

Abstract
Precise nucleosome-positioning patterns at promoters are thought to be crucial for faithful transcriptional regulation. However, the mechanisms by which these patterns are established, are dynamically maintained, and subsequently contribute to transcriptional control are poorly understood. The switch/sucrose non-fermentable chromatin remodeling complex, also known as the Brg1 associated factors complex, is a master developmental regulator and tumor suppressor capable of mobilizing nucleosomes in biochemical assays. However, its role in establishing the nucleosome landscape in vivo is unclear. Here we have inactivated Snf5 and Brg1, core subunits of the mammalian Swi/Snf complex, to evaluate their effects on chromatin structure and transcription levels genomewide. We find that inactivation of either subunit leads to disruptions of specific nucleosome patterning combined with a loss of overall nucleosome occupancy at a large number of promoters, regardless of their association with CpG islands. These rearrangements are accompanied by gene expression changes that promote cell proliferation. Collectively, these findings define a direct relationship between chromatin-remodeling complexes, chromatin structure, and transcriptional regulation.
In eukaryotic cells, DNA is tightly wrapped around a core of histone proteins to form nucleosomes, the basic units of chromatin structure. Because nucleosomes can impede transcription factors binding to DNA, dynamic regulation of nucleosome positioning is thought to play a critical role in transcriptional control and, in turn, numerous biological processes. Consequently, elucidating the mechanisms that modulate chromatin structure has been of great interest and has the potential to provide fundamental insight into the control of gene regulation.
Nucleosomes are assembled, modified, and repositioned with the assistance of chromatin remodeling complexes. Two broad classes of such complexes are known: those that covalently modify histones and those that use the energy of ATP hydrolysis to mobilize nucleosomes and remodel chromatin. The Swi/Snf complex was one of the first chromatin remodeling complexes to be identified, with many of its subunits conserved from yeast to humans. In mammalian cells, the Swi/Snf complex comprises 11–15 protein subunits that include SNF5 (SMARCB1) and one of the two mutually exclusive ATPases, BRG1 (SMARCA4) or BRM (SMARCA2) (1–3). The Swi/Snf complex is capable of facilitating both gene activation and repression and contributes to the regulation of lineage specificity and cell fate determination (4, 5).
Growing evidence indicates that the Swi/Snf complex serves a widespread role in tumor suppression. SNF5 was the first subunit linked to cancer and is inactivated in nearly all childhood malignant rhabdoid tumors as well as some cases of familial schwannomatosis, meningiomas, and epithelioid sarcomas (6–10). Recently, frequent and specific inactivating mutations in at least six other SWI/SNF subunits have been identified in a variety of cancers, including ARID1A, ARID1B, ARID2, PBRM1, BRD7, and BRG1 (1, 11). In mouse models, inactivation of Snf5 leads to rapid development of lethal cancers with 100% penetrance, and Brg1 haploinsufficient mice are tumor prone, establishing these subunits of the complex as bona fide tumor suppressors (1, 12–17). It is noteworthy that recent exome sequencing of 35 human SNF5-deficient rhabdoid tumors identified a remarkably low rate of mutations, with loss of SNF5 being essentially the sole recurrent event (18). Indeed, in two of the cancers, there were no other identified mutations. These results suggest that the rapid onset of cancer caused by SNF5 loss is driven not by consequent DNA damage but rather by epigenetic alterations resulting from loss of this chromatin remodeling subunit (18, 19).
Despite substantial effort in recent years, the molecular mechanisms underlying such a wide range of biological functions of Swi/Snf complex remain unclear (20, 21). In vitro studies using reconstituted nucleosomes have shown that the Swi/Snf complex can unwrap, slide, and eject nucleosomes as well as produce DNA loops on the nucleosome surface (22–24). In vivo, the complex was reported to bind preferentially to promoters and other regulatory regions (25). Interestingly, recent studies indicated that ATP-dependent chromatin remodelers are required for establishing the regular nucleosome organization at the 5′ end of genes (26). These findings suggest that Swi/Snf complex may affect transcription by mobilizing nucleosomes in promoters and altering accessibility of DNA for transcription factors. However, the extent to which it remodels nucleosomes in vivo and whether it serves any role in the establishment of the canonical nucleosome patterns are unknown.
In this study, we sought to investigate the in vivo functions of the mammalian Swi/Snf complex in the establishment and maintenance of nucleosome landscapes at transcription start sites (TSS). We generated primary mouse cells in which key subunits of the Swi/Snf complex (Snf5 or Brg1) are genetically deleted and compared nucleosome profiles in mutant and WT cells. We also mapped the locations of Swi/Snf complex in WT cells as well as examining the impact of its inactivation on gene expression. Our results show that the complex is essential for the establishment of both occupancy and phasing of the nucleosomes at a large number of promoters, and that the disruption of the canonical nucleosome patterns at TSS leads to downstream changes in gene expression.
Results
Inactivation of Swi/Snf Leads to Reduced Nucleosome Occupancy at Peri-TSS Regions.
Nucleosomal profiles at TSS of active genes in mammals consist of a nucleosome-depleted region (NDR) located immediately upstream of the TSS flanked by two well-positioned nucleosomes, referred to as the −1 and +1 nucleosomes (27–29) (Fig. 1A). Downstream of the TSS, several additional nucleosomes are also present in stably phased positions. In contrast, silent genes generally lack positioned nucleosomes at TSS, with the exception of a moderately well-positioned nucleosome at the +1 position (29). We considered three aspects of nucleosome landscape in the regions surrounding TSS (hereafter referred to as peri-TSS regions) that might be controlled by Swi/Snf (Fig. 1A). The first is the positioning of individual nucleosomes relative to the TSS, such as the precisely positioned −1 and +1 nucleosomes. The second is nucleosome occupancy, which reflects the frequency with which a nucleosome is present at a particular location within cell population. The third is the presence of regularly spaced nucleosomal arrays, referred to as nucleosome phasing, downstream of the TSS in active promoters.
