease comes from a well-studied cohort of men and women who were exposed in utero to the Dutch famine of 1944–1945. Comparison of individuals who had been exposed during early gestation and their unexposed siblings showed subtle blood methylation changes in several imprinted, i.e. the insulin-like growth factor 2 (IGF2), and non-imprinted, i.e. the leptin (LEP), genes (Heijmans et al., 2008; Tobi et al., 2009). The peptide hormone leptin plays an essential role in the regulation of body weight. The prenatally exposed individuals suffered from increased risks for metabolic and cardiovascular diseases, accelerated cognitive ageing and schizophrenia (Susser et al., 1996; Painter et al., 2006; Stein et al., 2007; De Rooij et al., 2010). At least some of the observed epigenetic and phenotypic effects depended on the timing of exposure during gestation (with early gestation being the most vulnerable period) and sex of the exposed individual (Roseboom et al., 2006; Tobi et al., 2009). The observation of an increased neonatal adiposity in the offspring of exposed women suggests possible transgenerational effects of maternal undernutrition (Painter et al., 2008).
During the rainy season in rural Gambia, pregnant women experience deficiencies in several micronutrients and the resultant offspring display a high incidence of low birthweight, and childhood morbidity and mortality. Maternal micronutrient supplementation (with vitamins and minerals) around the time of conception had widespread and sex-specific effects on the epigenome of the offspring. Some of the observed DNA methylation changes were shown to persist into early infancy (Cooper et al., 2012; Khulan et al., 2012). Similar to fetal adaption to maternal malnutrition, neonates with intrauterine growth restriction exhibited specific changes in DNA methylation, in particular the hepatocyte factor 4 alpha (HNF4A) gene (Einstein et al., 2010). Consistent with evidence from animal models, these studies in humans suggest that the nutritional environment in which the human embryo and fetus develops can influence the epigenetic programming of gene activity in later life.
Epigenetic effects of gestational diabetes
Using bisulfite pyrosequencing, we have analyzed the methylation levels of seven imprinted genes involved in pre- and post-natal growth, four genes involved in energy metabolism, one anti-inflammatory gene, one tumor suppressor gene, one pluripotency gene and two repetitive DNA families in umbilical cord blood and placenta of neonates of mothers with dietetically treated or insulin-dependent GDM and controls without GDM (El Hajj et al., 2013). The maternally imprinted mesoderm-specific transcript (MEST) and the non-imprinted glucocorticoid receptor NR3C1 genes showed subtly (in the order of several percentage points) but significantly decreased methylation levels in both GDM groups, compared with controls, in both analyzed tissues. In a conceptually related study, a lower DNA methylation level of the adiponectin (ADIPOQ) gene in fetal placenta tissue was correlated with maternal hyperglycemia (Bouchard et al., 2012).
Mouse knockout experiments have shown that the paternally expressed Mest gene plays a role in growth of the embryo and placenta as well as in development of a ‘social brain’ after birth (Lefebvre et al., 1998). Transgenic Mest overexpression led to an enlargement of adipocytes and fat mass expansion (Kamei et al., 2007). The endogenous Mest gene can be up-regulated by early overnutritional environment (Kozak et al., 2010). Variations of Mest expression in genetically identical mice (before overnutrition) correlated with the development of diet-induced adiposity (Koza et al., 2006). The glucocorticoid receptor NR3C1, which was also susceptible to metabolic programming, is a transcription factor involved in many cellular processes, including proliferation and differentiation. In the rat model, environmentally (i.e. by maternal diet or early care) induced epigenetic changes in Nr3c1 have been associated with long-term effects on metabolism, stress response and behavior (Weaver et al., 2004; Lillycrop et al., 2007). Similarly in humans, prenatal (Oberlander et al., 2008) and childhood experiences (McGowan et al., 2009; Tyrka et al., 2012) of adverse parental behavior were associated with blood and brain NR3C1 methylation changes, increased stress reactivity and risk for adult psychopathology. The circulating hormone ADIPOQ is secreted abundantly by adipocytes and has anti-inflammatory, anti-atherogenic and anti-diabetic (insulin-sensitizing) effects (Pyrzak et al., 2010).
In addition to single copy genes, interspersed ALU repeats also showed a slightly (1% point) lower methylation level in cord blood and placenta of neonates who were exposed to GDM in utero (El Hajj et al., 2013). Transposable ALU elements comprise ∼10% of the human genome and, when inserted into promoters/first exons, can affect the regulation of gene expression (International Human Genome Sequencing Consortium, 2004). This suggests that hyperglycemia during pregnancy affects multiple loci in the fetal epigenome, which is not unexpected considering the manifold long-term phenotypic effects of an adverse fetal environment.
In humans, it is difficult to study the long-term consequences of altered methylation patterns in blood and/or placenta of neonates. We found significantly decreased blood MEST methylation levels in adults with morbid obesity, compared with normal-weight controls (El Hajj et al., 2013). The observation that MEST is specifically up-regulated in fat tissue of obese individuals (Kosaki et al., 2000) supports the view that intrauterine malprogramming of MEST contributes to obesity predisposition throughout life. Godfrey et al. (2011) analyzed the methylation levels of five candidate genes (selected on the basis of animal studies and methylation array data) in umbilical cord tissue of healthy newborns and found an association of perinatal promoter methylation of the retinoid receptor α (RXRA) and endothelial nitric oxide synthetase (NOS3) genes with childhood adiposity at 9 years of age.
Limitations
In general, epigenetic changes due to maternal nutritional exposure are relatively small at the single gene level but appear to be widespread (Tobi et al., 2009; Khulan et al., 2012; Salbaum and Kappen, 2012; El Hajj et al., 2013). Similar to the genetic variants that have been identified by genome-wide association studies (GWASs), the effect sizes of epigenetic markers that have been associated with an adverse periconceptional and prenatal environment may be small. Thus, consistent with the multifactorial threshold model for complex diseases, stochastic and environmentally induced epigenetic variations in many genes and pathways may determine the disease risk later in life. Highly penetrant disease-causing epimutations, i.e. in imprinted genes are very rare (Ivanova et al., 2012; El Hajj and Haaf, 2013) and, evidently, cannot explain the fetal programming of adult disease.
An additional shortcoming is that DNA methylation patterns are cell type and tissue-specific. At least in humans, it is not possible to study the target tissues for metabolic disease, i.e. the hypothalamic–pituitary–adrenal axis, fat, liver, skeletal muscle and/or pancreatic islets. It is generally assumed that the observed epigenetic changes in accessible tissues such as blood or placenta can identify genes susceptible to fetal programming in the target tissues. Even animal studies so far largely ignored that the cell composition of a tissue may vary between groups, i.e. exposed and unexposed individuals.
