

Significance
Recent innovations facilitate collecting genome-wide data from organisms, tissues, or individual cells. Analysis of the data commonly produce long lists of genomic intervals whose meaning is context-dependent. For example, an interval may signify a genomic region of cancer-related DNA copy number variation. We propose a method of explaining such data in terms of a much smaller number of fundamental recurrent intervals called “cores.” Cores are useful, first, as a basis for studying subpopulation structure in a given primary tumor or metastasis; and, second, in delineating patterns of copy number aberrations in a given cancer type.
Keywords: genome analysis, interval data, statistical inference
Abstract
Finding regions of the genome that are significantly recurrent in noisy data are a common but difficult problem in present day computational biology. Cores of recurrent events (CORE) is a computational approach to solving this problem that is based on a formalized notion by which “core” intervals explain the observed data, where the number of cores is the “depth” of the explanation. Given that formalization, we implement CORE as a combinatorial optimization procedure with depth chosen from considerations of statistical significance. An important feature of CORE is its ability to explain data with cores of widely varying lengths. We examine the performance of this system with synthetic data, and then provide two demonstrations of its utility with actual data. Applying CORE to a collection of DNA copy number profiles from single cells of a given tumor, we determine tumor population phylogeny and find the features that separate subpopulations. Applying CORE to comparative genomic hybridization data from a large set of tumor samples, we define regions of recurrent copy number aberration in breast cancer.
Large collections of intervals are a common form of data generated by high-throughput genomics. For example, DNA copy number analysis yields intervals of the genome corresponding to gains or losses of DNA segments. Likewise, chromatin structure is often reported as intervals of the genome. In such cases a common goal is inference of contiguous genomic target regions, which under certain model assumptions, generates the observed patterns in the data. Such target regions are termed “cores” in the following text. The typical evidence for such cores is the presence of “recurrent” observations, suitably defined.
The following two examples illustrate how cores arise in a specific genomic setting. In the case of nucleosome positioning problems, the input interval set consists of DNA fragments obtained by micrococcal nuclease digestion. For an appropriate digestion regime, fragments protected by nucleosomes will dominate the set. Cores derived from these input data will correspond to nucleosome positions. As a second example, consider copy number analysis performed on a collection of individual DNAs. In this case, each interval in the input represents an observed region of copy number variation observed in a person. We can ask, “What are the common genomic copy number polymorphisms?” Rare variants, spurious segments created by observation and data processing, and the blurring of interval boundaries by noise in the observation protocol make an otherwise easy problem quite difficult. Cores will then represent genomic regions where copy number variation recurs in the population with a frequency unexpected by chance events.
We sought a solution that admits cores with a wide range of lengths, as is appropriate for data comprised of intervals with a wide range of lengths. We also sought to avoid explicit probability models in favor of more general set-theoretic and combinatorial methods, to which specific probabilistic assumptions could be added. We use the term “cores of recurrent events” (CORE) for our method. Central to CORE is the notion of explanatory power. A core is a proposed interval that “explains” an observed interval event by assigning a measure of geometric association between the two. The task is to find a set of cores that jointly provide an optimized explanation of the observed events. We show that certain association measures are more favorable than others with regard to algorithmic complexity. Next, we show how to subject the collection of cores to statistical tests for significance, so that a minimum number of justified cores, the “depth” of the solution, can be set. This approach is described and illustrated with synthetic data. We then demonstrate two applications with actual data sets: one that facilitates phylogenetic analysis and feature extraction for tumor subpopulations from single-cell copy number profiles and another that identifies regions of recurrent copy number aberration in breast cancer made from a large collection of profiles of tumor samples.
Results
Example of the Problem.
We first provide one concrete case, both to provoke intuitions and to clarify language that we will use in the following sections. Consider genome copy number analysis, performed for a collection of tumor biopsies. Whether determined by microarray hybridization (1) or sequence counting (2), the yield is a set of copy number profiles, one per sample, describing the amplifications and deletions within the genome of the tumor of each patient. For a type of cancer, for example breast or prostate cancer, these events presumably arise rather randomly throughout the genome of an unstable cell, but are selected for retention in the successful tumor clones at least in part by the presence of cancer genes, oncogenes in the amplified regions, and tumor suppressors in the deleted regions. The profiles can be further reduced to a set of intervals, regions of the genome where the amplifications or deletions took place. We refer to this data-reduction step as “slicing” (see again below). Some of the intervals may contain the oncogenes and tumor suppressors that provided selective advantage, and some intervals are present by chance. Intervals of the first class will, in some sense, share recurrent elements, and intervals of the second class will not. Sets of genomic intervals that explain many of the observed intervals, for example because they contain cancer genes, are what we call cores. There are various types of explanation. A putative core might explain an interval if the interval contains the core. Alternatively, a core might explain an interval if they significantly overlap. Any number of quantitative relations between core and interval can be postulated to accommodate a variety of biological notions. In the end, one wishes to have a minimal set of cores that “best” explain the data, and that can be subject to some form of statistical testing for significance. We refer to this process as CORE.
Formulation of the General Case.