Fig. 1.

Snf5- and Brg1-deficient cells display loss of nucleosome occupancy around the TSS. (A) Key parameters of the nucleosome landscape. We considered the possibility that loss of Swi/Snf could affect position (orange line), occupancy (green line), or spacing of phased nucleosomes (pink line). Genomewide nucleosome occupancy profiles at peri-TSS regions are shown for all genes (B), expressed genes (C), and silent genes (D). Profiles for WT and Snf5- and Brg1-deficient cells are shown with blue, pink, and green lines, respectively. (E) Immunoblot for nonchromatin-associated Histone H3 in WT and Snf5-deficient cells.
To examine the in vivo functions of Swi/Snf, we deleted either Snf5 or Brg1 in primary cells. We transduced murine embryonic fibroblasts (MEFs) derived from Snf5-conditional (Snf5f/f), Brg1-conditional (Brg1f/f), and control WT mice with retrovirus containing the bacterial Cre recombinase gene, which results in the deletion of the conditionally targeted genes, and achieved nearly complete elimination of Snf5 or Brg1 protein (SI Appendix, Fig. S1 A and B). We used micrococcal nuclease (MNase) digestion assays to profile nucleosome occupancy in these cells. Because promoters are enriched for easily removable, “fragile” nucleosomes (30–32), we used mild MNase digestion conditions to obtain the most informative nucleosome profiles at these regions. We isolated mononucleosomes (comprising ∼10% of entire chromatin) and sequenced the purified DNA on the Helicos platform (Materials and Methods). This platform does not require PCR amplification, thus significantly reducing the GC bias (33). This is particularly relevant for our analysis given the affinity bias of nucleosomes for GC-rich sequences (34, 35).
We observed that in WT cells, active genes display high levels of nucleosome occupancy immediately upstream of the NDR with prominent and precisely positioned −1 and +1 nucleosomes (Fig. 1B), consistent with previous reports (29). Upon Snf5 loss, nucleosome occupancy was markedly reduced across the peri-TSS region. In particular, the high nucleosome occupancy upstream of the NDR was completely abolished and the occupancy at the +1 nucleosome was substantially reduced. These effects were particularly prominent at expressed genes (Fig. 1C). Interestingly, the positions of the NDR and nucleosomes +1 through +5 remained roughly the same as in WT cells and were readily identifiable. At silent genes, the changes were similar but less pronounced (Fig. 1D). When Brg1 was inactivated, nucleosome occupancy was further depleted across the TSS (Fig. 1 B–D). The prominent −1 nucleosome position was eliminated and the occupancy of the +1 position was severely reduced such that its peak was even below baseline occupancy levels distant from the TSS. When individual genes were examined, similar effects were observed but with considerable diversity, indicating that the genes were not uniformly affected (SI Appendix, Fig. S2 A, D, and E).
We next examined whether the alteration in nucleosome occupancy was dependent upon promoter type, CpG island (CpGi)-containing versus non-CpGi, because analysis of Brg1 shRNA knock-down in macrophages concluded that CpGi-containing genes were largely Swi/Snf-independent (36). We observed that inactivation of Snf5 or Brg1 led to a marked reduction in nucleosome occupancy at both CpGi and non-CpGi promoters (SI Appendix, Fig. S3), suggesting that, at least in MEFs, the Swi/Snf complex contributed the establishment of nucleosome occupancy profiles at both types of promoters. When genes were further stratified by expression status in addition to the presence of CpGi, Swi/Snf inactivation led to reduced peri-TSS occupancy, particularly at the −1 and +1 nucleosomes in all subclasses.
We also performed a number of checks to ensure that the observed differences in the nucleosome occupancy between WT and mutant samples did not originate from experimental or data processing artifacts. In particular, to confirm that the observed defects in Snf5- and Brg1-deficient cells were not due to their inability to express histones, we measured total nonchromatin-associated histone H3 in WT and Snf5-deficient cells by immunoblotting. Snf5-deficient cells displayed increased levels of unincorporated histone H3, suggesting that impaired deposition of nucleosomes resulted in an increase in the pool of free histones (Fig. 1E). We also repeated the nucleosome profiling experiments using the Illumina platform for two concentrations of MNase (SI Appendix, Fig. S4). With paired-end sequencing, we verified that the distributions of the nucleosome fragment sizes were similar for all samples at each MNase concentration (SI Appendix, Fig. S4 A and B), which is indicative of the same level of digestion (37). We observed that the TSS profiles were similar for the WT and mutant samples in the case of the light digestion (SI Appendix, Fig. S4C). At the moderate digestion level, however, the occupancy at TSS decreased substantially in the mutant cells compared with the WT, consistent with our Helicos data (SI Appendix, Fig. S4D). Furthermore, we analyzed data from a recent independent study that evaluated the role of Brg1 in differentiation using human CD36+ cells (38) and found a similar decrease in nucleosome occupancy at TSS induced by Brg1 inactivation (SI Appendix, Fig. S4F). Based on these observations, we conclude that Swi/Snf inactivation leads to reduction of nucleosome occupancy at a substantial fraction of promoters of diverse types.
Inactivation of Swi/Snf Results in Altered Nucleosome Phasing.
Previous studies have shown that ATP-dependent chromatin remodelers can modulate the spacing between individual nucleosomes and that even subtle changes in linker lengths can substantially affect transcription factor binding (38). We sought to evaluate whether the Swi/Snf complex also contributes to the internucleosomal spacing. We found that the position of the +1 nucleosome remained constant in all cell types, but the phasing of nucleosomes downstream of the +1 position (+2, +3, and +4) was altered, with a slight shift toward the TSS (Fig. 2A). To quantify this change, we performed Fourier analysis, which is often used to quantify the changes in nucleosome phasing (39). This analysis revealed that the average internucleosomal distance of 182 bp in peri-TSS regions in WT MEFs was reduced to 174 bp in both Snf5- and Brg1-deficient cells (Fig. 2B). Thus, our results indicate that in addition to the regulation of nucleosome occupancy, the Swi/Snf complex affects nucleosome phasing in the peri-TSS region in vivo.