Because intrauterine exposures are nearly impossible to quantify in time and space and each exposure has the potential to induce numerous changes with small effect sizes, most likely interacting with each other, it is difficult to prove that the epigenetic changes that have been associated with different developmental experiences are directly involved in the pathogenesis of chronic disease later in life and not just a consequence of the pathology. One of the most impressive examples of the phenotypic consequences of aberrant methylation patterns is epigenetic silencing of tumor suppressor genes such as BRCA1 that plays an important role in breast and ovarian cancer development (Esteller et al., 2000; Hansmann et al., 2012). In vitro methylation and transfection assays showed that repression of the BRCA1 promoter directly depends on the number of methylated CpGs (Magdinier et al., 2000). It is usually the density of methylated CpGs rather than individual CpGs that turns a promoter or an imprinting control region off (Weber et al., 2007). Similarly, inactivation of the somatostatin gene in gastric cancers is characterized by an inverse correlation between promoter methylation level and mRNA expression (Jackson et al., 2011). It is plausible to assume that the methylation changes that have been linked to different nutritional exposures also alter gene expression and, thus, phenotypic information.
Conclusions
So far, the main focus of reproductive medicine has been on pregnancy rates and the outcome at birth. The long-term consequences of environmental factors around the time of conception and during pregnancy are largely neglected. The epigenome is most plastic in early stages of embryo development when parent-specific genome reprogramming occurs (Mayer et al., 2000; Reik et al., 2001). This plasticity is then steadily decreasing during prenatal and post-natal life (Gluckman et al., 2009). If gestational hyperglycemia can permanently increase an individual's risk of chronic disorders in later stages of life, we should be more concerned about the possible adverse effects of assisted reproductive technologies (ART), in particular embryo culture on adult health and disease (El Hajj and Haaf, 2013). In in vitro produced lambs and calves, suboptimal embryo culture has been linked to aberrant expression of imprinted genes, causing large offspring syndrome (Young et al., 1998, 2001). Similarly, mouse studies provided clear evidence that embryo culture conditions can affect epigenetic gene regulation and cause adverse phenotypic changes, including organomegaly, glucose intolerance, as well as neurodevelopmental and behavioral alterations (Khosla et al., 2001; Ecker et al., 2004; Fernández-Gonzalez et al., 2004; Mann et al., 2004; Calle et al., 2012).
A systematic comparison of mouse embryo responses to different human ART culture protocols showed significant differences in blastocyst and fetal developmental rates, demonstrating the dramatic impact of the culture system on somatic differentiation (Market-Velker et al., 2010; Schwarzer et al., 2012). Because gametogenesis and embryonic development differ considerably in rodents and humans, mouse oocyte and embryo assays do not necessarily allow one to extrapolate to the human situation. Owing to the striking similarities with human development, bovine oocytes and embryos are increasingly used as models for human ART (Menezo and Herubel, 2002; Wrenzycki et al., 2005; Heinzmann et al., 2011). For legal and ethical reasons, it is not possible to use large numbers of human ART and non-ART embryos to systematically study the epigenetic and phenotypic effects of early human life conditions. Because it is problematic to assess the epigenetic safety of human ART protocols using rodent or large animal models, manipulation of oocyte and embryo should be restricted to a minimum or to the advantage of a specific technique (i.e. selection during embryo culture and blastocyst transfer) and must outweigh possible negative epigenetic effects (i.e. during extended embryo culture). Therefore, long-term epidemiologic studies on the lifelong risks of different ART protocols for metabolic, cardiovascular, neurodevelopmental, behavioral and other complex diseases are urgently needed. One important prerequisite for such studies is that future medical records of an individual also include information on parental infertility, mode of conception, and in the case of ART, which protocols have been used.
It is now widely accepted that an adverse periconceptional and intrauterine environment is associated with epigenetic malprogramming of the fetal metabolism and predisposition to chronic, in particular, metabolic disorders later in life (Barker et al., 2002; Gillman, 2005; Fernández-Morera et al., 2010; Hanson et al., 2011; Plagemann, 2011). Considering that in highly industrialized Western countries, up to 20% of women are obese at the start of pregnancy and up to 10% develop GDM during late pregnancy (Ben-Haroush et al., 2004; Kim et al., 2007), fetal overnutrition with glucose, free fatty acids and amino acids and the resulting hyperinsulinism may play a major role in the etiopathogenesis and current epidemics of obesity and type 2 diabetes (Fig. 1). The genetic variants that have been identified so far in large GWASs explain only a small fraction of the considerable heritability of type 2 diabetes and obesity (26 and 70%, respectively; Imamura and Maeda, 2011; Manco and Dallapiccola, 2012). In our opinion, modulations of the fetal epigenome by maternal diabetes and/or obesity provide the most reasonable mechanism for non-genetic intergenerational transmission of the phenotype. The missing heritability of complex disorders may be largely attributable to epigenetic inheritance.
Figure 1.
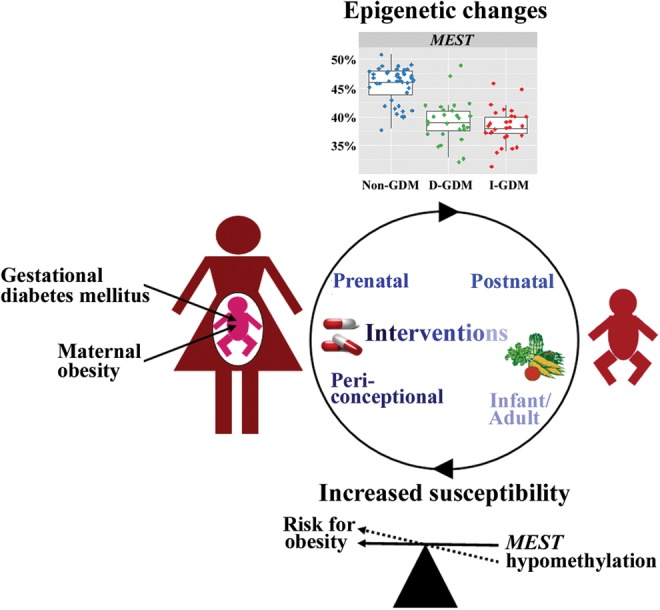
The cycle of metabolic disease epidemics. Fetal overnutrition due to GDM and/or maternal obesity leads to epigenetic changes in the exposed offspring. As an example, the box plot diagrams on top show the significantly different distribution of placenta MEST methylation values in newborns of mothers with dietetically treated D-GDM and insulin-treated I-GDM, compared with newborns of mothers without GDM. MEST hypomethylation may foreshadow diet-induced obesity. Similarly, epigenetic changes in numerous other genes may increase the lifelong metabolic disease susceptibility and, thus, the likelihood for a new generation of mothers with GDM and/or obesity, feeding the vicious cycle. Consistent with the decreasing plasticity of the epigenome, the effect of possible interventions to break the cycle can be expected to be larger in the periconceptional and prenatal period than after birth, during infancy and in adulthood.
