

Abstract
With the rapidly growing wealth of genomic data, experimental inquiries on the functional significance of important divergence sites in protein evolution are becoming more accessible. Here we trace the evolution of dihydrofolate reductase (DHFR) and identify multiple key divergence sites among 233 species between humans and bacteria. We connect these sites, experimentally and computationally, to changes in the enzyme’s binding properties and catalytic efficiency. One of the identified evolutionarily important sites is the N23PP modification (∼mid-Devonian, 415–385 Mya), which alters the conformational states of the active site loop in Escherichia coli dihydrofolate reductase and negatively impacts catalysis. This enzyme activity was restored with the inclusion of an evolutionarily significant lid domain (G51PEKN in E. coli enzyme; ∼2.4 Gya). Guided by this evolutionary genomic analysis, we generated a human-like E. coli dihydrofolate reductase variant through three simple mutations despite only 26% sequence identity between native human and E. coli DHFRs. Molecular dynamics simulations indicate that the overall conformational motions of the protein within a common scaffold are retained throughout evolution, although subtle changes to the equilibrium conformational sampling altered the free energy barrier of the enzymatic reaction in some cases. The data presented here provide a glimpse into the evolutionary trajectory of functional DHFR through its protein sequence space that lead to the diverged binding and catalytic properties of the E. coli and human enzymes.
Keywords: phylogenetic, EVB
Enzymes are responsible for greatly accelerating chemical transformations that are critical for all cellular processes (1). Thus, it is not surprising that the evolutionary divergence of homologous proteins is highly regulated/restricted (2), where divergences occur slowly through a rugged protein fitness landscape imposed by Darwinian pressure (3, 4). However, the ability to identify key discrepancies among homologous enzymes from different organisms has the practical consequences of suggesting how to selectively modulate enzyme activities (e.g., antibacterial and antifungal applications) for pharmaceutical purposes. Here we take advantage of the wealth of sequence data available for dihydrofolate reductase (DHFR) to explore evolutionarily significant divergences in DHFR and determine how these changes affect the properties of the enzyme.
DHFR is a ubiquitous enzyme that catalyzes the NADPH-dependent conversion of 7,8-dihydrofolate (DHF) to 5,6,7,8-tetrahydrofolate (THF) (5), which is involved in subsequent metabolic reactions such as thymidylate and purine nucleotide biosynthesis. Because of its biological role, DHFR is an important therapeutic target for anticancer and antibacterial drugs (6). One of the inhibitors of DHFR is trimethoprim (TMP), which exhibits potent antibacterial properties because of its heavily biased selectivity toward bacterial DHFRs over mammalian DHFRs (6, 7). Aside from trimethoprim inhibition, there are many catalytic/kinetic discrepancies between the human (8, 9) and the Escherichia coli (5) enzymes. These differences in the behaviors between the human (hsDHFR) and the E. coli (ecDHFR) enzymes are likely consequences of important divergences through countless rounds of evolutionary selection. ecDHFR has low primary sequence agreement with the human enzyme (26% identity alignment shown in SI Appendix), yet a common overall structural scaffold has been retained over the billions of years since divergence. In view of the trillions of generations that E. coli has undergone since its divergence from human (10), the 26% identity may represent a floor to the divergence possible with retention of structure and function. All divergence nodes of species living today are represented by multiple determined sequences. Although these sequences are not ancestral themselves, at conserved sites we can reliably infer the ancestral sequence for each node. This gives a sequence time series during which DHFR was always functional (because it is an essential enzyme). However, the binding and the catalytic properties of DHFR have diverged over time because E. coli and human DHFRs are quite different today despite their common ancestry. It is not clear whether the observed differences in enzymatic properties in contemporary DHFRs arose gradually from the cumulative effect of numerous near-neutral point substitutions or are better attributed to a few major events that presumably provided selective advantage to the affected lineage.
We devised strict evolutionary criteria for analyzing DHFR amino acid sequences from 233 species ranging from human to bacteria and found three evolutionarily important sequence divergent sites, defined herein as phylogenetically coherent events (PCEs). Next we experimentally probed the catalytic consequences of the different PCEs as they were introduced into WT ecDHFR. Empirical valence bond (EVB) molecular dynamics (MD) simulations (11) were able to reproduce the kinetic data of various PCEs and provided further comparisons among ecDHFR, hsDHFR, and the ecDHFR variants with PCE components artificially added. One of the identified PCEs drastically altered the native ecDHFR’s binding affinity to its cofactor NADPH, product NADP+, and TMP to more closely resemble its human counterpart. Guided by these results, we then were able to engineer a human-like ecDHFR variant by introducing these three PCEs into native ecDHFR through mutagenesis.
Results and Discussion
Evolutionary Analysis: Phylogenetically Coherent Events.
DHFR is an attractive target for this demonstration because of the wealth of sequence, kinetics, and structural data of the enzyme from various organisms that covers a large segment of evolutionary time. We analyzed DHFR amino acid sequences from 233 species (99 vertebrates and 14 bacteria) ranging from humans to bacteria to identify evolutionarily important modifications. We first exhaustively searched GenBank, manually curating proteins orthologous to hsDHFR, emphasizing multiple representatives for each divergence node of the current consensus phylogenetic tree to mitigate errors in individual sequencing projects (12). Fig. 1 shows a subset aligned in phylogenetic divergence order from human to E. coli (genus species abbreviations and the full set of full-length sequences are provided at http://genomewiki.ucsc.edu/index.php/DHFR_dihydrofolate). The ancestral sequence at each divergence node can be reconstructed using a parsimony principle, in which conservation at an amino acid position is observed at a site over two or more consecutive divergence nodes, as is the case here for significant events in DHFR evolution. Using these considerations, we examined the 233 aligned sequences for PCEs, defined as changes at an amino acid position at which both the newly “altered site” and the unaltered long-conserved “ancestral site” remained fixed for a significant amount of subsequent geological time. Such events have only become identifiable in the large-scale genomic era (13) because of the number of species required to establish pre- and postinvariance with adequate confidence. Because the time scale of conservation of strongly supported PCEs is much longer than that of site turnover by genetic drift, PCE retentions/changes require strong (but probably differently based) selective pressure acting at the site of change in both descendent lineages. Note that evolutionarily significant divergence sites represent PCEs that are difficult to find because of challenges such as insufficient phylogenetic coverage, gaps and discrepancies across species, incomplete genes, and considerations for paralogs and pseudogenes. In addition, the identified PCEs are coherent changes with respect to the phylogenetic tree, as distinct from random wobbling within a fixed reduced alphabet with preference. Because of these stringent requirements, it was not known (a priori) whether DHFR would possess any PCEs at the start of this study. However, we hypothesized that some dramatic functional/mechanistic transitions occur at the PCEs.