The input into CORE is a set of N intervals dj, j = 1, … , N of a given type (for example, amplification or deletion events) derived from the observations. The domain Δ in which these observed intervals reside depends on the origin of the data. For data originating from genome-wide analysis, Δ consists of multiple disjoint intervals of the real line, each representing a chromosome. The objective of CORE is to find an optimal explanation of the intervals, the solution of a problem formulated as follows.
For an observed interval dj and an explanatory interval s in Δ, we define an “explanation” of dj by s as a function E(dj,s) with values in [0,1]. The specific functional form of E(dj,s) is dictated by biological considerations. For example, a useful form of E(dj,s) that reflects the degree of overlap of the two intervals is the Jaccard index:
In this case, s explains dj completely if and only if the two coincide and not at all if the two are disjoint. However, a specific form for E is not required for a general formulation of the method. We also refer to E(dj,s) as an association measure. In the following, we use P(dj,s) ≡ 1–E(dj,s), the portion of dj that s leaves unexplained.
Next, to generalize this concept to a set of explanatory intervals S = {s1,s2, … ,sK}, we define the portion P(dj,SK) of dj left unexplained by S as:
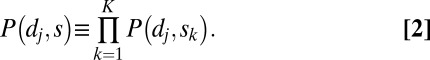 |
Finally, to generalize even further, we write the unexplained portion of the entire observed interval set D = {d1,d2, … ,dN} as defined by summation over the events:
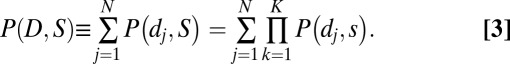 |
For a fixed number of explanatory intervals K, we seek to minimize P(D,S) over all possible sets S of K explaining intervals. Any such solution set of explaining intervals, CK = {c1,c2, … ,cK} will be called “optimal” and the individual elements cores. Note that we have not so far specified the appropriate number K of cores to be sought. This question is addressed later when we consider the statistical assessment of cores.
Forms of Explanation.
The computational complexity of the minimization problem depends on the form of explanation. From now on, we consider important restricted cases of explanation in which P(D,S) cannot attain a minimum unless each boundary of the cores sk coincides with that of one of the observed intervals. With this proviso, minimization of P(D,S) requires considering only a finite set of explaining intervals, namely those bound by O(N2) pair-wise combinations of the boundaries of the N events. Consequently, the quantities Pjk ≡ P(dj,sk) form a finite matrix of N rows and O(N2) columns, and the problem amounts to a choice of K columns such that Eq. 3 is minimized—that is, the minimizaton becomes a combinatorial problem.
To permit such minimization by a finite search, it is sufficient for P(D,S) to be concave or linear as a function of either boundary position of sk for all k, in any interval between adjacent event boundaries in D. In particular, this condition is satisfied for the following three special forms of association measures, E(d,s): (first) E(d,s) = 1 if s⊆d and otherwise E(d,s) = 0; (second) the Jaccard index J(d,s) raised to a power P ≥ 1; (third) E(d,s) = f(|s|/|d|), where f is any strictly convex or linear function on the interval [0,1] with a range contained in [0,1] when s⊆d and otherwise E(d,s) = 0.
These three forms of explanation capture different aspects of recurrence. The first form is especially simple and is designed to seek the genomic positions with the highest possible combined event count. However, this form of explanation ignores the degree of overlap among events explained by a given s and emphasizes regions where events overlap. The ability to detect clustering of broad events is thus reduced, especially when the broad events contain regions of narrow events that can be recurrent. On the other hand, the second and third explanation forms favor explanatory intervals at the intersection of multiple events with approximately coincident boundaries. Each core will therefore tend to be representative of a large number of similar genomic lesions.
Minimization of the Unexplained Portion.
The minimization problem defined by the first form of explanation as defined above is an instance of the p-coverage location problem, exactly solvable by dynamic programming in O(KN2) time (3), making this form of explanation computationally advantageous. To our knowledge, however, no general algorithm with execution time polynomial in K has been found for the exact minimization problem as posed in Eq. 3, even if P(D,S) permits combinatorial minimization. In the absence of such a solution, we offer an iterative greedy procedure for finding cores that has a polynomial time complexity.
We initialize at i = 0 by setting C0 = ∅ and P(dj,C0) = 1 for all j. Then, at the i-th iteration, ci = argmins ∑j P(dj,Ci–1)P(dj,s) is found, and Ci is formed by adding ci to Ci–1. To continue the iteration efficiently, P(dj,Ci) is stored for each j, computed as in Eq. 1 above: P(dj,Ci) = P(dj,Ci–1)P(dj,ci). The execution time of an individual iteration is independent of i, and the total execution time is proportional to K. Moreover, with any of the three explanatory forms, only a finite number of explanatory intervals need be searched at each iteration, and the greedy solution must search no more than O(N2) candidate explaining intervals. As the unexplained portion is a sum over N terms, the execution time is not greater than O(KN3). We will consider only greedy solutions for the remainder of this work.