Fig. 2.

Snf5- and Brg1-deficient cells display altered nucleosomal phasing. (A) Alignment of WT, Snf5-deficient, and Brg1-deficient nucleosomal profiles. Thin vertical lines mark stable nucleosome positions in WT (solid lines) and Snf5 mutant cells (dotted lines). (B) Fourier analysis reveals internucleosomal distance of 182 bp in WT cells (blue line) and 174 bp in Snf5- and Brg1-deficient cells (pink and green lines, respectively).
Swi/Snf Binds to a Subset of Promoters and Controls Nucleosome Occupancy.
Given the marked effect of Snf5 and Brg1 loss on the nucleosome landscape at promoters, we sought to determine the localization of Swi/Snf and to classify genes based upon the degree of Swi/Snf binding. We performed Brg1 ChIP combined with MNase digestion to achieve nucleosome-resolution mapping of the complex (40). Consistent with a prior report (25), Brg1 preferentially bound promoter regions and the level of binding was positively correlated with expression level of the target genes (R = 0.55, Fig. 3A). Although binding was greatest at the +1 nucleosome position, we found that Brg1 was also enriched at the NDR.
Fig. 3.

Brg1 ChIP reveals enrichment of Swi/Snf at peri-TSS regions. (A) Brg1 binding is prominent at TSS. Results are shown for all (black), expressed (red), and silent (blue) genes. (B) Brg1 enrichment in the TSS region (+/−2 kb) displays a bimodal distribution. The red line indicates the threshold of Brg1 enrichment used to identify groups of genes with low and high Brg1 levels. (C and D) Nucleosomal occupancy profiles for Brg1 high and Brg1 low genes.
Across the genome, Brg1 enrichment at peri-TSS regions in WT cells exhibited a bimodal distribution, indicating clear binding at a subset of promoters and low or no binding at the rest (Fig. 3B). For the subset of genes with high Brg1 enrichment, the nucleosome profile displayed prominent occupancy at the −1 and +1 positions, a pronounced NDR and readily identifiable nucleosome phasing (Fig. 3C). In contrast, the average occupancy profile for genes with low Brg1 enrichment was nearly flat, except for a slight increase at the +1 position (Fig. 3D). The two groups also differed in composition: genes with high levels of Brg1 binding were enriched for CpGs, whereas the majority of genes with low levels of Brg1 binding lacked CpGs (75% vs. 20%; SI Appendix, Table S1). Furthermore, although the loss of Brg1 led to reduced nucleosome occupancy at TSS region in both groups, we found that higher levels of Brg1 enrichment indeed correlated with a greater degree of nucleosome depletion following Brg1 deletion (R = −0.32, P < 10−32; SI Appendix, Figs. S2 B and C and S5). Collectively, these findings strongly suggest that the loss of nucleosome occupancy at TSS in the mutant cells is a direct rather than secondary effect of Swi/Snf inactivation.
Effect of Snf5/Brg1 Inactivation on Gene Expression.
To determine how changes in nucleosome occupancy driven by inactivation of Snf5 or Brg1 affect gene expression, we profiled mRNAs from the same samples on Affymetrix microarrays. Overall, changes in gene expression were relatively modest and variable in magnitude and direction for individual genes, and most genes had similar expression levels in mutant and WT cells. However, a subset of genes displayed altered expression (SI Appendix, Fig. S6 A and B), with more genes being significantly up-regulated than down-regulated (793 vs. 491 following Snf5 loss, and 1,226 vs. 782 genes following Brg1 loss; P < 0.01 in both cases, Fisher’s exact test). There was a high degree of overlap between the genes affected by Snf5 loss and those affected by Brg1 loss (Table 1 and SI Appendix, Figs. S7 and S8 A–D), suggesting similar but not identical impact in the two cases. Overall, we observed a low but statistically significant negative correlation between gene expression and level of nucleosome occupancy at the NDR in both WT and mutant MEFs (SI Appendix, Fig. S9 A–C). When we examined the relationship between changes in nucleosome occupancy and changes in gene expression caused by Snf5 or Brg1 loss, there was only a modest association between greater increase in nucleosome occupancy with stronger gene down-regulation and vice versa (SI Appendix, Figs. S8 E–H and S9 D and E). These findings suggest that the average transcription rate at the majority of genes is maintained even when nucleosome occupancy is altered around the TSS. It is noteworthy that similar findings have been made recently in yeast, where inactivation of related chromatin remodelers revealed that changes in nucleosome organization did not correlate with changes in transcription (41).
Table 1.
Relationship between the sets of genes up- or down-regulated in the absence of Snf5 or Brg1
| Gene set | Brg1 up | Brg1 no change | Brg1 down |
| Snf5 up | 510 | 272 | 6 |
| Snf5 no change | 710 | 10,410 | 454 |
| Snf5 down | 1 | 168 | 322 |
Numbers of genes in each overlapping group are shown. The table cells corresponding to up- and down-regulated genes are shaded in pink and blue, respectively.
To examine whether there existed specific features of nucleosome organization that may play a role in regulating gene expression, we examined the relative nucleosome density at different positions near the TSS, which has been shown to correlate with transcription rate (42, 43). For example, a high level of transcription correlates with high relative occupancy at the −1 and +1 positions accompanied by low occupancy at the NDR. To quantify such effects, we calculated a “relative occupancy score” by subtracting occupancy at the NDR from occupancy at the −1 and +1 positions and normalizing it by the baseline tag density for each TSS (Fig. 4A; Materials and Methods). In WT cells, higher relative occupancy scores were found in genes that had higher expression levels. These include gene sets that have CpGs in their promoters as well as those having high Brg1 binding (SI Appendix, Fig. S10A). This finding confirms that a pronounced pattern of NDR flanked by two positioned nucleosomes (high score) correlates with high level of expression.
