

Abstract
Glucokinase (GK) is a monomeric allosteric enzyme and plays a pivotal role in blood glucose homeostasis. GK is regulated by GK regulatory protein (GKRP), and indirectly by allosteric effectors of GKRP. Despite the critical roles of GK and GKRP, the molecular basis for the allosteric regulation mechanism of GK by GKRP remains unclear. We determined the crystal structure of Xenopus GK and GKRP complex in the presence of fructose-6-phosphate at 2.9 Å. GKRP binds to a super-open conformation of GK mainly through hydrophobic interaction, inhibiting the GK activity by locking a small domain of GK. We demonstrate the molecular mechanism for the modulation of GK activity by allosteric effectors of GKRP. Importantly, GKRP releases GK in a sigmoidal manner in response to glucose concentration by restricting a structural rearrangement of the GK small domain via a single ion pair. We find that GKRP acts as an allosteric switch for GK in blood glucose control by the liver.
Keywords: hexokinase, sigmoidicity I conformational restriction, type 2 diabetes
Glucokinase (GK), a hexokinase isozyme, is a monomeric allosteric enzyme and mainly expressed in hepatocytes and pancreatic β-cells (1, 2). Through its unique kinetic character, GK plays a central role in blood glucose homeostasis by converting glucose to glucose-6-phosphate (G6P), enhancing glycogen synthesis, in hepatocytes (3), and sensing glucose for insulin secretion in pancreatic β-cells (4). Defect and mutations in GK are directly associated with type 2 diabetes and maturity-onset diabetes of the young type 2 (5, 6). Thus, GK has been an important molecular target for studying blood glucose homeostasis and developing antidiabetes drugs (5, 7, 8). The crystal structure of human GK (hGK) revealed that GK undergoes a slow and energetically unfavorable structural rearrangement of the small domain in response to glucose during the transition from a super-open conformation into an open conformation (9). As a result, GK exhibits a sigmoidal activity curve with respect to glucose concentration as a monomeric allosteric enzyme (9).
Unlike pancreatic β-cells, blood glucose control by the hepatocytes is more complicated. Hepatocytes remove high amount of exogenous glucose after a meal in a fast and efficient manner (3). At the same time, hepatocytes also produce endogenous glucose to blood stream to maintain blood glucose level in a fasting state (3, 10). GK must be fully active for fast glucose clearance after a meal (3), whereas it should be turned off during a fasting state to prevent futile cycling of endogenous glucose to G6P (10). Therefore, GK in hepatocytes is presumed be regulated in a different way from that in pancreatic β-cells. In hepatocytes, GK is regulated by GK regulatory protein (GKRP) that is located mainly in hepatocytes with an excess ratio to GK (11, 12). GKRP allosterically regulates the activity and subcellular localization of GK (13, 14). GKRP inhibits and sequesters GK into the nucleus of hepatocytes and releases GK into the cytoplasm in response to glucose concentration (13–15). GK is also indirectly regulated by allosteric effectors of GKRP, such as fructose-1-phosphate (F1P) and fructose-6-phosphate (F6P) (11, 16). F1P is derived from fructose and sorbitol in a meal, and F6P is an intermediate product of glycolysis (16). Recent studies have characterized the biophysical features regarding the interaction between GK and GKRP and a modulation through the effectors (17, 18). Despite the intensive studies on GK and GKRP, the molecular basis for the allosteric regulation mechanism of GK by GKRP remains poorly understood because of the lack of structural information of the GK/GKRP complex.
Here, to demonstrate the molecular mechanism for the allosteric regulation of GK by GKRP and effectors, we determined the crystal structure of a Xenopus laevis GK and GKRP complex in the presence of F6P. Structural analysis of the complex and mutational studies with human GK and GKRP revealed that GKRP interacts with GK mainly through hydrophobic interaction and inhibits GK activity through a single ion pair between GKRP and the small domain of GK. We also show the molecular mechanism by which F1P and F6P modulate the activity of GK through molecular dynamics simulations and mutational analysis. Importantly, GKRP was revealed to release GK in a sigmoidal manner in response to glucose concentration by restricting a structural rearrangement of the GK small domain through a single ion pair, acting as an allosteric switch for GK.
Results
Determination of Crystal Structure of GK/GKRP Complex.