To prevent metabolic disease epidemics, it is important to break the vicious cycle (Fig. 1) in which mothers with GDM and/or obesity have babies with epigenetic changes who are prone to develop metabolic disease later in life and, thus, give rise to a new generation of mothers with GDM/obesity. Considering the enormous plasticity of the epigenome during late stages of oocyte and early stages of embryo development (El Hajj and Haaf, 2013), one could hypothesize that the best time for action is periconceptionally. However, the true challenge resides in finding a solution as to how we can optimize the periconceptional environment with our currently limited knowledge on the underlying mechanisms and the optimum supply with macro- and micronutrients around conception, both in vivo and in vitro. In the sheep model it was shown that micronutrient deficiencies, such as folic acid and vitamin B12 during the periconceptional period cause widespread epigenetic alterations and long-term health implications for the offspring (Sinclair et al., 2007). Dietary restriction of folic acid, methionine, choline, and/or vitamin B12 can perturb transmethylation pathways (i.e. for DNA methylation) by decreasing the availability of methyl groups (Niculescu and Zeisel, 2002). One interesting study on a relatively small number of Gambian women (Khulan et al., 2012) suggests that micronutritional supplementation around the time of conception can affect the epigenome of the offspring (Khulan et al., 2012), possibly reducing fetal maladaption to inadequate nutrition during gestation. This is consistent with animal studies demonstrating that methyl supplementation of the maternal diet (i.e. with folic acid) can at least to some extent prevent a metabolic phenotype in the offspring (Waterland and Jirtle, 2003; Lillycrop et al., 2005, 2007; Burdge et al., 2008).
From a clinical point of view, it is important to note that effective treatment of maternal diabetes (Dörner et al., 2000; Plagemann, 2011) as well as large weight loss of obese women before pregnancy (Kral et al., 2006; Smith et al., 2009) appears to reduce the metabolic disease risk in the offspring. To prevent materno-fetal hyperglycemia and fetal hyperinsulinism, it is important to screen all pregnant women for disturbed glucose tolerance and to normalize a diabetic intrauterine environment by dietetic measures or insulin therapy. At the same time, we need biomarkers that help to assess adverse intrauterine exposures and lasting disease predispositions. This requires a more systematic comparison of fetal epigenomes using genome-wide methylation arrays or reduced representation bisulfite sequencing. Since the effect size of a single methylation change, i.e. in the MEST gene is low, we have to develop fetal and perinatal methylation profiles with more predictive power.
In contrast to genetic mutations/variants, epimutations are in principle reversible. This offers the exciting possibility to eventually compensate the epigenetic effects of an adverse intrauterine environment by pharmacological, dietary or behavioral interventions after birth. In the rat model of maternal undernutrition, neonatal leptin treatment and pre-weaning growth hormone treatment could prevent fetal programming of adult obesity, hypertension and vascular dysfunction (Vickers et al., 2005; Gray et al., 2013). Similarly, increasing folic acid intake during the juvenile-pubertal period was shown to reverse the negative phenotypic effects of prenatal undernutrition (Burdge et al., 2009). So far, in humans conclusive studies on the reversibility of fetal programming by specific post-natal treatments are missing. Breast feeding is generally recommended to avoid post-natal overfeeding (Plagemann, 2011). However, much more research is needed toward an optimal post-natal environment/management to correct adverse prenatal experiences.
Authors' roles
All authors provided substantial contributions to the review conception and data interpretation. T.H. wrote and H.L. and U.Z. critically revised the manuscript. All authors approved the submitted manuscript.
Funding
T.H. and U.Z. were supported by research grants (HA 1374/15-1 and ZE 442/5-2) from the German Research Foundation. Funding to pay the Open Access publication charges for this article was provided by the operating revenues of the Würzburg Institute of Human Genetics.
Conflict of interest
None declared.
References
- Aagaard-Tillery KM, Grove K, Bishop J, Ke X, Fu Q, McKnight R, Lane RH. Developmental origins of disease and determinants of chromatin structure: maternal diet modifies the primate fetal epigenome. J Mol Endocrinol. 2008;41:91–102. doi: 10.1677/JME-08-0025. doi:10.1677/JME-08-0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabet Care. 2006;29:S43–S48. [PubMed] [Google Scholar]
- Anderson OS, Nahar MS, Faulk C, Jones TR, Liao C, Kannan K, Weinhouse C, Rozek LS, Dolinoy DC. Epigenetic responses following maternal dietary exposure to physiologically relevant levels of bisphenol A. Environ Mol Mutagen. 2012;5:334–342. doi: 10.1002/em.21692. doi:10.1002/em.21692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armitage JA, Poston L, Taylor PD. Developmental origins of obesity and the metabolic syndrome: the role of maternal obesity. Front Horm Res. 2008;36:73–84. doi: 10.1159/000115355. doi:10.1159/000115355. [DOI] [PubMed] [Google Scholar]
- Baker JL, Olsen LW, Sørensen TI. Weight at birth and all-cause mortality in adulthood. Epidemiology. 2008;19:197–203. doi: 10.1097/EDE.0b013e31816339c6. doi:10.1097/EDE.0b013e31816339c6. [DOI] [PubMed] [Google Scholar]
- Barker DJ, Hales CN, Fall CH, Osmond C, Phipps K, Clark PM. Type 2 (non-insulin-dependent) diabetes mellitus, hypertension and hyperlipidaemia (syndrome X): relation to reduced fetal growth. Diabetologia. 1993;36:62–67. doi: 10.1007/BF00399095. doi:10.1007/BF00399095. [DOI] [PubMed] [Google Scholar]