Fig. 4.
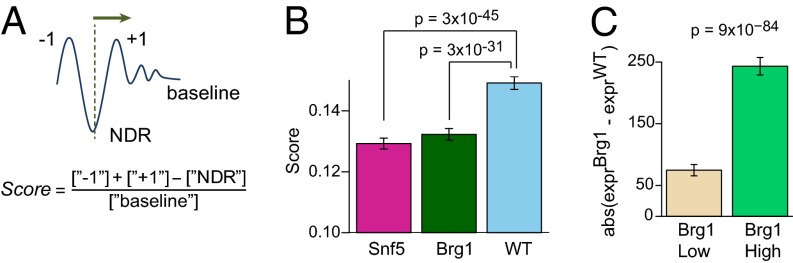
Effects of nucleosome occupancy and Brg1 binding on transcription. (A) Schematic illustration of the relative nucleosome occupancy score definition. (B) Comparison of the score in WT (blue), Snf5 (pink), and Brg1 (green) samples. (C) Average absolute changes in gene expression upon Brg1 inactivation are compared for genes with low and high Brg1 enrichment. The 95% confidence intervals and P values are indicated.
For the Snf5−/− and Brg1−/− samples, a significant reduction in the score was observed compared with the WT samples (Fig. 4B, P < 10−30). This suggests a complex relationship with two seemingly opposing effects of Swi/Snf inactivation. On the one hand, subunit inactivation results in a decrease in overall nucleosome occupancy at the TSS (Fig. 1), which would be predicted to correlate with up-regulation. On the other hand, it results in a reduction in relative occupancy at the −1 and +1 nucleosomes (Fig. 4), which would be predicted to correlate with down-regulation. Moreover, although actively expressed genes exhibit significantly higher scores than silent genes on average (SI Appendix, Fig. S10A), changes in the relative and absolute occupancy at TSS were poorly correlated at the individual gene level (|R| < 0.1 for both mutant samples; SI Appendix, Fig. S10 B and C). In facilitating coordinated gene expression responses such as during differentiation, the Swi/Snf complex has been shown capable of interacting with both coactivators and corepressors. Consequently, expression changes at individual genes may depend upon whether the gene is normally bound predominantly by activators or repressors.
Because the effect of Brg1 inactivation might be either activating or repressive, we next evaluated whether enrichment in Brg1 correlates with the magnitude of change in gene expression without accounting for the direction of this change. We observed that the genes with high Brg1 enrichment displayed significantly larger absolute change in expression than genes with low levels of Brg1 enrichment (Fig. 4C). In comparison, when direction of the expression change was considered, this difference was considerably reduced (SI Appendix, Fig. S10D). Although modest in magnitude, the mean change was again significantly greater than zero (P < 0.023) for genes highly enriched for Brg1, indicating that Brg1 loss slightly favored activation over repression, consistent with the overall effects we identified on gene expression.
Functions Regulated by Swi/Snf.
Identification of the genes that are regulated by Swi/Snf in MEFs allowed us to investigate whether specific cellular functions or pathways were affected by Swi/Snf mutation. Genes up-regulated in the absence of Snf5 demonstrated enrichment for four Gene Ontology (GO) sets (P < 0.01): cell-cycle process, cell-cycle phase, mitotic cell cycle, and cell division (SI Appendix, Table S2). Brg1 loss yielded a larger number of up-regulated gene sets, but the same theme emerged, with the top 5 and 21 of the 67 gene sets specific for cell-cycle progression. For the genes down-regulated following Snf5 loss, no annotated gene sets were significant. For Brg1 loss, down-regulated genes were significantly enriched for 20 gene sets, several of which were associated with extracellular interactions and motility. Thus, our findings indicate that the loss of Swi/Snf complex primarily results in stimulation of cell proliferation pathways, consistent with the links between Swi/Snf mutation and tumorigenesis.
Discussion
Our data demonstrate that the Swi/Snf complex serves specific roles in vivo in establishing and defining the nucleosome landscape at target promoters. Specifically, we find that the complex is highly enriched not only at the −1 and +1 positions, but also over the NDR where there is a paucity of nucleosomes. We further show that the complex is essential for broad establishment of high levels of nucleosome occupancy across target promoters relative to their flanking regions.
In principle, there is a possibility that the complex affects nucleosome occupancy at TSS through secondary effects (e.g., mediated by expression changes). However, our observation of strong correlation between the presence of the complex and its effect on nucleosome occupancy at promoters (Fig. 3) as well as high similarity of the differentially expressed gene lists in Snf5- and Brg1-deficient cells (Table 1 and SI Appendix, Figs. S7 and S8) argue in favor of the direct involvement of the complex. Consequently, activity of the complex is quite specific in that it serves to sculpt the landscape at active promoters by building occupancy at the −1 and +1 positions while ensuring relative depletion of nucleosomes at the NDR to establish the classic high peaks and low trough landscape (Fig. 5). Our study is focused upon nucleosomes easily released with MNase; thus, changes in chromatin structure that decrease accessibility or increase stability of nucleosomes in peri-TSS regions could, in principle, contribute to the observed differences. Although we cannot completely exclude at least a partial contribution of such a mechanism, this scenario seems unlikely given the positive correlation between the observed effect and transcription activity (SI Appendix, Fig. S9 D and E). We also note that the sequencing depth achieved in this study allows making confident conclusions about influence of Swi/Snf complex on average for a large set of genes but not for individual genes; variability of the Swi/Snf effects at the individual gene level may be substantial.
Fig. 5.

Swi/Snf sculpts the promoter-associated nucleosome landscape. Swi/Snf functions to establish the nucleosome landscape at the peri-TSS region by promoting occupancy at the −1 and +1 positions while ensuring relative depletion at the NDR.