Fig. 1.

Representative segment of phylogenetically aligned DHFR sequences from hsDHFR to ecDHFR. The phylogenetic tree (not to scale) on the left side illustrates the diverging relationship between species. The values at each divergence node represent the divergence for each evolutionary split in units of million years ago (Mya) (12). The hsDHFR and ecDHFR residue numberings are at the top and bottom of the figure, respectively. Strong PCE components [hsDHFR numberings: 24-PWPP-27, 62-PEKN-65, and F32 (L28 in ecDHFR)] are highlighted in pink. Genus species abbreviations and the full set of full length sequences can be found at http://genomewiki.ucsc.edu/index.php/DHFR_dihydrofolate.
Restricted to deuterostomes, we identified three strong PCEs that signal important evolutionary changes to DHFR between WT ecDHFR and hsDHFR. The locations of the PCE sites in the protein are illustrated in Fig. 2. The most recent PCE is a proline-rich (24-PWPPLRNEF-32 in hsDHFR as shown in Fig. 1, referred to as the PWPP region herein) region of the Met20 loop (residues 9–24 in ecDHFR; in hsDHFR residues 11–27). The PWPP region represents a unique evolutionary hotspot with a well-defined deletional/insertional history. Therefore, the timing for the development of the PWPP region in hsDHFR is very clear: after fish and before amphibians [Fig. 1; early to mid-Devonian, ∼415–385 Mya (12)].The second strong PCE found is the transition from L28 in ecDHFR to F32 in hsDHFR, which occurred around the same time and has persisted as phenylalanine ever since. Coevolution among these PCEs is possible, but we have found no evidence to support this.
Fig. 2.
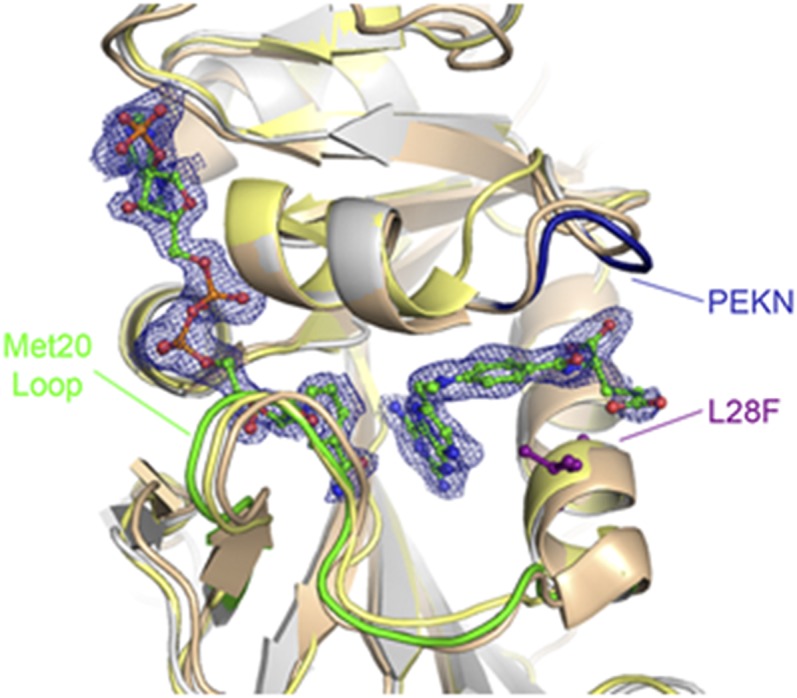
Superposition of N23PP/G51PEKN E. coli DHFR double mutant with human and E. coli DHFRs. The ecDHFR double mutant (silver, PDB ID 4GH8) is shown superimposed onto native human DHFR (bronze, PDB ID 1U72) (27) and native E. coli DHFR (gold, PDB ID 1RH3) (16). Shown in green, blue, and purple are the Met20 loop, the PEKN region, and the ecDHFR L28 residue, respectively. The methotrexate and NADPH ligands in the ecDHFR active site are shown in ball and stick representation (carbon atoms are green, oxygen atoms are red, nitrogen atoms are blue, and phosphate atoms are orange). The electron density is from an Fo–Fc map contoured at 3σ that was calculated before adding the ligands to the model.
The most ancient PCE identified occurs around G51 in ecDHFR and PEKN 62–65 in hsDHFR (referred to as the PEKN region herein). Structurally, the PEKN segment forms a flexible (14) lid-like portion of the folate-binding site. This is also the region of the enzyme where TMP binds (15). Tracing the last common ancestor for the PEKN region is less straightforward because this sequence differs in length across the three main clades (eukaryotes, bacteria, and archaea), suggesting that this is a very ancient divergence (∼2.4 Gya) (10, 12).
Kinetic Implications of PCEs.