Note that the Eqs. 2 and 3 can be generalized by the inclusion of weights for each event. In particular, the i-th minimization step of the greedy procedure may be interpreted as finding a single optimal core for the observed interval set D, but with each event dj of D assigned a weight Wj,i–1 = P(dj,Ci–1), namely the portion of dj left unexplained by previous cores. We view the set of intervals with their weights P(dj,Ci–1) as the remaining unexplained data after the i-th iteration. This interpretation is used next in assessing the statistical significance of a new core.
Statistical Criteria for Depth.
We tackle now a way to determine the depth of analysis, the lowest number of intervals that give a sufficient explanation of the data. Such a determination is made by seeking the lowest value for K such that the remaining unexplained data no longer display an unexpected amount of recurrence—that is, there is no new interval with a surprising amount of explanatory power. To determine this, we use a score, the amount of explanation gained from unexplained data by adding a new core, and compare this score to the scores obtained after the randomization of the unexplained data.
The total explanation provided by the core set CK is N–P(D,CK). The gain in explanation from the K-th core is then GK = P(D,CK–1) – P(D,CK). For an exact solution of the problem, it is generally not true that CK is obtained by adding one core to CK–1. However, this is an intrinsic property for our greedy solution to the problem, so for the greedy case we can define the score of the optimal interval, cK, as:
We seek to evaluate the statistical significance of this score, judging thereby the significance of the core itself. Significance is determined by testing the null hypothesis that the K-th observed score is not improbably high in the set of weighted events with the event randomly placed in the genome.
More specifically, we sample from the null distribution of the score. After m iterations of CORE, we generate multiple independent trials. In each trial, each event dj is transformed into an event d′j by a random placement, while its weight Wj,m–1 is left unchanged. We then estimate the probability of a value Gm or larger would be drawn from the distribution of G′m = maxs∑j Wj,m–1 E(d′j,s) generated from the multiple trials. Typically we perform 1,000 trials. If M+1 is the smallest m for which the null hypothesis cannot be rejected, the first M cores are retained.
Because events occur on chromosomes, and the events can themselves be large, on the order of the size of chromosomes, we must modify the above random translation scheme. The human chromosomes have broadly varying lengths, and a large event on chromosome 1, for example, cannot be translated to chromosome 21, restricting drastically our ability to randomize its placement. Therefore, when the observed interval data are randomly placed onto human chromosomes, we consider not the absolute length of an event but its length relative to the length of the chromosome on which it occurs. As we see next, this scheme appears to behave as expected.
Synthetic Data.
To evaluate the performance of CORE and to examine the statistical test for depth, we first simulated sets of interval events with built-in recurrence. Multiple parameters are required to specify such sets. Exploration of this multidimensional parameter space in a systematic way is not feasible, and so we provide one illustrative simulation, representative of several others we conducted, which was created as follows. First, we chose R = 5 recurrent regions with integer-valued lengths in the interval I = [0,2000]. The lengths of the regions were sampled at random from an exponential distribution with a mean value of Λ = 500 and rounded to the nearest integer. Given the length of a region, its position in I was chosen at random, among all possible positions with integer boundaries. Next, the total of NR = 200 events were assigned to these recurrent regions at random, with equal probability for all possible five-way partitions of 200. For each event, boundaries were set so that the event contained the region to which it was assigned, drawing the distance between the left (right) boundary of the event and that of the region from an exponential distribution with a mean value equal to σ = 1/4 of the length of the region and rounded to the nearest integer. Finally, NB = 100 background events were added to the set, by choosing 100 lengths from the set of 200 recurrent events with replacement and placing events of those lengths at random in the [0,2000] interval. A set of 300 events with integer boundary positions between 0 and 2000 was thus generated.
The resulting set of events was analyzed by CORE, with the third form of explanation and with f(x) = x. An analysis of one instance of a simulated event set, arbitrarily chosen, is illustrated in Figs. 1 and 2 and Dataset S1. CORE is able to accurately recover four out of the five recurrent regions, failing to recover a region whose small event count makes it indistinguishable from the background.
Fig. 1.

CORE analysis of a simulated set of events. The red-hashed rectangles indicate the positions and event counts of the five recurrent regions used to simulate data. The dashed line gives the event count for 200 events assigned to recurrent regions and the solid line the joint event count for these 200 and additional 100 background events. The blue Pi shapes indicate the positions and CORE scores of the five significant (P < 0.05) cores.
Fig. 2.

Significance analysis of cores derived from a simulated set of 300 events, as illustrated in Fig. 1. The observed scores are shown in red, plotted against the unexplained portion as defined by Eq. 3. Each of the box plots represents the corresponding empirical null distribution of the scores.
Significance analysis of the cores derived from the simulated set was performed as described above. The result of this analysis is shown in Fig. 2 for the interval data and the cores illustrated in Fig. 1. The top five scores are found to be significant. These correspond to the four underlying regions of highest recurrence, with the most recurrent region giving rise to two nearly coincident high-scoring cores. Note that the scores of the superfluous cores from the simulation appear to match well the empirical null distribution of the scores.
The success of this example, and many others we created, gave us sufficient confidence both in our formulations and coding to proceed with applications to existing empirical data sets. We focus on the third explanatory form in all of the following.