- Barker DJ, Eriksson JG, Forsén T, Osmond C. Fetal origins of adult disease: strength of effects and biological basis. Int J Epidemiol. 2002;31:1235–1239. doi: 10.1093/ije/31.6.1235. doi:10.1093/ije/31.6.1235. [DOI] [PubMed] [Google Scholar]
- Ben-Haroush A, Yogev Y, Hod M. Epidemiology of gestational diabetes mellitus and its association with type 2 diabetes. Diabet Med. 2004;21:103–113. doi: 10.1046/j.1464-5491.2003.00985.x. doi:10.1046/j.1464-5491.2003.00985.x. [DOI] [PubMed] [Google Scholar]
- Bogdarina I, Haase A, Langley-Evans S, Clark AJ. Glucocorticoid effects on the programming of AT1b angiotensin receptor gene methylation and expression in the rat. PLoS One. 2010;5:e9237. doi: 10.1371/journal.pone.0009237. doi:10.1371/journal.pone.0009237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boney CM, Verma A, Tucker R, Vohr BR. Metabolic syndrome in childhood: association with birth weight, maternal obesity, and gestational diabetes mellitus. Pediatrics. 2005;115:e290–e296. doi: 10.1542/peds.2004-1808. doi:10.1542/peds.2004-1808. [DOI] [PubMed] [Google Scholar]
- Bouchard L, Hivert MF, Guay SP, St-Pierre J, Perron P, Brisson D. Placental adiponectin gene DNA methylation levels are associated with mothers' blood glucose concentration. Diabetes. 2012;61:1272–1280. doi: 10.2337/db11-1160. doi:10.2337/db11-1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdge GC, Lillycrop KA, Jackson AA, Gluckman PD, Hanson MA. The nature of the growth pattern and of the metabolic response to fasting in the rat are dependent upon the dietary protein and folic acid intakes of their pregnant dams and post-weaning fat consumption. Br J Nutr. 2008;99:540–549. doi: 10.1017/S0007114507815819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdge GC, Lillycrop KA, Phillips ES, Slater-Jefferies JL, Jackson AA, Hanson MA. Folic acid supplementation during the juvenile-pubertal period in rats modifies the phenotype and epigenotype induced by prenatal nutrition. J Nutr. 2009;139:1054–1060. doi: 10.3945/jn.109.104653. doi:10.3945/jn.109.104653. [DOI] [PubMed] [Google Scholar]
- Calle A, Miranda A, Fernandez-Gonzalez R, Pericuesta E, Laguna R, Gutierrez-Adan A. Male mice produced by in vitro culture have reduced fertility and transmit organomegaly and glucose intolerance to their male offspring. Biol Reprod. 2012;87:34. doi: 10.1095/biolreprod.112.100743. doi:10.1095/biolreprod.112.100743. [DOI] [PubMed] [Google Scholar]
- Caughey RW, Michels KB. Birth weight and childhood leukemia: a meta-analysis and review of the current evidence. Int J Cancer. 2009;124:2658–2670. doi: 10.1002/ijc.24225. doi:10.1002/ijc.24225. [DOI] [PubMed] [Google Scholar]
- Chiang EP, Wang YC, Chen WW, Tang FY. Effects of insulin and glucose on cellular metabolic fluxes in homocysteine transsulfuration, remethylation, S-adenosylmethionine synthesis, and global deoxyribonucleic acid methylation. J Clin Endocrinol Metab. 2009;94:1017–1025. doi: 10.1210/jc.2008-2038. doi:10.1210/jc.2008-2038. [DOI] [PubMed] [Google Scholar]
- Clausen TD, Mathiesen ER, Hansen T, Pedersen O, Jensen DM, Lauenborg J, Schmidt L, Damm P. Overweight and the metabolic syndrome in adult offspring of women with diet-treated gestational diabetes mellitus or type 1 diabetes. J Clin Endocrinol Metab. 2009;94:2464–2470. doi: 10.1210/jc.2009-0305. doi:10.1210/jc.2009-0305. [DOI] [PubMed] [Google Scholar]
- Cooper WN, Khulan B, Owens S, Elks CE, Seidel V, Prentice AM, Belteki G, Ong KK, Affara NA, Constância M, et al. DNA methylation profiling at imprinted loci after periconceptional micronutrient supplementation in humans: results of a pilot randomized controlled trial. FASEB J. 2012;26:1782–1790. doi: 10.1096/fj.11-192708. doi:10.1096/fj.11-192708. [DOI] [PubMed] [Google Scholar]
- Corrigan N, Brazil DP, McAuliffe F. Fetal cardiac effects of maternal hyperglycemia during pregnancy. Birth Defects Res A Clin Mol Teratol. 2009;85:523–530. doi: 10.1002/bdra.20567. doi:10.1002/bdra.20567. [DOI] [PubMed] [Google Scholar]
- Coustan DR, Lowe LP, Metzger BE, Dyer AR International Association of Diabetes and Pregnancy Study Groups. The hyperglycemia and adverse pregnancy outcome (HAPO) study: paving the way for new diagnostic criteria for gestational diabetes mellitus. Am J Obstet Gynecol. 2010;202:654.e1–6. doi: 10.1016/j.ajog.2010.04.006. doi:10.1016/j.ajog.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crudo A, Petropoulos S, Moisiadis VG, Iqbal M, Kostaki A, Machnes Z, Szyf M, Matthews SG. Prenatal synthetic glucocorticoid treatment changes DNA methylation states in male organ systems: multigenerational effects. Endocrinology. 2012;153:3269–3283. doi: 10.1210/en.2011-2160. doi:10.1210/en.2011-2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabelea D, Hanson RL, Lindsay RS, Pettitt DJ, Imperatore G, Gabir MM, Roumain J, Bennett PH, Knowler WC. Intrauterine exposure to diabetes conveys risks for type 2 diabetes and obesity: a study of discordant sibships. Diabetes. 2000;49:2208–2211. doi: 10.2337/diabetes.49.12.2208. doi:10.2337/diabetes.49.12.2208. [DOI] [PubMed] [Google Scholar]
- De Rooij SR, Wouters H, Yonker JE, Painter RC, Roseboom TJ. Prenatal undernutrition and cognitive function in late adulthood. Proc Natl Acad Sci USA. 2010;107:16881–16886. doi: 10.1073/pnas.1009459107. doi:10.1073/pnas.1009459107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai M, Beall M, Ross MG. Developmental origins of obesity: programmed adipogenesis. Curr Diab Rep. 2013;13:27–33. doi: 10.1007/s11892-012-0344-x. doi:10.1007/s11892-012-0344-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dörner G, Plagemann A, Neu A, Rosenbauer J. Gestational diabetes as possible risk factor for type I childhood-onset diabetes in the offspring. Neuro Endocrinol Lett. 2000;21:355–359. [PubMed] [Google Scholar]