We then examined the catalytic implications of the PCE regions through pre–steady-state and steady-state kinetic analyses. The ecDHFR catalytic cycle comprises five major complexes that can be separated into two groups based on differences in Met20 loop conformation (16, 17). The Met20 loop of the enzyme (E) can adopt either a closed conformation (E:NADPH, E:NADPH:DHF) or an occluded conformation (E:NADP+:THF, E:THF, E:NADPH:THF) (16, 18). NMR relaxation dispersion experiments found a rate constant of ∼1,200 s−1 at 300 K (17) for the transition from the Michaelis-Menten complex (E:NADPH:DHF) to the initial product complex (E:NADP+:THF). This value is consistent with the pre–steady-state hydride transfer rate (khyd = 950 s−1 at 298 K) (5) for the WT enzyme. Sequence analysis showed that the most recent strong PCE in DHFR is the development of a proline-rich PWPP region in the Met20 loop in hsDHFR. This PWPP region was modeled into WT ecDHFR by engineering in the N23PP mutation. The presence of this proline-rich segment in hsDHFR prevents the active site Met20 loop from undergoing the large-scale conformational changes observed along the catalytic cycle of ecDHFR (16–19). The presence of this proline-rich segment prevents the active site Met20 loop from undergoing the large-scale conformational changes observed along the catalytic cycle of ecDHFR (16–19) and reduces the catalyzed hydride transfer rate by ∼30 times. For comparison, hsDHFR (20) remains solely in the closed conformation throughout the catalytic cycle (21) and is more catalytically efficient than both WT ecDHFR and the N23PP ecDHFR mutant, illustrating that other modifications are responsible for offsetting the catalytic influence of the PWPP development.
The portion of the DHFR active site loop (24-PWPPLRNEF-32 in hsDHFR) that includes the PWPP segment exhibited a well-defined deletional/insertional history (Fig. 1 and SI Appendix, section 8.2). Sequence analysis indicates that the terminus of this region, F32 in hsDHFR, has been mostly occupied by either a methionine or a leucine, and the transition away from a persisting phenylalanine occurred around the same time as, or maybe slightly preceding, the PWPP event. There is evidence (22, 23) that the L28F mutation in ecDHFR can influence enzyme catalysis as well as the stability of the ternary DHFR:NADPH:DHF complex (decreasing the Michaelis-Menten value of the ternary complex). However, at pH 7, the N23PP/L28F mutant (khyd = 85 s−1 at 298 K) was unable to recover the WT catalytic rate (khyd = 220 s−1 at 298 K) (5), nor was the double mutant able to reach the enzymatic hydride transfer rate of WT hsDHFR (9).
We then examined the third and most ancient PCE identified, which is the addition of the PEKN lid-domain in the folate-binding site. By mutagenesis, the addition of the PEKN lid-domain into WT ecDHFR completely negates the negative catalytic influence of the PWPP element. By itself, the G51PEKN ecDHFR mutant exhibited a hydride transfer rate (khyd = 1,100 s−1 at 298 K) and a turnover rate (kcat = 8.9 s−1 at 298 K) that are essentially identical to the values found in the WT enzyme (5). However, the N23PP/G51PEKN double mutant (khyd = 1,100 s−1 and kcat = 22.3 s−1 at 298 K) strikingly retains the WT enzymatic activity by circumventing the negative impact of the N23PP mutation previously observed (19) (Table 1). The chronological order (PEKN before PWPP) of DHFR evolution might have been important for maintaining a certain threshold of enzyme activity. Furthermore, the N23PP/L28F/G51PEKN ecDHFR triple mutant exhibits an enhanced catalytic efficiency (khyd = 5,200 ± 1,200 s−1 at 298 K) that is commonly found in human and other vertebrate DHFRs (20) (Table 1).
Table 1.
Summary of experimental pre–steady-state khyd, ∆G≠, pKa values, and kcat for various DHFR species at 298 K
| khyd max, s−1 | Expt khyd ∆G≠, kcal/mol | Theoretical ∆G≠ for khyd, kcal/mol | Kinetic pKa | kcat, s−1 | |
| WT ecDHFR (5) | 950 ± 50 | 13.4 | 13.4 | 6.5 ± 0.1 | 12 |
| N ecDHFR (19) | ∼35 | 15.3 | 14.7 | 6.6 ± 0.1 | 2.5 ± 1 |
| G ecDHFR | 1,100 ± 80 | 13.3 | NA | 6.77 ± 0.07 | 8.9 ± 0.4 |
| NG ecDHFR | 1,100 ± 100 | 13.3 | 13.2 | 6.20 ± 0.06 | 26.9 ± 1.6 |
| NLG ecDHFR | 5,100 ± 1200 | 12.4 | NA | 5.9 ± 0.1 | 17.6 ± 5.1 |
| WT mouse (22) | ∼2,400–9,000 | 12.1–12.8 | NA | 6.40 ± 0.05 | 17 ± 2 |
| WT human (20) | 3,000 | 12.7 | 13.1 | 5.9–6.2 | 12.5 |
The EVB potential was parameterized to reproduce the experimentally determined free energy barrier for WT ecDHFR, so these values agree by construction. The free energy barriers were determined assuming a transition state theory rate constant expression with a transmission coefficient of unity. This assumption was shown to be valid for DHFR in previous studies (24). The SE in the calculated ΔG≠ value is ∼1 kcal/mol. G, G51PEKN; L, L28F; N, N23PP; NA, not available.
Other Mechanistic Considerations.
The kinetic data in Table 1 were derived from fit pH/rate profiles (SI Appendix, Fig. S2) for both the pre–steady-state khyd step and the steady-state kcat for each mutant. The data fit well to equations derived from a simple mechanistic scheme (5) with one ionization event. The pKa values are the same within experimental error (Table 1) and comparable to the reported WT human (20), E. coli (5), and mouse (22) DHFRs. The near identity in this pKa value across all mutants suggests that the differences in the khyd values are not due to dissimilar pKa values.