Applications to Cancer.
All of our subsequent applications are performed on copy number profiles derived from cancer DNA, either biopsies or single cells from biopsies. A copy number profile is a piecewise constant function of genomic position, with everywhere a value of two for a normal genome without copy number polymorphisms. The inputs to CORE in all cases are not the profiles themselves, but the joint collection of intervals of constant value from which each profile can be constructed. These intervals are the input to CORE and are derived by a process we call slicing, which is described in the Methods section. We show two types of application, the first application being to phylogenetic analysis of single-cell data and the second to regions of common aberration in breast cancers.
Phylogeny from Single-Cell Data.
We previously used single-cell cancer copy number data to distinguish subpopulations with shared but distinct genomic history (2). The universally shared copy number lesions mark the ancestral trunk, while each main branch of the phylogenetic tree has its own distinctive copy number events. Within the branches there are presumably newer events that are unique to the single cells in the sample. Our previous methods were provisional, and here we explore using CORE as the analysis engine for such data, with the idea of identifying the recurrent events—those common to the trunk and those common to particular branches—while ignoring events that are not shared or insignificantly recurrent. We use the cores to build a tree, and then use supervised clustering to identify the distinguishing events. We look at two cases.
In the first case, tumor T10, single cells came from a primary breast tumor. FACS analysis of cells sampled from this tumor identified four distinct populations by ploidy (mean copy number): two aneuploid populations, one hypo-diploid, and one diploid. Dataset S2A provides the set of events derived by slicing in this case. CORE analysis of this set yields 172 amplification cores and 182 deletion cores at P = 0.05 level of significance. Dataset S2 B and C lists the position, significance, and scores for each of these cores. We next compute an incidence table of the overlap of each profile with the significant cores, using the procedure described in the Methods section. The table is presented in full in Dataset S2D, and its image is shown in Fig. 3 as a heat map. The rows are organized in a manner we will soon describe. It is clearly seen that some of the cells in the set correspond to sparse and some to dense—that is, event-rich—rows of the table. The former derive from the diploid population, while the latter are from the hypo-diploid population rich in deletions and the aneuploid populations, which are richer in amplifications.
Fig. 3.

Heat map of T10 incidence table, with rows corresponding to cells and columns to CORE cores. The amplification and deletion subtables are indicated by the blue and red horizontal bars. The order of cores in each subtable are left to right by the descending value of their CORE scores. Darker shades of gray correspond to higher values in the table. The order of the cells is clustered by the phylogenetic tree (on left) with horizontal distance related to distance. The tree yields a perfect separation of the (pseudo)diploid, hypodiploid, and the two aneuploid populations. The vertical color bar encodes the cell subpopulation label: yellow for aneuploid A, red for aneuploid B, blue for the hypodiploid, and green for diploid and pseudodiploid.
To examine the phylogeny of cell populations, we previously used two different metrics, one a Hamming distance based on shared interval breakpoints and another based on the absolute difference in the copy number (2). These metrics were used to compute phylogenetic trees. We returned to this problem using an incidence table (Methods). The incidence table is a J×K matrix, with J being the number of profiles and K the number of cores. Each row of the matrix can be viewed as a compression of the profile, decomposing the profile into the core elements. The matrix element, Tjk, is computed as the maximum explanation over all of the intervals of profile j by core k. All matrix elements of T are therefore in the [0,1] range.
We explored the phylogeny based on the Euclidean distance between rows (i.e., single cells) of the incidence table. Each core contributes equally, irrespective of its length, and so this metric more closely resembles the breakpoint method but incorporates information about intervals, namely the pairs of breakpoints (the breakpoints that define intervals). We generated a phyogenetic tree using hierarchical clustering with Ward linkage. It is this tree that defines the order of the rows in Fig. 3. The phylogeny cleanly separates the cells in the samples by ploidy. Essentially the same separation was accomplished by neighbor-joining method of phylogeny reconstruction (Fig. S1).
In our next example, tumor T16, the single cells came from a primary and a metastatic site. Unlike T10, which has a pattern we call polygenomic, only one profile dominates in the aneuploid cells from each site, a pattern we call monogenomic. Dataset S3A provides the set of events derived by slicing in this case. The resulting amplification and deletion cores are listed, respectively, in Dataset S3 B and C. Dataset S3D is the incidence table for this case. The image of the incidence table is shown in Fig. 4, with the dense aneuploid and sparse diploid sectors clearly visible.
Fig. 4.

Heat map of T16 incidence table with rows corresponding to cells and columns to cores. The amplification and deletion subtables and row and column order and shading of the heat map are as described for Fig. 3. The vertical color bar encodes the cell subpopulation label: red for the primary aneuploid, black for the metastatic aneuploid, green for the primary diploid and pseudodiploid, and blue for the metastatic diploid. The phylogenetic tree for the cells is shown on the left, with a perfect separation of the primary and metastatic aneuploid populations.