- Drake AJ, Reynolds RM. Impact of maternal obesity on offspring obesity and cardiometabolic disease risk. Reproduction. 2010;140:387–398. doi: 10.1530/REP-10-0077. doi:10.1530/REP-10-0077. [DOI] [PubMed] [Google Scholar]
- Ecker DJ, Stein P, Xu Z, Williams CJ, Kopf GS, Bilker WB, Abel T, Schultz RM. Long-term effects of culture of preimplantation mouse embryos on behavior. Proc Natl Acad Sci USA. 2004;101:1595–1600. doi: 10.1073/pnas.0306846101. doi:10.1073/pnas.0306846101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Einstein F, Thompson RF, Bhagat TD, Fazzari MJ, Verma A, Barzilai N, Greally JM. Cytosine methylation dysregulation in neonates following intrauterine growth restriction. PLoS One. 2010;5:e8887. doi: 10.1371/journal.pone.0008887. doi:10.1371/journal.pone.0008887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Hajj N, Haaf T. Epigenetic disturbances in in vitro cultured gametes and embryos: implications for human assisted reproduction. Fertil Steril. 2013;99:632–641. doi: 10.1016/j.fertnstert.2012.12.044. doi:10.1016/j.fertnstert.2012.12.044. [DOI] [PubMed] [Google Scholar]
- El Hajj N, Pliushch G, Schneider E, Dittrich M, Müller T, Korenkov M, Aretz M, Zechner U, Lehnen H, Haaf T. Metabolic programming of MEST DNA methylation by intrauterine exposure to gestational diabetes mellitus. Diabetes. 2013;62:1320–1328. doi: 10.2337/db12-0289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteller M, Silva JM, Dominguez G, Bonilla F, Matias-Guiu X, Lerma E, Bussaglia E, Prat J, Harkes IC, Repasky EA, et al. Promoter hypermethylation and BRCA1 inactivation in sporadic breast and ovarian tumors. J Natl Cancer Inst. 2000;92:564–569. doi: 10.1093/jnci/92.7.564. doi:10.1093/jnci/92.7.564. [DOI] [PubMed] [Google Scholar]
- Faulk C, Dolinoy DC. Timing is everything: the when and how of environmentally induced changes in the epigenome of animals. Epigenetics. 2011;6:791–797. doi: 10.4161/epi.6.7.16209. doi:10.4161/epi.6.7.16209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feil R, Fraga MF. Epigenetics and the environment: emerging patterns and implications. Nat Rev Genet. 2012;13:97–109. doi: 10.1038/nrg3142. [DOI] [PubMed] [Google Scholar]
- Fernández-Gonzalez R, Moreira P, Bilbao A, Jiménez A, Pérez-Crespo M, Ramírez MA, Rodríguez de Fonseca F, Pintado B, Gutiérrez-Adán A. Long-term effect of in vitro culture of mouse embryos with serum on mRNA expression of imprinting genes, development, and behavior. Proc Natl Acad Sci USA. 2004;101:5880–5885. doi: 10.1073/pnas.0308560101. doi:10.1073/pnas.0308560101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández-Morera JL, Rodríguez-Rodero S, Menéndez-Torre E, Fraga MF. The possible role of epigenetics in gestational diabetes: cause, consequence, or both. Obstet Gynecol Int. 2010;2010:605163. doi: 10.1155/2010/605163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillman MW. Developmental origins of health and disease. N Engl J Med. 2005;353:1848–1850. doi: 10.1056/NEJMe058187. doi:10.1056/NEJMe058187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill-Randall R, Adams D, Ollerton RL, Lewis M, Alcolado JC. Type 2 diabetes mellitus—genes or intrauterine environment? An embryo transfer paradigm in rats. Diabetologia. 2004;47:1354–1359. doi: 10.1007/s00125-004-1464-x. doi:10.1007/s00125-004-1464-x. [DOI] [PubMed] [Google Scholar]
- Gilmartin AB, Ural SH, Repke JT. Gestational diabetes mellitus. Rev Obstet Gynecol. 2008;1:129–134. [PMC free article] [PubMed] [Google Scholar]
- Gluckman PD, Hanson MA, Buklijas T, Low FM, Beedle AS. Epigenetic mechanisms that underpin metabolic and cardiovascular diseases. Nat Rev Endocrinol. 2009;5:401–408. doi: 10.1038/nrendo.2009.102. doi:10.1038/nrendo.2009.102. [DOI] [PubMed] [Google Scholar]
- Godfrey KM, Sheppard A, Gluckman PD, Lillycrop KA, Burdge GC, McLean C, Rodford J, Slater-Jefferies JL, Garratt E, Crozier SR, et al. Epigenetic gene promoter methylation at birth is associated with child's later adiposity. Diabetes. 2011;60:1528–1534. doi: 10.2337/db10-0979. doi:10.2337/db10-0979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray C, Li M, Reynolds CM, Vickers MH. Pre-weaning growth hormone treatment reverses hypertension and endothelial dysfunction in adult male offspring of mothers undernourished during pregnancy. PloS One. 2013;8:e53505. doi: 10.1371/journal.pone.0053505. doi:10.1371/journal.pone.0053505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene MF. Spontaneous abortions and major malformations in women with diabetes mellitus. Semin Reprod Endocrinol. 1999;17:127–136. doi: 10.1055/s-2007-1016220. doi:10.1055/s-2007-1016220. [DOI] [PubMed] [Google Scholar]
- Hansmann T, Pliushch G, Leubner M, Kroll P, Endt D, Gehrig A, Preisler-Adams S, Wieacker P, Haaf T. Constitutive promoter methylation of BRCA1 and RAD51C in patients with familial ovarian cancer and early-onset sporadic breast cancer. Hum Mol Genet. 2012;21:4669–4679. doi: 10.1093/hmg/dds308. doi:10.1093/hmg/dds308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson MA, Low FM, Gluckman PD. Epigenetic epidemiology: the rebirth of soft inheritance. Ann Nutr Metab. 2011;58(Suppl 2):8–15. doi: 10.1159/000328033. doi:10.1159/000328033. [DOI] [PubMed] [Google Scholar]
- Harder T, Rodekamp E, Schellong K, Dudenhausen JW, Plagemann A. Birth weight and subsequent risk of type 2 diabetes: a meta-analysis. Am J Epidemiol. 2007;165:849–857. doi: 10.1093/aje/kwk071. doi:10.1093/aje/kwk071. [DOI] [PubMed] [Google Scholar]