The reaction scheme involves the binding of E:NADPH with DHF to form the ternary Michaelis-Menten complex, E:NADPH:DHF, followed by a fast hydride transfer event to generate the initial product state E:NADP+:THF, with turnover at a slower rate. The measured hydride transfer rates exhibit a normal primary kinetic isotope effect [KIE = khyd(NADPH)/khyd(NADPD) ∼2.7], confirming that the hydride transfer rate was being measured. Because all of the ecDHFR mutants in this study exhibit similar turnover rates to WT ecDHFR, it is likely that the release of THF from the product complex (5) is still rate-limiting. For the turnover rates, the KIEs [kcat(NADPH)/kcat(NADPD)] were unity at low pH (SI Appendix, Table S1), indicating that the rate-limiting step in the catalytic cycle is independent of the hydride transfer step.
Computational Investigation of Enzyme Reaction.
Further mechanistic insights were provided by EVB MD simulations (11, 24) of the hydride transfer reaction in WT ecDHFR, the N23PP mutant, the N23PP/G51PEKN mutant, and WT hsDHFR. The calculated relative free energy barriers agree with the experimental values (Table 1). The calculations found a 1.3 kcal/mol increase in the activation free energy barrier for hydride transfer (∆G≠) from WT ecDHFR to the N23PP mutant, consistent with the 1.9 kcal/mol increase found experimentally (19). The computed ∆G≠ values for the N23PP/G51PEKN ecDHFR and hsDHFR are also similar to the experimental values. The agreement between theoretical and experimental data provides validation for the computational methodology applied.
The donor-acceptor distance (DAD) is known to strongly influence the free energy barriers of proton and hydride transfer reactions (24, 25). To assess whether the PCEs alter the DAD, we first examined the superposition of the crystal structures of the ternary Michaelis-Menten model complexes (E:NADPH:MTX; MTX = methotrexate) (16, 26) of the N23PP/G51PEKN ecDHFR mutant with WT ecDHFR (16) and human DHFR (27) (Fig. 2), which exhibit a very similar active site geometry in each variant. The inserted G51PEKN section of the double mutant closely traces the human PEKN region. However, small sub-Angstrom differences in the DAD can significantly impact the hydride transfer rate constant. EVB simulations indicate that the thermally averaged DADs along the collective reaction coordinate for hydride transfer are similar across the three ecDHFR variants as well as hsDHFR (SI Appendix, Fig. S4). More specifically, the thermally averaged DAD decreases to 2.7 Å at the transition state (TS), defined as zero energy gap reaction coordinate (11), in all cases. These data suggest that the differences in the calculated free energy barriers are not due to altered thermally averaged DADs.
The root-mean-square fluctuations (RMSFs), which measure the thermal motions of the atoms in the enzyme, are also very similar among all three species in both the reactant state (RS) and the TS (SI Appendix, Fig. S5). For comparison, the RMSFs of the three ecDHFR variants are evaluated against hsDHFR (Fig. 3A), showing that the extra amino acid insertions in the hsDHFR sequence are generally more flexible, whereas the homologous regions are qualitatively similar. Fig. 3B shows that the added PEKN lid domain in the N23PP/G51PEKN ecDHFR variant represents one of the more flexible regions in the structure. The thermal motions observed around the PCEs introduced into ecDHFR mimic the respective regions in WT hsDHFR (Fig. 3C).
Fig. 3.
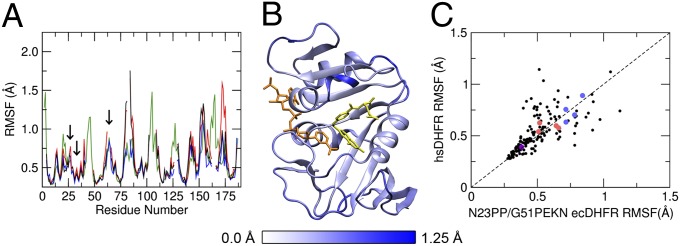
RMSFs of Cα atoms in the reactant state for WT ecDHFR (black), N23PP ecDHFR (red), N23PP/G51PEKN ecDHFR (blue), and hsDHFR (green). Residue numbering corresponds to hsDHFR; the locations of the PCEs are identified with arrows (A). The crystal structure of N23PP/G51PEKN ecDHFR (PDB ID 4GH8) with each residue colored according to its Cα RMSF, with folate [aligned based on RMSD minimization with hsDHFR (PDB ID 2W3M)] shown in yellow and NADP+ shown in orange (B). A comparison of the RMSFs of Cα atoms for residues in N23PP/G51PEKN ecDHFR to the RMSFs of the corresponding residues in hsDHFR (see SI Appendix, Table S5 for a full list of residues compared and the differences in the RMSF values). Residues in PCE regions are distinguished by color: PWPP (pink), PEKN (sky blue), and L28F (purple) (C).
To gain further insight into the catalytic differences, we also examined the average inter-Cα equilibrium distance changes between the RS and TS for all residue pairs (Fig. 4). We observed small (<1 Å) equilibrium conformational changes along the hydride transfer reaction coordinate. The magnitudes of the conformational changes between RS and TS are somewhat enhanced in the N23PP mutant relative to WT ecDHFR. We speculate that the reactive complex may need to undergo greater conformational changes to reach the TS in the N23PP mutant than in the WT enzyme (Fig. 4 A and B). Because the free energy barrier is related to the relative probabilities of sampling TS and RS configurations, the necessity of greater conformational changes to reach the TS would be consistent with the increased free energy barrier for the hydride transfer process in the N23PP ecDHFR mutant. The magnitudes of these conformational changes from RS to TS in the N23PP/G51PEKN mutant are similar to those in WT ecDHFR (Fig. 4 A and C), coinciding with recovery of the WT hydride transfer rate constant (Table 1). The observations described here are consistent with our previous view (28, 29) that stochastic thermal motions help to facilitate equilibrium conformational changes that are important to the progression from RS to TS. However, as discussed previously (28–31), these thermal motions do not represent special promoting vibrational modes (32) that are dynamically coupled to the chemical reaction itself.
Fig. 4.