Nucleosome sliding in cis and nucleosome transfer have been identified as the main or sole activities of the Swi/Snf complex in a number of studies (23, 44–46). Importantly, despite the ability of the complex to slide nucleosomes in vitro, our data demonstrate that it appears to have no role in establishing the positions of the major TSS-flanking −1 and +1 nucleosomes. Although deletion of the complex subunits results in reduction of occupancy at these positions, the peaks remain sharp, indicating that the residual nucleosomes are well positioned and unchanged in location. Given the overall reduction in occupancy, these findings suggest that transfer of nucleosomes onto DNA is the principal mechanism of action for the Swi/Snf complex at these positions. However, downstream of the +1 position, Swi/Snf is likely to contribute to the phasing via a sliding activity and/or affecting “statistical” nucleosome positioning (27), as evidenced by subtle alterations in nucleosome spacing there. This is also supported by a recent study showing that the yeast Swi/Snf complex can bind and shift promoter nucleosomes away from the TSS (47), although no pronounced loss of nucleosome occupancy upon Swi/Snf perturbation was reported there. Whether this reflects a functional difference between yeast and mammalian Swi/Snf complexes is unclear. Further studies are warranted to address this question. In addition, SWI/SNF effects may vary at individual targets or under specific conditions. For example, following recruitment by an activator, Swi/Snf was found to be required for a nucleosome sliding event that blocks transcription from the IFN-β promoter upon viral infection (48). Collectively, our data demonstrate a central role for the Swi/Snf complex both in depositing nucleosomes and in shifting the peri-TSS nucleosomes, either directly or via interaction with other chromatin remodelers such as those from the Imitation Switch (ISWI) family, which were shown to have nucleosome sliding activity in vivo (41, 47).
Although our studies were primarily focused on the effect of the Swi/Snf complex at promoters, several lines of evidence suggest that the complex may also have roles outside the promoters. First, although Swi/Snf binding was markedly enriched at promoters, we found that binding also occurred in gene bodies—and to some extent in intergenic regions as well—consistent with a prior report (25). Second, the total level of unincorporated histone H3 was increased in Snf5-deficient cells. Finally, we noted a decrease in baseline levels of nucleosome occupancy that occurred even at substantial distances from the TSS. Accordingly, Swi/Snf may contribute to the establishment of nucleosome occupancy outside the promoters.
Preferential binding of transcription factors at open chromatin regions of active promoters has fostered the notion that reduced nucleosome occupancy leads to increased transcription rate (49). Indeed, we observed negative correlation of nucleosome density at TSS, and at the NDR in particular, with gene expression level (SI Appendix, Fig. S9 A–C). However, we found that there is great variability in this relationship even in WT cells (SI Appendix, Fig. S2A), consistent with the variable effect on the expression of individual genes that result from a widespread loss of nucleosome occupancy in Swi/Snf mutant cells. We speculate that although reduced occupancy at the NDR may favor factor binding, the reduced prominence of the −1 and +1 nucleosomes may impinge upon recognition of promoter sites and impair factor recruitment. Overall, these findings are reminiscent of transcriptional regulation in yeast where deletion of the ISWI and chromodomain-helicase-DNA-binding protein 1 (CHD1) chromatin remodelers results in substantial changes in chromatin structure that do not directly correlate with changes in transcription rate (41). We note that Swi/Snf, which is broadly present at many promoters, can facilitate binding of lineage specification factors, which include both transcriptional activators and repressors. We speculate that defects in this regulation caused by SWI/SNF subunit mutation may contribute to the specificity of cancer types associated to some degree with mutation of individual subunits. Further, because some Swi/Snf subunits are expressed in a restricted manner, the activity of the complex itself may be controlled in a lineage-specific fashion.
Although the Swi/Snf complex is one of the most studied chromatin-remodeling complexes, the mechanism by which it controls nucleosome organization and function of individual subunits has not been well understood. Previous studies have provided some clues to the relationship between the core subunits BRG1 and SNF5. First, BRG1 does not require SNF5 to remodel nucleosomes in vitro, although yeast SNF5 has been shown to interact with activators (50), raising the possibility that SNF5 could contribute to targeting in vivo. Second, Brg1+/− mice display a different spectrum of tumors than Snf5+/− mice. Third, Brg1 is essential in Snf5-deficient cancers because its inactivation both halts proliferation of Snf5-deficient cancer cell lines and blocks tumor formation in Snf5-conditional mice (51). Together, these observations raise two possibilities. Snf5 could be an antagonist of Brg1 required to modulate its activity such that loss of Snf5 results in hyperactivity of the residual Brg1-containing complex. Alternatively, the functional effects of Snf5 and Brg1 loss could be similar such that loss of either one substantially impairs, but does not eliminate, activity of the residual complex. In the latter scenario, some residual activity of the complex may be essential so that subsequent loss of a second subunit is not compatible with cell survival. Our data are consistent only with the latter scenario. Although Brg1 loss resulted in a more prominent effect on nucleosome occupancy and altered the expression of more genes, we found that the functional roles of Snf5 and Brg1 are highly similar in the control of gene expression. Consequently, we speculate that therapeutic targets for Snf5-deficient cancers may well be effective against Brg1-deficient cancers and vice versa.
Our analyses of GO terms demonstrate that, despite the fact that MEFs undergo cell-cycle arrest following inactivation of either Snf5 or Brg1, loss of these subunits promotes cell-cycle progression. Thus, the aberrant proliferative drive caused by Swi/Snf mutation may trigger arrest at a cell-cycle checkpoint. Consistent with this, we previously found that inactivation of p53 dramatically accelerates the onset of cancers caused by Snf5 inactivation (52). Consistent with its role as a potent tumor suppressor, our data thus support Swi/Snf as a key regulator of cellular proliferation and that its loss stimulates activation of proliferation-associated genes. Further, at least in the case of Brg1, its loss may lead to impaired sensing of intercellular signaling and disrupted control of migration.