- Hawdon JM. Babies born after diabetes in pregnancy: what are the short- and long-term risks and how can we minimise them? Best Pract Res Clin Obstet Gynaecol. 2011;25:91–104. doi: 10.1016/j.bpobgyn.2010.10.005. doi:10.1016/j.bpobgyn.2010.10.005. [DOI] [PubMed] [Google Scholar]
- Heijmans BT, Tobi EW, Stein AD, Putter H, Blauw GJ, Susser ES, Slagboom PE, Lumey LH. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc Natl Acad Sci USA. 2008;105:17046–17049. doi: 10.1073/pnas.0806560105. doi:10.1073/pnas.0806560105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinzmann J, Hansmann T, Herrmann D, Wrenzycki C, Zechner U, Haaf T, Niemann H. Epigenetic profile of developmentally important genes in bovine oocytes. Mol Reprod Dev. 2011;78:188–201. doi: 10.1002/mrd.21281. doi:10.1002/mrd.21281. [DOI] [PubMed] [Google Scholar]
- Imamura M, Maeda S. Genetics of type 2 diabetes: the GWAS era and future perspectives. Endocr J. 2011;58:723–739. doi: 10.1507/endocrj.ej11-0113. doi:10.1507/endocrj.EJ11-0113. [DOI] [PubMed] [Google Scholar]
- International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature. 2004;31:931–945. doi: 10.1038/nature03001. [DOI] [PubMed] [Google Scholar]
- Ivanova E, Chen JH, Segonds-Pichon A, Ozanne SE, Kelsey G. DNA methylation at differentially methylated regions of imprinted genes is resistant to developmental programming by maternal nutrition. Epigenetics. 2012;7:1200–1210. doi: 10.4161/epi.22141. doi:10.4161/epi.22141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson K, Soutto M, Peng D, Hu T, Marshal D, El-Rifai W. Epigenetic silencing of somatostatin in gastric cancer. Dig Dis Sci. 2011;56:125–130. doi: 10.1007/s10620-010-1422-z. doi:10.1007/s10620-010-1422-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. 2003;33:245–254. doi: 10.1038/ng1089. doi:10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- Kamei Y, Suganami T, Kohda T, Ishino F, Yasuda K, Miura S, Ezaki O, Ogawa Y. Peg1/Mest in obese adipose tissue is expressed from the paternal allele in an isoform-specific manner. FEBS Lett. 2007;581:91–96. doi: 10.1016/j.febslet.2006.12.002. doi:10.1016/j.febslet.2006.12.002. [DOI] [PubMed] [Google Scholar]
- Khosla S, Dean W, Brown D, Reik W, Feil R. Culture of preimplantation mouse embryos affects fetal development and the expression of imprinted genes. Biol Reprod. 2001;64:918–926. doi: 10.1095/biolreprod64.3.918. doi:10.1095/biolreprod64.3.918. [DOI] [PubMed] [Google Scholar]
- Khulan B, Cooper WN, Skinner BM, Bauer J, Owens S, Prentice AM, Belteki G, Constancia M, Dunger D, Affara NA. Periconceptional maternal micronutrient supplementation is associated with widespread gender related changes in the epigenome: a study of a unique resource in the Gambia. Hum Mol Genet. 2012;21:2086–2101. doi: 10.1093/hmg/dds026. doi:10.1093/hmg/dds026. [DOI] [PubMed] [Google Scholar]
- Kim SY, Dietz PM, England L, Morrow B, Callaghan WM. Trends in prepregnancy obesity in nine states, 1993-2003. Obesity (Silver Spring) 2007;15:986–993. doi: 10.1038/oby.2007.621. doi:10.1038/oby.2007.621. [DOI] [PubMed] [Google Scholar]
- Kosaki K, Kosaki R, Craigen WJ, Matsuo N. Isoform-specific imprinting of the human PEG1/MEST gene. Am J Hum Genet. 2000;66:309–312. doi: 10.1086/302712. doi:10.1086/302712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koza RA, Nikonova L, Hogan J, Rim JS, Mendoza T, Faulk C, Skaf J, Kozak LP. Changes in gene expression foreshadow diet-induced obesity in genetically identical mice. PLoS Genet. 2006;2:e81. doi: 10.1371/journal.pgen.0020081. doi:10.1371/journal.pgen.0020081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak LP, Newman S, Chao PM, Mendoza T, Koza RA. The early nutritional environment of mice determines the capacity for adipose tissue expansion by modulating genes of caveolae structure. PLoS One. 2010;5:e11015. doi: 10.1371/journal.pone.0011015. doi:10.1371/journal.pone.0011015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kral JG, Biron S, Simard S, Hould FS, Lebel S, Marceau S, Marceau P. Large maternal weight loss from obesity surgery prevents transmission of obesity to children who were followed for 2 to 18 years. Pediatrics. 2006;118:e1644–e1649. doi: 10.1542/peds.2006-1379. doi:10.1542/peds.2006-1379. [DOI] [PubMed] [Google Scholar]
- Lefebvre L, Viville S, Barton SC, Ishino F, Keverne EB, Surani MA. Abnormal maternal behaviour and growth retardation associated with loss of the imprinted gene Mest. Nat Genet. 1998;20:163–169. doi: 10.1038/2464. doi:10.1038/2464. [DOI] [PubMed] [Google Scholar]
- Lillycrop KA, Phillips ES, Jackson AA, Hanson MA, Burdge GC. Dietary protein restriction of pregnant rats induces and folic acid supplementation prevents epigenetic modification of hepatic gene expression in the offspring. J Nutr. 2005;135:1382–1386. doi: 10.1093/jn/135.6.1382. [DOI] [PubMed] [Google Scholar]
- Lillycrop KA, Slater-Jeffries JL, Hanson MA, Godfrey KM, Jackson AA, Burdge GC. Induction of altered epigenetic regulation of the hepatic glucocorticoid receptor in the offspring of rats fed a protein-restricted diet during pregnancy suggests that reduced DNA methyltransferase-1 expression is involved in impaired DNA methylation and changes in histone modifications. Br J Nutr. 2007;97:1064–1073. doi: 10.1017/S000711450769196X. doi:10.1017/S000711450769196X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magdinier F, Billard LM, Wittmann G, Frappart L, Benchaïb M, Lenoir GM, Guérin JF, Dante R. Regional methylation of the 5′ end CpG island of BRCA1 is associated with reduced gene expression in human somatic cells. FASEB J. 2000;14:1585–1594. doi: 10.1096/fj.14.11.1585. doi:10.1096/fj.14.11.1585. [DOI] [PubMed] [Google Scholar]
- Manco M, Dallapiccola B. Genetics of pediatric obesity. Pediatrics. 2012;130:123–133. doi: 10.1542/peds.2011-2717. doi:10.1542/peds.2011-2717. [DOI] [PubMed] [Google Scholar]