Computed thermally averaged Cα-Cα distance changes from the RS to the TS for all pairs of residues in E. coli WT DHFR (A), the N23PP mutant (B), and the N23PP/G51PEKN mutant (C). Distances that increase from the RS to the TS are shown in red; distances that decrease from the RS to the TS are shown in blue. A color scale that corresponds to the magnitude and the direction of changes is shown at the bottom. Although each matrix is symmetrical, for clarity distances that increase are shown on the left side and distances that decrease are shown on the right.
G51PEKN Alters Cofactor and Inhibitor Bindings.
Further “human-like” characteristics generated from the G51PEKN mutation are revealed by a series of isothermal titration calorimetry (ITC) experiments (Table 2).The enzyme’s binding affinity (defined as dissociation constants, Kd) for NADPH and NADP+ shows that the partition between E:NADPH ↔ E ↔ E:NADP+ [defined as Kp = Kd(E:NADPH)/Kd(E:NADP+)] has dramatically shifted from Kp ∼ 0.0075 (favors NADPH) in WT ecDHFR (5) to a more “human-like” value (Kp ∼ 11.6; favors NADP+) (9) for all G51PEKN-containing mutants. There are two important points to consider. First, the differences in the Kp values between different ecDHFR mutants are within the uncertainty of the data, and it is likely that all three G51PEKN ecDHFR variants share the same Kp value. Second, although the G51PEKN mutation was able to shift the Kp value by ∼100-fold toward hsDHFR, other amino acid sequence divergence(s) is (are) responsible for the other 20-fold differences in the Kp values between the “humanized” ecDHFR in this study and the hsDHFR.
Table 2.
Dissociation constants (Kd; reciprocal of the binding constants) of binary DHFR complexes of E:TMP, E:NADPH, and E:NADP+ in aqueous medium at pH 7.0 and 298 K
| TMP Kd, M | NADPH Kd, M | NADP+ Kd, M | Kp = Kd(NADPH)/ Kd(NADP+) | |
| WT ecDHFR (5, 7) | 6 × 10−9 | 1.75 × 10−7 | 2.3 × 10−5 | 0.0076 |
| G ecDHFR | (1.65 ± 0.2) × 10−6 | (9.2 ± 0.8) × 10−7 | (1.1 ± 0.2) × 10−6 | 0.88 |
| NG ecDHFR | (1.2 ± 0.5) × 10−6 | (2.6 ± 0.4) × 10−6 | (4.0 ± 0.3) × 10−6 | 0.67 |
| NLG ecDHFR | (5.0 ± 0.7) × 10−6 | (3.0 ± 0.1) × 10−6 | (7 ± 1) × 10−6 | 0.45 |
| WT human (9, 23) | 10−6 | 2.2 × 10−5 | 1.9 × 10−6 | 11.6 |
G, G51PEKN; L, L28F; N, N23PP.
Close inspection shows that the G51PEKN mutation lowers ecDHFR’s intrinsic affinity for NADPH while increasing the enzyme’s binding affinity for NADP+. It has been noted that in prokaryotes (such as E. coli) the concentrations of NADPH and NADP+ are similar, whereas in eukaryotic cells the concentration of NADP+ is typically 100 times smaller than the NADPH concentration (33). Therefore, a large Kp value in E. coli would significantly stall the DHFR catalytic cycle through a greater degree of product inhibition, where [NADPH] ≈ [NADP+]. However, a large Kp value would not be as inhibitory for human DHFR because the cellular [NADP+] is much less than [NADPH]. It is interesting to note that these mutations do not appear to affect the binding of DHF to the E:NADPH complex (SI Appendix, Fig. S3); therefore, it is possible that the introduction of the PEKN domain into eukaryotic sequences might be a response to this change in the cellular environment. The subsequent additions of the N23PP and L28F mutations lead to at least a twofold increase in both the turnover (kcat) and the hydride transfer (khyd) rate constants. The evolutionary pressure on the hydride transfer rate is probably not significant, because the conversion of DHF to THF is gated (rate-limited) by the enzymatic turnover process (product dissociation). The enhanced turnover cycle resulting from the additions of these PCEs might reflect increased metabolic demands in higher species. At the same time, it is important to note that in vivo DHFR is not saturated by its substrates. Thus, mutations that affect the enzyme’s ability to bind its substrates can alter the flux (V/K rates) of the enzymatic reactions and influence the fitness of an organism, especially if there is a significant change in the availability of the substrates in the environment that the organisms inhabit. Mutations, especially those on the surface of protein, may also be epistatic. However, analysis of the reaction flux between the various ecDHFR variants studied here would be arbitrary without acknowledging specific protein partners or precise historical data on the environments and the organisms.
Finally, the binding constant between ecDHFR and trimethoprim is greatly weakened because of the G51PEKN mutation (Table 2). Trimethoprim is an antibiotic that is a million times more selective for bacterial DHFRs (E:TMP has Kd = 6 pM) (7) over mammalian DHFRs (Kd ∼1–10 mM for DHFRs from bovine liver, murine, and human) (7, 22). All ecDHFRs containing the G51PEKN mutation bind TMP with Kd values between 1 and 6 mM, which is comparable to values observed in mammalian DHFRs. It has been shown that the binding of inhibitors, such as TMP to ecDHFR, can affect the conformational states of the protein (34). Because the flexible PEKN domain resides over the TMP binding pocket [Protein Data Bank (PDB) ID2W3A] (15), the local conformational fluctuations in this region should affect the binding and dissociation of ligands (17, 33, 35). Again, within experimental errors, all PEKN-containing ecDHFR variants show the same binding affinity for TMP, and neither the L28F nor the N23PP mutations exhibit any additional impacts on TMP binding.