Based on the incidence table of T16 (Fig. 4), we achieve clear separation into three subpopulations, one the separation by ploidy and among the aneuploid a separation of metastatic and primary sites. These populations also are nearly perfectly separated by the neighbor-joining algorithm (Fig. S2), the only exception being a primary aneuploid cell (A17), which clusters with the metastatic aneuploid population. In our previous phylogenetic analysis of T16 (2), we had difficulty distinguishing the metastatic and primary cells.
With phylogeny determined by CORE, we observe a separation in the diploid population that we had previously not appreciated. As can be seen in Fig. 4, there are two populations of diploid cells, drawn roughly equally from the primary and metastatic sites. Upon closer analysis, we found that the distinction between these two populations is the frequency of deletions near the ends of chromosomes. Two such populations are not seen in the diploid cells from T10, which closely resemble the subpopulation of T16 with frequent deletions. We do not know at this time whether this represents an artifact of single-cell copy number measurement or interesting biology, but we expect the former.
The genomic distinctions between metastatic and primary cells are of biological importance. We therefore sought to detect cores that are markers of the primary or metastatic location within the aneuploid population. For this purpose, we applied the Random Forests (RF) classifier (Methods). Indeed, we could readily separate by RF the cells of the two populations with 100% accuracy. In Table 1 we list the four cores found to be the most important classifying features by RF. Three of the four are better matched by intervals in the metastatic than in the primary cells, and one the reverse. To quantify the observations statistically, we examined, for each of these cores, the significance of association between the discriminating core and the label (primary or metastatic) using Fisher’s exact test, adjusting for multiple hypothesis correction as explained in the Methods section. The resulting P values are reported in Table 1.
Table 1.
Four important cores (RF importance score of at least 1) for distinguishing primary and metastatic aneuploid cells in T16
| Core | Importance | Chromosome | Start | End | Score | Cutpoint | M– | P– | M+ | P+ | Fisher P |
| A48 | 1.7 | 11 | 120556477 | 134452384 | 10.9 | 0.0 | 22 | 0 | 0 | 16 | 5.40 × 10−10 |
| A13 | 1.41 | 3 | 98933466 | 154624504 | 36.3 | 0.87 | 1 | 16 | 21 | 0 | 1.61 × 10−8 |
| D30 | 1.24 | 10 | 42735344 | 135374737 | 20.9 | 0.0 | 1 | 16 | 21 | 0 | 5.35 × 10−9 |
| D8 | 1.01 | 9 | 70584473 | 140273252 | 46.5 | 0.779 | 2 | 16 | 20 | 0 | 9.63 × 10−8 |
For each, the genomic position (“start” and “end”), CORE “score,” and its optimal threshold, “cutpoint,” are tabulated together with the corrected Fisher P value (Methods) for association with the subpopulation label. The numbers of metastatic aneuploid cells scoring above and at/below the optimal cutpoint are given in the M+ and M– columns. The corresponding numbers of the primary aneuploid cells are given in the P+ and P– columns.
In a completely analogous fashion, we applied the RF classifier to separate diploid from aneuploid cells in T16. Here again, the algorithm separated the two with 100% accuracy. While the distinction between these two populations is strikingly apparent in Fig. 4, it is still of interest to identify cores that best distinguish between the two. We find 27 cores of the highest importance by RF (Table S1). It is noteworthy that some of these cores harbor validated cancer genes. For example, core D13 delineates a narrow region of chromosome 9 containing cyclin-dependent kinase inhibitor 2A (CDKN2A) and is homozygously deleted in all of the aneuploid cells.
Common Cores of Aberration in Breast Cancers.
This last application (distinctive differences in populations of cancer cells within the same person) has an important generalization—namely, the example we proposed at the beginning of the Results section. Can we use CORE to identify the boundaries of significantly recurring amplifications and deletions within the cancers of many different patients? We attempted this with two published sets of copy number profiles of breast tumors, both from Scandinavia and both analyzed on the same array hybridization platform (1,4). Although we had previously noted regions of recurrent aberrations, we did not have until now a single method that we trusted to describe events, both narrow and broad.
Profiles were processed as described in the Methods section. Amplification and deletion events derived by slicing are listed in Dataset S4A. Cores were then derived separately for amplification and deletion events. Analysis of significance was performed (Methods), resulting in 44 amplification and 22 deletion cores at P = 0.05 level of significance. The observed scores are compared with their empirical null distributions in Fig. 5. The significant amplification and deletion cores explained about one-fifth of the data, and are reported, respectively, in Dataset S4 B and C and shown in Fig. 6. The amplification cores span a broad range of widths, from 165 Kb to over 95 Mb. The top-scoring broad cores correspond to well-known features of breast cancer copy number landscape such as whole-arm amplifications of 1q, 8q, and 16p. On the opposite end of the width spectrum are 11 narrow cores, each containing fewer than 35 genes. Ten of these 11 cores contain known driver genes, notably v-erb-b2 erythroblastic leukemia viral oncogene homolog 2 (ERBB2), cyclin D1 (CCND1), and insulin-like growth factor 1 receptor (IGF1R). The gene content of the narrow cores is reported in detail in Dataset S4D. The narrowest core bracketing v-myc myelocytomatosis viral oncogene homolog (avian) (MYC) is 11.8 Mb wide and contains 54 genes.