- Mann MR, Lee SS, Doherty AS, Verona RI, Nolen LD, Schultz RM, Bartolomei MS. Selective loss of imprinting in the placenta following preimplantation development in culture. Development. 2004;131:3727–3735. doi: 10.1242/dev.01241. doi:10.1242/dev.01241. [DOI] [PubMed] [Google Scholar]
- Market-Velker BA, Fernandes AD, Mann MR. Side-by-side comparison of five commercial media systems in a mouse model: suboptimal in vitro culture interferes with imprint maintenance. Biol Reprod. 2010;83:938–950. doi: 10.1095/biolreprod.110.085480. doi:10.1095/biolreprod.110.085480. [DOI] [PubMed] [Google Scholar]
- Mayer W, Niveleau A, Walter J, Fundele R, Haaf T. Demethylation of the zygotic paternal genome. Nature. 2000;403:501–502. doi: 10.1038/35000656. doi:10.1038/35000656. [DOI] [PubMed] [Google Scholar]
- McGowan PO, Sasaki A, D'Alessio AC, Dymov S, Labonté B, Szyf M, Turecki G, Meaney MJ. Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nat Neurosci. 2009;12:342–348. doi: 10.1038/nn.2270. doi:10.1038/nn.2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menezo YJ, Herubel F. Mouse and bovine models for human IVF. Reprod Biomed Online. 2002;4:170–175. doi: 10.1016/s1472-6483(10)61936-0. doi:10.1016/S1472-6483(10)61936-0. [DOI] [PubMed] [Google Scholar]
- Moore TR. Fetal exposure to gestational diabetes contributes to subsequent adult metabolic syndrome. Am J Obstet Gynecol. 2010;202:643–649. doi: 10.1016/j.ajog.2010.02.059. doi:10.1016/j.ajog.2010.02.059. [DOI] [PubMed] [Google Scholar]
- Niculescu MD, Zeisel SH. Diet, methyl donors and DNA methylation: interactions between dietary folate, methionine and choline. J Nutr. 2002;132(8 Suppl):2333S–2335S. doi: 10.1093/jn/132.8.2333S. [DOI] [PubMed] [Google Scholar]
- Nolan CJ, Damm P, Prentki M. Type 2 diabetes across generations: from pathophysiology to prevention and management. Lancet. 2011;378:169–181. doi: 10.1016/S0140-6736(11)60614-4. doi:10.1016/S0140-6736(11)60614-4. [DOI] [PubMed] [Google Scholar]
- Oberlander TF, Weinberg J, Papsdorf M, Grunau R, Misri S, Devlin AM. Prenatal exposure to maternal depression, neonatal methylation of human glucocorticoid receptor gene (NR3C1) and infant cortisol stress responses. Epigenetics. 2008;3:97–106. doi: 10.4161/epi.3.2.6034. doi:10.4161/epi.3.2.6034. [DOI] [PubMed] [Google Scholar]
- Painter RC, De Rooij SR, Bossuyt PM, Simmers TA, Osmond C, Barker DJ, Bleker OP, Roseboom TJ. Early onset of coronary artery disease after prenatal exposure to the Dutch famine. Am J Clin Nutr. 2006;84:322–327. doi: 10.1093/ajcn/84.1.322. [DOI] [PubMed] [Google Scholar]
- Painter RC, Osmond C, Gluckman P, Hanson M, Phillips DI, Roseboom TJ. Transgenerational effects of prenatal exposure to the Dutch famine on neonatal adiposity and health in later life. BJOG. 2008;115:1243–1249. doi: 10.1111/j.1471-0528.2008.01822.x. doi:10.1111/j.1471-0528.2008.01822.x. [DOI] [PubMed] [Google Scholar]
- Pinney SE, Simmons RA. Metabolic programming, epigenetics, and gestational diabetes mellitus. Curr Diab Rep. 2012;12:67–74. doi: 10.1007/s11892-011-0248-1. doi:10.1007/s11892-011-0248-1. [DOI] [PubMed] [Google Scholar]
- Plagemann A. Maternal diabetes and perinatal programming. Early Hum Dev. 2011;87:743–747. doi: 10.1016/j.earlhumdev.2011.08.018. doi:10.1016/j.earlhumdev.2011.08.018. [DOI] [PubMed] [Google Scholar]
- Plagemann A, Harder T, Brunn M, Harder A, Roepke K, Wittrock-Staar M, Ziska T, Schellong K, Rodekamp E, Melchior K, et al. Hypothalamic proopiomelanocortin promoter methylation becomes altered by early overfeeding: an epigenetic model of obesity and the metabolic syndrome. J Physiol. 2009;58:4963–4976. doi: 10.1113/jphysiol.2009.176156. doi:10.1113/jphysiol.2009.176156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plagemann A, Roepke K, Harder T, Brunn M, Harder A, Wittrock-Staar M, Ziska T, Schellong K, Rodekamp E, Melchior K, et al. Epigenetic malprogramming of the insulin receptor promoter due to developmental overfeeding. J Perinat Med. 2010;38:393–400. doi: 10.1515/jpm.2010.051. [DOI] [PubMed] [Google Scholar]
- Pyrzak B, Ruminska M, Popko K, Demkow U. Adiponectin as a biomarker of the metabolic syndrome in children and adolescents. Eur J Med Res. 2010;15(Suppl 2):147–151. doi: 10.1186/2047-783X-15-S2-147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reik W, Dean W, Walter J. Epigenetic reprogramming in mammalian development. Science. 2001;293:1089–1093. doi: 10.1126/science.1063443. doi:10.1126/science.1063443. [DOI] [PubMed] [Google Scholar]
- Ridderstråle M, Groop L. Genetic dissection of type 2 diabetes. Mol Cell Endocrinol. 2009;297:10–17. doi: 10.1016/j.mce.2008.10.002. doi:10.1016/j.mce.2008.10.002. [DOI] [PubMed] [Google Scholar]
- Robitaille J, Grant AM. The genetics of gestational diabetes mellitus: evidence for relationship with type 2 diabetes mellitus. Genet Med. 2008;10:240–250. doi: 10.1097/GIM.0b013e31816b8710. doi:10.1097/GIM.0b013e31816b8710. [DOI] [PubMed] [Google Scholar]
- Roseboom T, De Rooij S, Painter R. The Dutch famine and its long-term consequences for adult health. Early Hum Dev. 2006;82:485–491. doi: 10.1016/j.earlhumdev.2006.07.001. doi:10.1016/j.earlhumdev.2006.07.001. [DOI] [PubMed] [Google Scholar]
- Salbaum JM, Kappen C. Responses of the embryonic epigenome to maternal diabetes. Birth Defects Res A Clin Mol Teratol. 2012;94:770–781. doi: 10.1002/bdra.23035. doi:10.1002/bdra.23035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz R. Hyperinsulinemia and macrosomia. N Engl J Med. 1990;323:340–342. doi: 10.1056/NEJM199008023230512. doi:10.1056/NEJM199008023230512. [DOI] [PubMed] [Google Scholar]
- Schwarzer C, Esteves TC, Araúzo-Bravo MJ, Le Gac S, Nordhoff V, Schlatt S, Boiani M. ART culture conditions change the probability of mouse embryo gestation through defined cellular and molecular responses. Hum Reprod. 2012;27:2627–2640. doi: 10.1093/humrep/des223. doi:10.1093/humrep/des223. [DOI] [PubMed] [Google Scholar]