Conclusions
Evolutionary analysis of DHFR from 233 species identified three prominent PCEs, with strong implications regarding their importance in the preservation and divergence of enzyme functions through time. Here we show that despite only 26% sequence identity between ecDHFR and hsDHFR [and billions of years of subsequent divergence since the initial branch point (10)], the introduction of the three identified PCEs into the E. coli DHFR framework was able to transform the in vitro properties of the E. coli enzyme into the human counterpart. EVB MD simulations accurately reproduced the kinetic outcomes of a few cases where PCEs were engineered into ecDHFR. These simulations also indicated that the PCEs (N23PP and G51PEKN) do not significantly alter the equilibrium fluctuations (i.e., the RMSFs) in the ternary complex or the thermally averaged DAD during the hydride transfer reaction. In our view, equilibrium conformational motions facilitate the optimization of the active site electrostatic environment (28–30, 36) as well as the proximity and orientation of the reactants, leading to configurations conducive to the chemical reaction. Subtle differences in conformational sampling alter the free energy landscape (i.e., the equilibrium conformational states or ensembles) in a manner that impacts the free energy barrier and therefore the hydride transfer rate constant. Nevertheless, the overall conformational motions of the protein within a common scaffold are retained throughout evolution.
Divergence within an orthologous protein family during evolution arises from fixation of small mutational substitutions that must avoid passing through nonfunctional intermediates in protein sequence space (2–4). Here we analyzed the primary amino acid sequences of functional DHFRs of living species in the context of established phylogeny and constructed a timeline for the divergence of functional DHFRs among investigated species. We identified three changes (PCEs) in the DHFR primary sequence that exhibit strong functional implications that might be responsible for the major discrepancies between hsDHFR and ecDHFR. Because a strongly supported PCE is likely to require positive (possibly differently driven) Darwinian selection to be operative at the PCE site in both descendent lineages, PCEs could represent important time markers in history where major changes occurred, such as shifts in climate or environmental resources. With the rapidly growing wealth of genomic data and capacity for whole proteome alignment, identification of all human PCEs has become accessible. This should lead to exciting studies aimed at understanding the likely functional consequences of important divergences due to countless rounds of evolutionary selection (37). Furthermore, this type of sequence analysis approach can be highly efficient at identifying useful discrepancies among homologous proteins from different organisms. These discrepancies can provide guidance for exploiting selective modulation of homologous proteins.
Materials and Methods
Chemicals.
NADPH, NADP+, trimethoprim, methotrexate, folic acid, PEG 400, Hepes, MES, Tris, methotrexate-agarose, DTT, and ethanolamine were purchased from Sigma-Aldrich and used without further purification. DHF (38) and [(4′R)-2H]NADPH (NADPD) (39) were prepared according to published procedures.
Site-Directed Mutagenesis.
G51PEKN, N23PP/G51PEKN, N23PP/L28F, and N23PP/L28F/G51PEKN E. coli DHFR mutants were generated using the Stratagene QuikChange site-directed mutagenesis kit and the WT ecDHFR template as described (40). Primer sequences were 5′-GAA AAC GCC ATG CCA TGG CCG CCG CTG CCT GCC GAT CTC GCC-3′ (N23PP), 5′-G AAC CTG CCT GCC GAT TTC GCC TGG TTT AAA CG-3′ (L28F), and 5′-C TGG GAA TCA ATC CCT GAG AAG AAT AGG CCT TTG CCC-3′ (G51PEKN) (underlining represents mutations). Plasmid construction, protein expression, and purification were performed following previous publications (40).
Sequence Analysis.
Sequence analysis was achieved by manual curation of GenBank data as described in greater detail in the SI Appendix, section 1.
Kinetic Measurements.
Both the pre–steady-state and steady-state kinetic experiments were performed using an Applied Photophysics stopped-flow spectrophotometer at 25 °C. The reactions were carried out in MTEN buffer (composed of 50 mM MES, 25 mM Tris, 25 mM ethanolamine, and 100 mM NaCl) following the published procedures (5, 40). The final concentrations of the individual species in the reaction chamber were (10 µM enzyme, 125 μM NADPH, 100 μM DHF, 2 mM DTT, and 50 mM MTEM buffer). For the pre–steady-state kinetics, the progress of the DHFR-catalyzed hydride transfer reaction was monitored by excitation at 290 nm with the emission was measured using a 400-nm cutoff output filter (5). Steady-state kinetics experiments were performed following similar experimental conditions as described previously with the exception that the reaction progress was monitored at 340 nm. KIE experiments were conducted according to the conditions listed above using NADPH or NADPD. Further data analyses and experimental details are described in the SI Appendix, sections 2–4.
ITC.
ITC experiments were done using MicroCal Auto-iTC200 (GE) and the procedures are described in the SI Appendix, section 8.
Structure Determination.
Crystals of E. coli N23PP/G51PEKN ecDHFR variant were obtained by the hanging-drop vapor diffusion method using conditions similar to those previously described (16). Detailed methods, data collection, and refinement statistics are included in the SI Appendix, section 5.
Empirical Valence Bond MD Simulations.
Classical MD trajectories with a two-state EVB potential (23, 41) were used to simulate the hydride transfer reaction in WT ecDHFR, N23PP ecDHFR mutant, N23PP/G51PEKN ecDHFR mutant, and WT hsDHFR. The free energy profiles were generated along a collective reaction coordinate defined as the difference in energy between the two valence bond states. The computational details of the EVB potential can be found in the SI Appendix, section 6. The free energy profiles were generated from a series of 19 trajectories with different mapping potentials (i.e., windows) and combined using the weighted histogram analysis method (42). Three independent sets of trajectories were propagated for WT ecDHFR and N23PP/G51PEKN ecDHFR, and two independent sets of trajectories were propagated for N23PP ecDHFR and WT hsDHFR. The trajectory for each window was propagated for 100 ps of equilibration and 500 ps of production for all DHFR variants. Independent datasets were combined to obtain a total of 28.5 ns for WT ecDHFR and N23PP/G51PEKN ecDHFR and a total of 19.0 ns for N23PP ecDHFR and WT hsDHFR.