Fig. 5.

Significance analysis of amplification and deletion cores derived from the combined WZ and MicMa set of 257 copy number profiles of breast tumors. The core scores are plotted against the unexplained portion as defined by Eq. 3. The observed scores are shown in red. Each of the box plots represents the corresponding empirical null distribution of the scores. Portions of the plot to the right of the dashed lines correspond to statistically significant (P < 0.05) cores.
Fig. 6.

Significant (P < 0.05) cores derived from the combined WZ and MicMa set of copy number profiles of breast tumors, shown as color bars at the chromosomal locations where they occur. The 44 amplification (22 deletion) cores are shown above (below) the corresponding chromosomes in blue (red and orange). Darker colors correspond to higher CORE scores. Genomic positions of four accepted driver genes (MYC, CCND1, IGF1R, and ERBB2) are indicated by arrows.
On the other hand, we find no narrow deletion cores. The narrowest of these is 8.2 Mb wide and contains 46 genes. Similarly to amplification cores, the top-scoring deletion cores represent familiar features of somatic copy number variation in breast cancer, in particular whole-arm deletions of 16q, 8p, 17p (thus deleting TP53), and 11q.
Methods
Incidence Table for Profiles and Cores.
In the case of DNA copy number analysis discussed in the following, the input set of intervals is formed by copy number events (gains or losses), each originating in one of multiple copy number profiles. Each profile represents a biological entity such as a tissue sample or a cell. Having derived K cores from this joint input set, we construct an incidence table T that quantifies how well each core performs in each profile. The incidence table is thus an L×K matrix, L being the number of profiles and K the number of cores. Each of its matrix elements, Tlk, is computed as the maximum over all of the intervals in profile l of the explanations by core k. In other words, Tlk is the explanation of the best fit of core k to profile l. It follows from this definition that all matrix elements of T are in the [0,1] range.
Using the Incidence Table for Marker Detection.
If we have class labels for our copy number profiles, we may wish to know which of the derived cores are useful as markers for the class. To this end we use the RF classifier (5). Two properties of RF make it useful for this purpose. First of all, RF does not overfit, and its classification accuracy can be reliably judged from the training data. Secondly, RF is able to quantify the importance of each feature for classification. For marker detection we proceeded as follows. First, RF is trained to predict the class label on the entire incidence table. We then rank cores by their “importance” as defined by RF. For each of a small number of top-ranking cores, we perform the following additional assessment of its significance as a marker. For a given core, and hence a column in the incidence table, all of the distinct values in the column are found. Then, using each of the distinct values in turn as a threshold, Fisher’s exact test is applied to the contingency table built from the association between the class label and the value being above the threshold for the core in the incidence table. The number of tests conducted is thus equal to the number of distinct values in the column. The lowest Fisher P value thus found is corrected for this multiplicity of tests, and the result is taken as the P value for association between the core and the label.
Breast Cancer Data.
For analysis of individual tumor subpopulations, we use single-cell copy number data we previously described for human breast cancer tumors T10 and T16 (2). The data consist of bin counts of sequence reads, segmented, and then converted to integer copy number segments. A total of 50,009 bins cover the entire genome, laid out in the usual order of chromosomes: 1, … , 22, X, Y.
For analysis of the cores from profiles of tumors of populations of patients, we use a combination of two published sets of copy number profiles of breast tumors, the WZ set (1) and the MicMa set (4), both obtained by microarray hybridization. These profiles represent DNA copy number averaged over multiple tumor cells, likely being an admixture of different subpopulations and normal cells (“mixed cell population data”). Profiles are masked for common copy number variation as described in ref. 1. For comparison, we used normal individual genome profiles obtained from the same platform (6).
Processing Breast Cancer Data.
To use CORE, we must first extract interval events from segmented (7) copy number profiles. The method of transforming each profile into a set of intervals differs for single-cell data and for mixed-cell population data. In both cases, we use a process we call slicing. We then find the significant cores, and create an incidence table.
To slice profiles from single cells, we determine the median ploidy for each cell, defined as the median of integer copy numbers for all bins. Segments above the median ploidy are considered amplified, and those below deleted. There is no restriction on the segment lengths, and these range from the shortest detectable by the segmentation algorithm to an entire chromosome. For each integer value of copy number except the median ploidy, we determine a unique set of largest intervals that can be placed without disruption into the profile (illustrated for a portion of chromosome 1 in Fig. 7). In essence, this procedure is a simplified version of the ziggurat deconstruction algorithm (8). Note that the information about the degree of copy number change caused by an amplification or a deletion event is lost in this transformation. The input into CORE, separately for amplifications and deletions, is formed by pooling the intervals, with start and end positions specified as bin numbers.
Fig. 7.

Slicing. A portion of the integer copy number profile of chromosome 1 in the primary cell A5 of tumor T16 (in black) is sliced into four unique interval events (in red). The basal copy number for this cell is 4 (shown by a dashed line).