Our findings demonstrate a central role for the Swi/Snf complex in the precise control of gene expression by sculpting the nucleosomal landscape at promoters. The previously suggested role of Swi/Snf was mostly linked to nucleosome mobilization and thus gene activation. Our findings show that on a genome scale, the complex affects nucleosome landscape in an intricate way, slightly favoring gene silencing. Taken together, our data provide mechanistic insights into both the fundamental control of transcription and the mechanism of action of an epigenetic tumor suppressor.
Materials and Methods
Cell Culture.
Primary MEFs were harvested from embryonic day 13.5 embryos. Cre was introduced into cells via retroviral infection two times at 4-h intervals. Cells were stably selected in medium containing puromycin (2.5 µg/mL) 48 h after infection.
Preparation of Mononucleosomes.
WT, Snf5-deficient, or Brg1-deficient cells were harvested, washed in PBS, and resuspended in 2 mL low detergent buffer [0.3 M sucrose, 15 mM Tris⋅Cl (pH 7.6), 60 mM KCl, 15 mM NaCl, 5 mM MgCl2, 0.1 mM EDTA, 0.5 mM DTT, 0.4% Nonidet P-40] for 10 min to lyse cells. MEFs from multiple embryos were pooled. The cell solution was layered on 8 mL high sucrose buffer [1.2 M sucrose, 60 mM KCl, 15 mM NaCl, 5 mM MgCl2, 0.1 mM EDTA, 0.5 mM DTT] and spun at 10,000 × g to pellet nuclei. Isolated nuclei were resuspended in 1 mL micrococcal nuclease (MNase) buffer [0.32 sucrose, 50 mM Tris⋅Cl (pH 7.6), 4 mM MgCl2, 1 mM CaCl2] and digested with MNase (Worthington Biochemical). For the WT sample, 9.4 × 107 cells were digested; for the Snf5 sample, 5.9 × 107 cells were digested; and for the Brg1 sample, 6.4 × 107 cells were digested. All digestions were performed with MNase concentration of 15 U/mL DNA fragments (about 150 bp) were RNase-treated, precipitated, gel-extracted, end-repaired, and submitted for Helicos sequencing. Chromatin from MEFs of all genotypes (∼4 million cells/genotype) was also digested with MNase at two different concentrations (0.2 U or 4 U per 100-μL reaction volume) for sequencing on the Illumina platform. The mononucleosomal DNA was then isolated, end-repaired, and treated with Taq polymerase to generate a protruding A base for adaptor ligation. After ligation of a pair of Illumina adaptors, the DNA was size selected and amplified by PCR using the adaptor primers. The amplified DNA was then purified and used for cluster generation and sequencing.
Histone Solubility.
To evaluate total nonchromatin-associated histone H3 in WT and Snf5-deficient cells, equal numbers of nuclei were isolated by centrifugation through a sucrose gradient (as described in the preparation of mononucleosomes). Nuclei were then resuspended for 10 min in ice-cold Buffer 4 (0.32 M sucrose, 50 mM Tris pH 7.6, 4 mM MgCl2, 1 mM CaCl2, protease inhibitors) containing a final concentration of 1% (vol/vol) Nonidet P-40. The extract was then spun at 10,000 × g in a refrigerated microcentrifuge. Supernatants containing nonchromatin-associated proteins were run on SDS/PAGE and immunoblotted for Histone H3 (Abcam), Snf5 (Bethyl), and Actin (Sigma).
Brg1 ChIP.
Nuclei from WT MEFs were isolated and subjected to micrococcal nuclease digestion followed by brief sonication to release nucleosomal fragments. Brg1 ChIP was performed on the lysate and immunoprecipitated DNA was purified and sequenced.
Data Analysis.
A detailed description of the computational methods is provided in the SI Appendix. In brief, sequenced tags were mapped to the mm9 assembly of the mouse genome. Tags were filtered for possible artifacts before further analyses. The final tag counts for the data sets used in this study are summarized in SI Appendix, Table S3. The coordinates of genes and CpGs were taken from the University of California Santa Cruz annotations. Gene expression data were generated on GeneChip Mouse Genome 430A 2.0 Arrays (Affymetrix) and processed using the MAS 5.0 algorithm as implemented in the bioconductor package Affy (www.bioconductor.org). Different background correction and normalization methods were explored (e.g., robust multiarray analysis, quantile normalization) to ensure robustness of the results. The Gene Ontology Term Finder Web server (http://go.princeton.edu/cgi-bin/GOTermFinder/GOTermFinder) was used for gene ontology analysis.
Supplementary Material
Acknowledgments
We thank Xi Wang for critical review of the manuscript and Lee Whale for the artwork in Figs. 1A and 5. This work was supported in part by a Ruth L. Kirschstein National Research Service Award Fellowship 5F32 CA123776 from the National Cancer Institute (to C.G.S.), U01HG004258 (to M.Y.T.), R01GM082798 (to P.J.P.), R01CA113794 (to C.W.M.R.); an Innovative Research Grant from Stand Up 2 Cancer (to C.W.M.R.); and a U01 NCI Mouse Models of Cancer Consortium Award (to C.W.M.R.). The Sloan Research Fellowship (P.J.P.), Garrett B. Smith Foundation, Miles for Mary, and the foundation Cure AT/RT Now (C.W.M.R.) provided additional support.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The data reported in this paper have been deposited in the Gene Expression Omnibus (GEO) database, www.ncbi.nlm.nih.gov/geo (accession no. GSE46588).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1302209110/-/DCSupplemental.