Supplementary Material
Acknowledgments
This work was supported by postdoctoral fellowships from The Natural Sciences and Engineering Research Council of Canada and Canadian Institutes of Health Research (to C.T.L. and J.B.F., respectively) and National Institutes of Health (NIH) Grant GM56207 (to S.H.-S. and P.H.). Isothermal titration calorimetry experiments were performed at Penn State Automated Biological Calorimetry Facility, which is supported by National Science Foundation (NSF) Major Research Instrumentation Grant 0922974; X-ray diffraction data were collected at the Cornell High Energy Synchrotron Source (CHESS), which is supported by the NSF and the NIH/National Institute of General Medical Sciences (NIGMS) under NSF Award DMR-0936384, and using the Macromolecular Diffraction at CHESS facility, which is supported by NIH Grant GM103485 through its NIGMS.
Footnotes
The authors declare no conflict of interest.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 4GH8).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1307130110/-/DCSupplemental.
References
- 1.Wolfenden R, Snider MJ. The depth of chemical time and the power of enzymes as catalysts. Acc Chem Res. 2001;34(12):938–945. doi: 10.1021/ar000058i. [DOI] [PubMed] [Google Scholar]
- 2.Smith JM. Natural selection and the concept of a protein space. Nature. 1970;225(5232):563–564. doi: 10.1038/225563a0. [DOI] [PubMed] [Google Scholar]
- 3.Povolotskaya IS, Kondrashov FA. Sequence space and the ongoing expansion of the protein universe. Nature. 2010;465(7300):922–926. doi: 10.1038/nature09105. [DOI] [PubMed] [Google Scholar]
- 4.Weinreich DM, Delaney NF, Depristo MA, Hartl DL. Darwinian evolution can follow only very few mutational paths to fitter proteins. Science. 2006;312(5770):111–114. doi: 10.1126/science.1123539. [DOI] [PubMed] [Google Scholar]
- 5.Fierke CA, Johnson KA, Benkovic SJ. Construction and evaluation of the kinetic scheme associated with dihydrofolate reductase from Escherichia coli. Biochemistry. 1987;26(13):4085–4092. doi: 10.1021/bi00387a052. [DOI] [PubMed] [Google Scholar]
- 6.Schweitzer BI, Dicker AP, Bertino JR. Dihydrofolate reductase as a therapeutic target. FASEB J. 1990;4(8):2441–2452. doi: 10.1096/fasebj.4.8.2185970. [DOI] [PubMed] [Google Scholar]
- 7.Sasso SP, Gilli RM, Sari JC, Rimet OS, Briand CM. Thermodynamic study of dihydrofolate reductase inhibitor selectivity. Biochim Biophys Acta. 1994;1207(1):74–79. doi: 10.1016/0167-4838(94)90053-1. [DOI] [PubMed] [Google Scholar]
- 8.Appleman JR, Prendergast N, Delcamp TJ, Freisheim JH, Blakley RL. Kinetics of the formation and isomerization of methotrexate complexes of recombinant human dihydrofolate reductase. J Biol Chem. 1988;263(21):10304–10313. [PubMed] [Google Scholar]
- 9.Appleman JR, et al. Unusual transient- and steady-state kinetic behavior is predicted by the kinetic scheme operational for recombinant human dihydrofolate reductase. J Biol Chem. 1990;265(5):2740–2748. [PubMed] [Google Scholar]
- 10.Brocks JJ, Logan GA, Buick R, Summons RE. Archean molecular fossils and the early rise of eukaryotes. Science. 1999;285(5430):1033–1036. doi: 10.1126/science.285.5430.1033. [DOI] [PubMed] [Google Scholar]
- 11.Warshel A. Computer Modeling of Chemical Reactions in Enzymes and Solutions. New York: John Wiley & Sons, Inc.; 1991. [Google Scholar]
- 12.Hedges SB, Dudley J, Kumar S. TimeTree: A public knowledge-base of divergence times among organisms. Bioinformatics. 2006;22(23):2971–2972. doi: 10.1093/bioinformatics/btl505. [DOI] [PubMed] [Google Scholar]
- 13.Genome 10K Community of Scientists Genome 10K: A proposal to obtain whole-genome sequence for 10000 vertebrae species. J Hered. 2009;100(6):659–674. doi: 10.1093/jhered/esp086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ramanathan A, Agarwal PK. Evolutionarily conserved linkage between enzyme fold, flexibility, and catalysis. PLoS Biol. 2011;9(11):e1001193. doi: 10.1371/journal.pbio.1001193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leung AKW, et al. Structural basis for selective inhibition of Mycobacterium avium dihydrofolate reductase by a lipophilic antifolate. Available at www.rcsb.org/pdb/explore.do?structureId=2W3A. 10.2210/pdb2w3a/pdb. Accessed April 25, 2013.