Slicing mixed-cell population data are more complicated because the copy number need not be integer, and hence requires more preprocessing. We followed the protocol from a previous publication (9), with one addition. We determine a suitable threshold such that segments deviating by more than that threshold from the center (as defined in ref. 9) are considered copy number events. The threshold is derived from a set of normalized and segmented noncancer copy number profiles originating from the same platform as the cancer set in question and centered at the median. Segments where large deviations from the median are expected are removed from the noncancer set. These include segments in chromosomes X and Y and segments shorter than 5 Mb. The threshold for amplification (deletion) is then set by the top (bottom) 0.02% of all segmented log ratio probe values of the remaining segments. Microarray probe numbers were used to specify the start and end positions of copy number events derived by slicing.
Availability of Software.
An implementation of CORE as an R package is available upon request and includes tools for computing core positions and scores and for assessing the statistical significance of scores, with a choice among measures of association as described here. In addition, R software is available for the analysis of integer copy number data, both upstream and downstream of CORE, including the slicing procedure and the derivation of the incidence table that we used to examine the subpopulation structure of breast tumors. R code for generating a simulated event configuration for arbitrary R, I, Λ, NR , σ, NB (Results) will also be provided upon request.
Data Access.
Sequencing data for T10 and T16, mapped to version 18 of human genome, are available online from the Sequence Read Archive (www.ncbi.nlm.nih.gov/sra), accession code SPR002535. Tumor DNA copy number data for the WZ and MicMa cohorts, mapped to version 17 of human genome, are available online from the Gene Expression Omnibus (www.ncbi.nlm.nih.gov/geo/), record GSE19425, and the corresponding microarray annotation table is available as record GPL9776.
Discussion
CORE is a general approach to inference from interval data. Given a collection of observed events and a geometric association measure between events and explanatory intervals, CORE finds a given number of explanatory cores that maximizes the explanation. When the association measure is drawn from three broad varieties outlined in the text, for example the Jaccard index, we find a greedy solution with algorithmic complexity O(KN3), where N is the number of events and K is the number of cores. We believe our formulation of the problem is “natural” in the sense that it captures the manner in which a human observer seeks to find fundamental intervals behind a set of recurrent events in the presence of noisy events and boundaries.
A previous paper included an analysis of nucleosome positioning that used a version of CORE based on the first form of association measure (Methods) (10). For this form and problem, known as the p-coverage location problem (3), exactly optimal solutions can be found in O(KN3) time. However, this form of explanation does not usually capture the richness of an underlying interval data. We are continuing studies of nucleosome positioning with more powerful varieties of geometric association, using the algorithmic methods described here.
In this article we address the more common problem of interval events representing genome copy number variation. We apply the third form of explanation to two particular problem classes encountered in our research: (i) determining the subpopulation structure of a tumor and (ii) defining the regions of recurring aberration in large collections of breast cancers. For the first problem, we could have equally chosen as explanatory measure the Jaccard index raised to a power. For the second problem, our choice was motivated by the belief that amplification events have drivers, transcribed regions with objectively defined borders that are contained within the events.
Our method is greedy and iterative. For any such procedure, overinterpretation is a major concern. To provide a plausible depth of analysis, we implemented a permutation test. After each iteration through the data, each interval is reweighted to the extent that it remains unexplained. We then determine whether the reweighted, or residual, data have unexpected recurrence. We stop when the residual data no longer display a statistically unexpected amount of recurrence. We have no proof that our statistical method stops in the general case, but in simulations and actual data, it does.
In the first application, we use CORE for subpopulation analysis by reanalyzing single-cell data from two breast cancer patients, one with a primary cancer and one with both a primary and metastatic lesion. We transform the copy number profiles of each cell by slicing, a simplified version of ziggurat deconstruction (8) that produces a set of events from each single profile. The cores provide a reduced representation of the significantly recurrent events in the totality of cells, and an incidence table is computed that represents the copy number profile of each cell as the overlap of its events with the cores. We use the incidence table for supervised (RF) and unsupervised (hierarchical clustering and neighbor joining) learning of subpopulation structure. We learn the events that distinguish the metastatic from primary cells, and construct phylogenetic trees that are completely compatible with trees constructed by our previous methods. Using cores and incidence tables for phylogeny has the major theoretical advantage over our previous methods in that it gives rise to a metric between single cells that is continuously valued but based on pairs of recurrent boundaries.
In the second application, CORE is used to summarize recurrent events in tumor profiles of a given tissue type. For breast cancers, we find on the order of 70 statistically significant recurrently amplified or deleted cores, thus enabling us to represent a tumor in terms of this reduced set of elements. In so doing, we can overcome to a large extent the problem of multiple hypothesis testing when looking for biomarkers of sensitivity to chemotherapy (work in progress). Cores can be used for prediction of survival for ovarian cancer (also work in progress). In yet another utility, CORE provides an automated method to find candidate oncogenes or tumor suppressor genes in narrow events. In fact many of the narrow amplification cores do contain familiar oncogenes (Dataset S4 B and D and Fig. 6).