- Sinclair KD, Allegrucci C, Singh R, Gardner DS, Sebastian S, Bispham J, Thurston A, Huntley JF, Rees WD, Maloney CA, et al. DNA methylation, insulin resistance, and blood pressure in offspring determined by maternal periconceptional B vitamin and methionine status. Proc Natl Acad Sci USA. 2007;104:19351–19356. doi: 10.1073/pnas.0707258104. doi:10.1073/pnas.0707258104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slater-Jefferies JL, Lillycrop KA, Townsend PA, Torrens C, Hoile SP, Hanson MA, Burdge CC. Feeding a protein-restricted diet during pregnancy induces altered epigenetic regulation of peroxisomal proliferator-activated receptor-α in the heart of the offspring. J Dev Orig Health Dis. 2011;2:250–255. doi: 10.1017/S2040174410000425. doi:10.1017/S2040174410000425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith J, Cianflone K, Biron S, Hould FS, Lebel S, Marceau S, Lescelleur O, Biertho L, Simard S, Kral JG, et al. Effects of maternal surgical weight loss in mothers on intergenerational transmission of obesity. J Clin Endocrinol Metab. 2009;94:4275–4283. doi: 10.1210/jc.2009-0709. doi:10.1210/jc.2009-0709. [DOI] [PubMed] [Google Scholar]
- Stein AD, Kahn HS, Rundle A, Zybert PA, Van der Pal-de Bruin K, Lumey LH. Anthropometric measures in middle age after exposure to famine during gestation: evidence from the Dutch famine. Am J Clin Nutr. 2007;85:869–876. doi: 10.1093/ajcn/85.3.869. [DOI] [PubMed] [Google Scholar]
- Susser E, Neugebauer R, Hoek HW, Brown AS, Lin S, Labovitz D, Gorman JM. Schizophrenia after prenatal famine. Further evidence. Arch Gen Psychiatry. 1996;53:25–31. doi: 10.1001/archpsyc.1996.01830010027005. doi:10.1001/archpsyc.1996.01830010027005. [DOI] [PubMed] [Google Scholar]
- Tobi EW, Lumey LH, Talens RP, Kremer D, Putter H, Stein AD, Slagboom PE, Heijmans BT. DNA methylation differences after exposure to prenatal famine are common and timing- and sex-specific. Hum Mol Genet. 2009;18:4046–4053. doi: 10.1093/hmg/ddp353. doi:10.1093/hmg/ddp353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torloni MR, Betrán AP, Horta BL, Nakamura MU, Atallah AN, Moron AF, Valente O. Prepregnancy BMI and the risk of gestational diabetes: a systematic review of the literature with meta-analysis. Obes Rev. 2009;10:194–203. doi: 10.1111/j.1467-789X.2008.00541.x. doi:10.1111/j.1467-789X.2008.00541.x. [DOI] [PubMed] [Google Scholar]
- Tyrka AR, Price LH, Marsit C, Walters OC, Carpenter LL. Childhood adversity and epigenetic modulation of the leukocyte glucocorticoid receptor: preliminary findings in healthy adults. PloS One. 2012;7:e30148. doi: 10.1371/journal.pone.0030148. doi:10.1371/journal.pone.0030148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vickers MH, Gluckman PD, Coveny AH, Hofman PL, Cutfield WS, Gertler A, Breier BH, Harris M. Neonatal leptin treatment reverses developmental programming. Endocrinology. 2005;146:4211–4216. doi: 10.1210/en.2005-0581. doi:10.1210/en.2005-0581. [DOI] [PubMed] [Google Scholar]
- Waterland RA, Jirtle RL. Transposable elements: targets for early nutritional effects on epigenetic gene regulation. Mol Cell Biol. 2003;23:5293–5300. doi: 10.1128/MCB.23.15.5293-5300.2003. doi:10.1128/MCB.23.15.5293-5300.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver IC, Cervoni N, Champagne FA, D'Alessio AC, Sharma S, Seckl JR, Dymov S, Szyf M, Meaney MJ. Epigenetic programming by maternal behavior. Nat Neurosci. 2004;7:847–854. doi: 10.1038/nn1276. doi:10.1038/nn1276. [DOI] [PubMed] [Google Scholar]
- Weber M, Hellmann I, Stadler MB, Ramos L, Pääbo S, Rebhan M, Schübeler D. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat Genet. 2007;39:457–466. doi: 10.1038/ng1990. doi:10.1038/ng1990. [DOI] [PubMed] [Google Scholar]
- Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB. ATP-citrate lyase links cellular metabolism to histone acetylation. Science. 2009;324:1076–1080. doi: 10.1126/science.1164097. doi:10.1126/science.1164097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrenzycki C, Herrmann D, Lucas-Hahn A, Gebert C, Korsawe K, Lemme E, Carnwath JW, Niemann H. Epigenetic reprogramming throughout preimplantation development and consequences for assisted reproductive technologies. Birth Defects Res C Embryo Today. 2005;75:1–9. doi: 10.1002/bdrc.20035. doi:10.1002/bdrc.20035. [DOI] [PubMed] [Google Scholar]
- Wright CS, Rifas-Shiman SL, Rich-Edwards JW, Taveras EM, Gillman MW, Oken E. Intrauterine exposure to gestational diabetes, child adiposity, and blood pressure. Am J Hypertens. 2009;22:215–220. doi: 10.1038/ajh.2008.326. doi:10.1038/ajh.2008.326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CS, Nohr EA, Bech BH, Vestergaard M, Olsen J. Long-term health outcomes in children born to mothers with diabetes: a population-based cohort study. PLoS One. 2012;7:e36727. doi: 10.1371/journal.pone.0036727. doi:10.1371/journal.pone.0036727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoder JA, Walsh CP, Bestor TH. Cytosine methylation and the ecology of intragenomic parasites. Trends Genet. 1997;13:335–340. doi: 10.1016/s0168-9525(97)01181-5. doi:10.1016/S0168-9525(97)01181-5. [DOI] [PubMed] [Google Scholar]
- Young LE, Sinclair KD, Wilmut I. Large offspring syndrome in cattle and sheep. Rev Reprod. 1998;3:155–163. doi: 10.1530/ror.0.0030155. doi:10.1530/ror.0.0030155. [DOI] [PubMed] [Google Scholar]
- Young LE, Fernandes K, McEvoy TG, Butterwith SC, Gutierrez CG, Carolan C, Broadbent PJ, Robinson JJ, Wilmut I, Sinclair KD. Epigenetic change in IGF2R is associated with fetal overgrowth after sheep embryo culture. Nat Genet. 2001;27:153–154. doi: 10.1038/84769. doi:10.1038/84769. [DOI] [PubMed] [Google Scholar]