References
- 1.Wilson BG, Roberts CW. SWI/SNF nucleosome remodellers and cancer. Nat Rev Cancer. 2011;11(7):481–492. doi: 10.1038/nrc3068. [DOI] [PubMed] [Google Scholar]
- 2.Reisman D, Glaros S, Thompson EA. The SWI/SNF complex and cancer. Oncogene. 2009;28(14):1653–1668. doi: 10.1038/onc.2009.4. [DOI] [PubMed] [Google Scholar]
- 3.Weissman B, Knudsen KE. Hijacking the chromatin remodeling machinery: Impact of SWI/SNF perturbations in cancer. Cancer Res. 2009;69(21):8223–8230. doi: 10.1158/0008-5472.CAN-09-2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Narlikar GJ, Fan HY, Kingston RE. Cooperation between complexes that regulate chromatin structure and transcription. Cell. 2002;108(4):475–487. doi: 10.1016/s0092-8674(02)00654-2. [DOI] [PubMed] [Google Scholar]
- 5.Martens JA, Winston F. Recent advances in understanding chromatin remodeling by Swi/Snf complexes. Curr Opin Genet Dev. 2003;13(2):136–142. doi: 10.1016/s0959-437x(03)00022-4. [DOI] [PubMed] [Google Scholar]
- 6.Biegel JA, et al. Germ-line and acquired mutations of INI1 in atypical teratoid and rhabdoid tumors. Cancer Res. 1999;59(1):74–79. [PubMed] [Google Scholar]
- 7.Versteege I, et al. Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature. 1998;394(6689):203–206. doi: 10.1038/28212. [DOI] [PubMed] [Google Scholar]
- 8.Hulsebos TJ, et al. Germline mutation of INI1/SMARCB1 in familial schwannomatosis. Am J Hum Genet. 2007;80(4):805–810. doi: 10.1086/513207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Christiaans I, et al. Germline SMARCB1 mutation and somatic NF2 mutations in familial multiple meningiomas. J Med Genet. 2011;48(2):93–97. doi: 10.1136/jmg.2010.082420. [DOI] [PubMed] [Google Scholar]
- 10.Modena P, et al. SMARCB1/INI1 tumor suppressor gene is frequently inactivated in epithelioid sarcomas. Cancer Res. 2005;65(10):4012–4019. doi: 10.1158/0008-5472.CAN-04-3050. [DOI] [PubMed] [Google Scholar]
- 11.Garraway LA, Lander ES. Lessons from the cancer genome. Cell. 2013;153(1):17–37. doi: 10.1016/j.cell.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 12.Bultman SJ, et al. Characterization of mammary tumors from Brg1 heterozygous mice. Oncogene. 2008;27(4):460–468. doi: 10.1038/sj.onc.1210664. [DOI] [PubMed] [Google Scholar]
- 13.Roberts CW, Galusha SA, McMenamin ME, Fletcher CD, Orkin SH. Haploinsufficiency of Snf5 (integrase interactor 1) predisposes to malignant rhabdoid tumors in mice. Proc Natl Acad Sci USA. 2000;97(25):13796–13800. doi: 10.1073/pnas.250492697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roberts CW, Leroux MM, Fleming MD, Orkin SH. Highly penetrant, rapid tumorigenesis through conditional inversion of the tumor suppressor gene Snf5. Cancer Cell. 2002;2(5):415–425. doi: 10.1016/s1535-6108(02)00185-x. [DOI] [PubMed] [Google Scholar]
- 15.Klochendler-Yeivin A, et al. The murine SNF5/INI1 chromatin remodeling factor is essential for embryonic development and tumor suppression. EMBO Rep. 2000;1(6):500–506. doi: 10.1093/embo-reports/kvd129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guidi CJ, et al. Disruption of Ini1 leads to peri-implantation lethality and tumorigenesis in mice. Mol Cell Biol. 2001;21(10):3598–3603. doi: 10.1128/MCB.21.10.3598-3603.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tsikitis M, Zhang Z, Edelman W, Zagzag D, Kalpana GV. Genetic ablation of Cyclin D1 abrogates genesis of rhabdoid tumors resulting from Ini1 loss. Proc Natl Acad Sci USA. 2005;102(34):12129–12134. doi: 10.1073/pnas.0505300102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee RS, et al. A remarkably simple genome underlies highly malignant pediatric rhabdoid cancers. J Clin Invest. 2012;122(8):2983–2988. doi: 10.1172/JCI64400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McKenna ES, et al. Loss of the epigenetic tumor suppressor SNF5 leads to cancer without genomic instability. Mol Cell Biol. 2008;28(20):6223–6233. doi: 10.1128/MCB.00658-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Clapier CR, Cairns BR. The biology of chromatin remodeling complexes. Annu Rev Biochem. 2009;78:273–304. doi: 10.1146/annurev.biochem.77.062706.153223. [DOI] [PubMed] [Google Scholar]
- 21.Liu N, Balliano A, Hayes JJ. Mechanism(s) of SWI/SNF-induced nucleosome mobilization. ChemBioChem. 2011;12(2):196–204. doi: 10.1002/cbic.201000455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lorch Y, Maier-Davis B, Kornberg RD. Mechanism of chromatin remodeling. Proc Natl Acad Sci USA. 2010;107(8):3458–3462. doi: 10.1073/pnas.1000398107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kassabov SR, Zhang B, Persinger J, Bartholomew B. SWI/SNF unwraps, slides, and rewraps the nucleosome. Mol Cell. 2003;11(2):391–403. doi: 10.1016/s1097-2765(03)00039-x. [DOI] [PubMed] [Google Scholar]