Our emphasis in the present work is DNA copy number variation. A particularly challenging aspect common to such data are the broad range of lengths of recurrent events, extending from the smallest observable on a given platform to an entire chromosome. Other methods of recurrence analysis that are focused on position-wise event count are blind to the distinction between common narrow events and broad events. However, broad recurrent events may be critical in cancer progression, as the core regions of aberration may contain multiple tumor suppressor genes (11). Genome events cannot be assumed to target a single functionally important gene.
The genomic identification of significant targets in cancer (GISTIC) method addresses this problem by separating events into broad and focal using a fixed width threshold and by handling the two groups of events separately (8, 12). The significance testing for aberrant copy number (STAC) algorithm (13) takes into account the consistency of event pileups to assess recurrence, but the reported measure of recurrence is a local function of genomic position. By contrast, separation of event pileups into broad and narrow ones is not necessary in CORE. With association measures such as those used here, the output of CORE is a table of extended objects (cores), and the characteristic width of events defining a core is dictated by the observed data. These features of CORE are shared with the method presented by Ionita et al. (14), but unlike CORE, that method requires model assumptions about the distribution of event lengths and single point-like drivers.
An additional advantage of CORE compared with other methods of interval data analysis lies in its ability to offer a choice of options to suit the genomic problem at hand. This choice includes, in addition to a variety of core-to-event association measures, a freedom to weigh events according to their importance in a given context. For example, weights may be used to filter out certain types of events, or set as a decreasing function of the event lengths to emphasize focal events. In application to copy-number analysis, weights may be set as an increasing function of the magnitude of copy-number change caused by the events. We supply software to facilitate exploration of these options by the end user.
We do not wish to imply that all problems for analyzing interval data are solved with the tools we have provided. Indeed, that is not the case. We are continuing to examine additional avenues, such as association measures optimized for treatment of measurement error, unique protocols for preprocessing data, and recursive applications of CORE for phylogenetic analysis.
Supplementary Material
Acknowledgments
We acknowledge very informative discussions of computational methodology with Arie Tamir, Steven Skiena, and Gene Bryant. This work was supported by grants (to M.W.) from the Simons Foundation (Simons Foundation Autism Research Initiative Award SF51), the Breast Cancer Research Foundation, and the US Department of Defense (W81XWH-11-1-0747). A.K. acknowledges support by the National Institute of Health (Grants 5RC2CA148532-02 and 1U01CA168409-01) and by the US Department of Defense (Grant W81XWH-11-1-0747). M.W. is an American Cancer Society Research Professor.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1306909110/-/DCSupplemental.
References
- 1.Hicks J, et al. Novel patterns of genome rearrangement and their association with survival in breast cancer. Genome Res. 2006;16(12):1465–1479. doi: 10.1101/gr.5460106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Navin N, et al. Tumour evolution inferred by single-cell sequencing. Nature. 2011;472(7341):90–94. doi: 10.1038/nature09807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hassin R, Tamir A. Improved complexity-bounds for location-problems on the real line. Oper Res Lett. 1991;10(7):395–402. [Google Scholar]
- 4.Russnes HG, et al. Genomic architecture characterizes tumor progression paths and fate in breast cancer patients. Sci Transl Med. 2010;2(38):38ra47. doi: 10.1126/scitranslmed.3000611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Breiman L. Random forests. Mach Learn. 2001;45(1):5–32. [Google Scholar]
- 6.Sebat J, et al. Strong association of de novo copy number mutations with autism. Science. 2007;316(5823):445–449. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Venkatraman ES, Olshen AB. A faster circular binary segmentation algorithm for the analysis of array CGH data. Bioinformatics. 2007;23(6):657–663. doi: 10.1093/bioinformatics/btl646. [DOI] [PubMed] [Google Scholar]
- 8.Beroukhim R, et al. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011 doi: 10.1186/gb-2011-12-4-r41. 12(4):R41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xue W, et al. DLC1 is a chromosome 8p tumor suppressor whose loss promotes hepatocellular carcinoma. Genes Dev. 2008;22(11):1439–1444. doi: 10.1101/gad.1672608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Floer M, et al. A RSC/nucleosome complex determines chromatin architecture and facilitates activator binding. Cell. 2010;141(3):407–418. doi: 10.1016/j.cell.2010.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xue W, et al. A cluster of cooperating tumor-suppressor gene candidates in chromosomal deletions. Proc Natl Acad Sci USA. 2012;109(21):8212–8217. doi: 10.1073/pnas.1206062109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beroukhim R, et al. Assessing the significance of chromosomal aberrations in cancer: Methodology and application to glioma. Proc Natl Acad Sci USA. 2007;104(50):20007–20012. doi: 10.1073/pnas.0710052104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Diskin SJ, et al. STAC: A method for testing the significance of DNA copy number aberrations across multiple array-CGH experiments. Genome Res. 2006;16(9):1149–1158. doi: 10.1101/gr.5076506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ionita I, Daruwala RS, Mishra B. Mapping tumor-suppressor genes with multipoint statistics from copy-number-variation data. Am J Hum Genet. 2006;79(1):13–22. doi: 10.1086/504354. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.