- 16.Sawaya MR, Kraut J. Loop and subdomain movements in the mechanism of Escherichia coli dihydrofolate reductase: Crystallographic evidence. Biochemistry. 1997;36(3):586–603. doi: 10.1021/bi962337c. [DOI] [PubMed] [Google Scholar]
- 17.Boehr DD, McElheny D, Dyson HJ, Wright PE. The dynamic energy landscape of dihydrofolate reductase catalysis. Science. 2006;313(5793):1638–1642. doi: 10.1126/science.1130258. [DOI] [PubMed] [Google Scholar]
- 18.Venkitakrishnan RP, et al. Conformational changes in the active site loops of dihydrofolate reductase during the catalytic cycle. Biochemistry. 2004;43(51):16046–16055. doi: 10.1021/bi048119y. [DOI] [PubMed] [Google Scholar]
- 19.Bhabha G, et al. A dynamic knockout reveals that conformational fluctuations influence the chemical step of enzyme catalysis. Science. 2011;332(6026):234–238. doi: 10.1126/science.1198542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beard WA, Appleman JR, Delcamp TJ, Freisheim JH, Blakley RL. Hydride transfer by dihydrofolate reductase. Causes and consequences of the wide range of rates exhibited by bacterial and vertebrate enzymes. J Biol Chem. 1989;264(16):9391–9399. [PubMed] [Google Scholar]
- 21.Davies JF, 2nd, et al. Crystal structures of recombinant human dihydrofolate reductase complexed with folate and 5-deazafolate. Biochemistry. 1990;29(40):9467–9479. doi: 10.1021/bi00492a021. [DOI] [PubMed] [Google Scholar]
- 22.Thillet J, Adams JA, Benkovic SJ. The kinetic mechanism of wild-type and mutant mouse dihydrofolate reductases. Biochemistry. 1990;29(21):5195–5202. doi: 10.1021/bi00473a028. [DOI] [PubMed] [Google Scholar]
- 23.Prendergast NJ, Appleman JR, Delcamp TJ, Blakley RL, Freisheim JH. Effects of conversion of phenylalanine-31 to leucine on the function of human dihydrofolate reductase. Biochemistry. 1989;28(11):4645–4650. doi: 10.1021/bi00437a020. [DOI] [PubMed] [Google Scholar]
- 24.Agarwal PK, Billeter SR, Hammes-Schiffer S. Nuclear quantum effects and enzyme dynamics in dihydrofolate reductase catalysis. J Phys Chem B. 2002;106(12):3283–3293. [Google Scholar]
- 25.Kiefer PM, Hynes JT. Nonlinear free energy relations for adiabatic proton transfer reactions in a polar environment. II. Inclusion of the hydrogen bond vibration. J Phys Chem A. 2002;106(9):1850–1861. [Google Scholar]
- 26.McElheny D, Schnell JR, Lansing JC, Dyson HJ, Wright PE. Defining the role of active-site loop fluctuations in dihydrofolate reductase catalysis. Proc Natl Acad Sci USA. 2005;102(14):5032–5037. doi: 10.1073/pnas.0500699102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cody V, Luft JR, Pangborn W. Understanding the role of Leu22 variants in methotrexate resistance: Comparison of wild-type and Leu22Arg variant mouse and human dihydrofolate reductase ternary crystal complexes with methotrexate and NADPH. Acta Crystallogr D Biol Crystallogr. 2005;61(Pt 2):147–155. doi: 10.1107/S0907444904030422. [DOI] [PubMed] [Google Scholar]
- 28.Hammes GG, Benkovic SJ, Hammes-Schiffer S. Flexibility, diversity, and cooperativity: Pillars of enzyme catalysis. Biochemistry. 2011;50(48):10422–10430. doi: 10.1021/bi201486f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hammes-Schiffer S, Benkovic SJ. Relating protein motion to catalysis. Annu Rev Biochem. 2006;75:519–541. doi: 10.1146/annurev.biochem.75.103004.142800. [DOI] [PubMed] [Google Scholar]
- 30.Kamerlin SCL, Warshel A. At the dawn of the 21st century: Is dynamics the missing link for understanding enzyme catalysis? Proteins. 2010;78(6):1339–1375. doi: 10.1002/prot.22654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Loveridge EJ, Behiry EM, Guo J, Allemann RK. Evidence that a ‘dynamic knockout’ in Escherichia coli dihydrofolate reductase does not affect the chemical step of catalysis. Nat Chem. 2012;4(4):292–297. doi: 10.1038/nchem.1296. [DOI] [PubMed] [Google Scholar]
- 32.Schwartz SD, Schramm VL. Enzymatic transition states and dynamic motion in barrier crossing. Nat Chem Biol. 2009;5(8):551–558. doi: 10.1038/nchembio.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Weikl TR, Boehr DD. Conformational selection and induced changes along the catalytic cycle of Escherichia coli dihydrofolate reductase. Proteins. 2012;80(10):2369–2383. doi: 10.1002/prot.24123. [DOI] [PubMed] [Google Scholar]
- 34.Mauldin RV, Carroll MJ, Lee AL. Dynamic dysfunction in dihydrofolate reductase results from antifolate drug binding: Modulation of dynamics within a structural state. Structure. 2009;17(3):386–394. doi: 10.1016/j.str.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carroll MJ, et al. Evidence for dynamics in proteins as a mechanism for ligand dissociation. Nat Chem Biol. 2012;8(3):246–252. doi: 10.1038/nchembio.769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kosugi T, Hayashi S. Crucial role of protein flexibility in formation of a stable reaction transition state in an α-amylase catalysis. J Am Chem Soc. 2012;134(16):7045–7055. doi: 10.1021/ja212117m. [DOI] [PubMed] [Google Scholar]
- 37.Grossman SR, et al. 1000 Genomes Project Identifying recent adaptations in large-scale genomic data. Cell. 2013;152(4):703–713. doi: 10.1016/j.cell.2013.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Blakley RL. Crystalline dihydropteroylglutamic acid. Nature. 1960;188:231–232. [Google Scholar]
- 39.Jeong SS, Gready JE. A method of preparation and purification of (4R)-deuterated-reduced nicotinamide adenine dinucleotide phosphate. Anal Biochem. 1994;221(2):273–277. doi: 10.1006/abio.1994.1411. [DOI] [PubMed] [Google Scholar]
- 40.Cameron CE, Benkovic SJ. Evidence for a functional role of the dynamics of glycine-121 of Escherichia coli dihydrofolate reductase obtained from kinetic analysis of a site-directed mutant. Biochemistry. 1997;36(50):15792–15800. doi: 10.1021/bi9716231. [DOI] [PubMed] [Google Scholar]
- 41.Kumarasiri M, Baker GA, Soudackov AV, Hammes-Schiffer S. Computational approach for ranking mutant enzymes according to catalytic reaction rates. J Phys Chem B. 2009;113(11):3579–3583. doi: 10.1021/jp810363k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kumar S, Rosenberg JM, Bouzida D, Swendsen RH, Kollman PA. The weighted histogram analysis method for free-energy calculations on biomolecules. I. The method. J Comput Chem. 1992;13(8):1011–1021. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.