- 24.Dechassa ML, et al. SWI/SNF has intrinsic nucleosome disassembly activity that is dependent on adjacent nucleosomes. Mol Cell. 2010;38(4):590–602. doi: 10.1016/j.molcel.2010.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ho L, et al. An embryonic stem cell chromatin remodeling complex, esBAF, is essential for embryonic stem cell self-renewal and pluripotency. Proc Natl Acad Sci USA. 2009;106(13):5181–5186. doi: 10.1073/pnas.0812889106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang Z, et al. A packing mechanism for nucleosome organization reconstituted across a eukaryotic genome. Science. 2011;332(6032):977–980. doi: 10.1126/science.1200508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jiang C, Pugh BF. Nucleosome positioning and gene regulation: Advances through genomics. Nat Rev Genet. 2009;10(3):161–172. doi: 10.1038/nrg2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hartley PD, Madhani HD. Mechanisms that specify promoter nucleosome location and identity. Cell. 2009;137(3):445–458. doi: 10.1016/j.cell.2009.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schones DE, et al. Dynamic regulation of nucleosome positioning in the human genome. Cell. 2008;132(5):887–898. doi: 10.1016/j.cell.2008.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Henikoff S, Henikoff JG, Sakai A, Loeb GB, Ahmad K. Genome-wide profiling of salt fractions maps physical properties of chromatin. Genome Res. 2009;19(3):460–469. doi: 10.1101/gr.087619.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weiner A, Hughes A, Yassour M, Rando OJ, Friedman N. High-resolution nucleosome mapping reveals transcription-dependent promoter packaging. Genome Res. 2010;20(1):90–100. doi: 10.1101/gr.098509.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xi Y, Yao J, Chen R, Li W, He X. Nucleosome fragility reveals novel functional states of chromatin and poises genes for activation. Genome Res. 2011;21(5):718–724. doi: 10.1101/gr.117101.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Goren A, et al. Chromatin profiling by directly sequencing small quantities of immunoprecipitated DNA. Nat Methods. 2010;7(1):47–49. doi: 10.1038/nmeth.1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Peckham HE, et al. Nucleosome positioning signals in genomic DNA. Genome Res. 2007;17(8):1170–1177. doi: 10.1101/gr.6101007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kharchenko PV, Woo CJ, Tolstorukov MY, Kingston RE, Park PJ. Nucleosome positioning in human HOX gene clusters. Genome Res. 2008;18(10):1554–1561. doi: 10.1101/gr.075952.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ramirez-Carrozzi VR, et al. A unifying model for the selective regulation of inducible transcription by CpG islands and nucleosome remodeling. Cell. 2009;138(1):114–128. doi: 10.1016/j.cell.2009.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Johnson SM, Tan FJ, McCullough HL, Riordan DP, Fire AZ. Flexibility and constraint in the nucleosome core landscape of Caenorhabditis elegans chromatin. Genome Res. 2006;16(12):1505–1516. doi: 10.1101/gr.5560806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hu G, et al. Regulation of nucleosome landscape and transcription factor targeting at tissue-specific enhancers by BRG1. Genome Res. 2011;21(10):1650–1658. doi: 10.1101/gr.121145.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang J, et al. Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors. Genome Res. 2012;22(9):1798–1812. doi: 10.1101/gr.139105.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.He HH, et al. Nucleosome dynamics define transcriptional enhancers. Nat Genet. 2010;42(4):343–347. doi: 10.1038/ng.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gkikopoulos T, et al. A role for Snf2-related nucleosome-spacing enzymes in genome-wide nucleosome organization. Science. 2011;333(6050):1758–1760. doi: 10.1126/science.1206097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tirosh I, Barkai N. Two strategies for gene regulation by promoter nucleosomes. Genome Res. 2008;18(7):1084–1091. doi: 10.1101/gr.076059.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Batta K, Zhang Z, Yen K, Goffman DB, Pugh BF. Genome-wide function of H2B ubiquitylation in promoter and genic regions. Genes Dev. 2011;25(21):2254–2265. doi: 10.1101/gad.177238.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Whitehouse I, et al. Nucleosome mobilization catalysed by the yeast SWI/SNF complex. Nature. 1999;400(6746):784–787. doi: 10.1038/23506. [DOI] [PubMed] [Google Scholar]
- 45.Gutiérrez JL, Chandy M, Carrozza MJ, Workman JL. Activation domains drive nucleosome eviction by SWI/SNF. EMBO J. 2007;26(3):730–740. doi: 10.1038/sj.emboj.7601524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lorch Y, Cairns BR, Zhang M, Kornberg RD. Activated RSC-nucleosome complex and persistently altered form of the nucleosome. Cell. 1998;94(1):29–34. doi: 10.1016/s0092-8674(00)81218-0. [DOI] [PubMed] [Google Scholar]
- 47.Yen K, Vinayachandran V, Batta K, Koerber RT, Pugh BF. Genome-wide nucleosome specificity and directionality of chromatin remodelers. Cell. 2012;149(7):1461–1473. doi: 10.1016/j.cell.2012.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lomvardas S, Thanos D. Nucleosome sliding via TBP DNA binding in vivo. Cell. 2001;106(6):685–696. doi: 10.1016/s0092-8674(01)00490-1. [DOI] [PubMed] [Google Scholar]
- 49.Thurman RE, et al. The accessible chromatin landscape of the human genome. Nature. 2012;489(7414):75–82. doi: 10.1038/nature11232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Prochasson P, Neely KE, Hassan AH, Li B, Workman JL. Targeting activity is required for SWI/SNF function in vivo and is accomplished through two partially redundant activator-interaction domains. Mol Cell. 2003;12(4):983–990. doi: 10.1016/s1097-2765(03)00366-6. [DOI] [PubMed] [Google Scholar]
- 51.Wang X, et al. Oncogenesis caused by loss of the SNF5 tumor suppressor is dependent on activity of BRG1, the ATPase of the SWI/SNF chromatin remodeling complex. Cancer Res. 2009;69(20):8094–8101. doi: 10.1158/0008-5472.CAN-09-0733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Isakoff MS, et al. Inactivation of the Snf5 tumor suppressor stimulates cell cycle progression and cooperates with p53 loss in oncogenic transformation. Proc Natl Acad Sci USA. 2005;102(49):17745–17750. doi: 10.1073/pnas.0509014102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.