We determined the crystal structure of a X. laevis GK/GKRP (xGK/xGKRP) complex in the presence of F6P at 2.9 Å (Fig. 1A and Table S1). Our attempts to crystallize a hGK/human GKRP (hGKRP) complex have been unsuccessful. The crystals contain two xGK/xGKRP complexes in an asymmetric unit, and share 82% and 58% sequence identity with hGK and hGKRP, respectively (Fig. S1). The overall structure of the xGK/xGKRP complex reveals that xGKRP is bound to the super-open conformation of xGK (Fig. 1B), sharing an rmsd of 1.7 Å for 406 Cα atoms for the super-open conformation of apo-hGK [Protein Data Bank (PDB) ID code 1V4T] with an approximately 11° rotation of a small domain of xGK toward a large domain (Fig. S2). The structure of xGKRP comprises two sugar isomerase (SIS) superfamily domains and a C-terminal extended all-helical motif (Fig. 1A). The sugar-isomerase domains show typical αβα folds as reported elsewhere (19, 20) (Fig. S3). We identified the electron density of F6P as a linear keto form, as previously suggested (16), in SIS domain 1, where SIS domain 2 and the C-terminal motif meet together (Fig. 1 A and C). A stretch of four residues including Ser179, Ser258, Glu347, and Lys513 of xGKRP participate in the binding of F6P through hydrogen bonding (Fig. 1C). A previous study on rat GKRP also revealed that Ser179 and Lys513 interact with F6P (21).
Fig. 1.
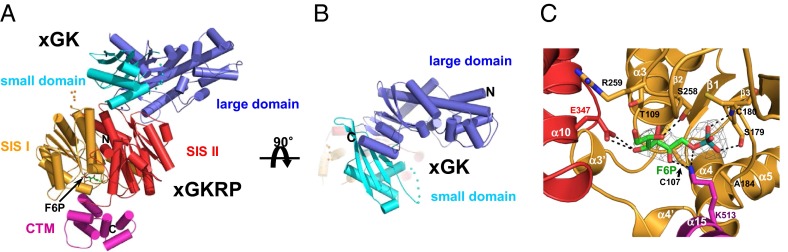
Overall structure of xGK/xGKRP complex. (A) Cartoon representation of the xGK/xGKRP complex. xGK is shown in blue (large domain) and cyan (small domain), and xGKRP is in orange (SIS domain 1), red (SIS domain 2), and pink [C-terminal all-α-helical motif (CTM)]. F6P is shown through a stick representation. (B) Cartoon representation of the xGK in xGK/xGKRP complex; as in A but rotated 90°. For clarity, xGKRP is faintly presented. (C) Close-up view of F6P binding site. Difference electron density contoured at 1.5σ is shown at F6P (green, stick model), which was excluded from the phase calculation. Oxygen, nitrogen, and phosphate atoms are shown in red, blue, and cyan, respectively. The dotted lines indicate intermolecular hydrogen bonds between F6P and xGKRP.
Analysis of Interface Between GK and GKRP.
Based on the crystal structure of the xGK/xGKRP complex, we analyzed the interface between GK and GKRP. The xGK/xGKRP complex shows that a wedge-shaped structure is formed by two loops (L1 and L2), two helices (α8 and α14), and one strand (β10) in SIS domain 2 of xGKRP, anchoring to the allosteric cleft in the hinge region of xGK (Fig. 2A). The interface between xGK and xGKRP is predominantly hydrophobic, with a total burial area of 1,913 Å2. The edge of the wedge formed by two loops, L1 and L2, of xGKRP creates multiple van der Waals contacts with the hydrophobic surface in the cleft formed by a strand (S1), loops L1 and L2 from the large domain, loop L3 from the small domain, and connecting region II of the xGK, generating major hydrophobic interactions (Fig. 2A). A hydrophobic surface is composed of Leu40, Lys49, Leu51, Pro52, Tyr54, Arg56, Lys136, Met231, Leu236, Val237, Glu238, and Met244 from the large domain, and Asp191, Val192, and Val193 from the small domain of xGK (Fig. 2 A and B). This hydrophobic surface interacts with Ala440, Gly441, and Tyr443 of loop L1 and Pro461, Ile462, Leu463, and Phe464 of loop L2 in xGKRP (Fig. 2 A and B). It is interesting to note that most of the hydrophobic surface of xGK is located in the large domain. To assess the importance and universality of the interactions in a GK/GKRP complex, we performed a mutational analysis for hGKRP. We mutated Leu463 and Phe465 in hGKRP to Ala, which are equivalent to Ile462 and Phe464 in xGKRP and share approximately one fourth of the hydrophobic contacts (481 Å2). In gel filtration analysis, the resulting hGKRPL463A/F465A mutant did not form a complex with hGK (Fig. S4), which indicates that hydrophobic interactions are crucial for the binding of GKRP to GK.
Fig. 2.

Interface of xGK/xGKRP complex. (A) Close-up view of the xGK/xGKRP interface. (Inset) Location of interface. The residues and secondary structures of the wedge in xGKRP (red) are shown to interact with the residues in the allosteric cleft of xGK (blue and cyan). The hydrophobic interface in the cleft of xGK is presented as a transparent surface. The dotted circle in red indicates the ion pair between xGKRP and xGK small domain. (B) Ligplot of the residues in the interface between xGK and xGKRP complex. The side chains of the small domain (cyan) and large domain (blue) of xGK and SIS domain 2 (red) of xGKRP are shown. The dotted circle is the same as in A. (C) Inhibition of hGK activity by various hGKRP mutants. The activity shows average and SDs from triplicate experiments.
The interface is further stabilized by conserved two ion pairs and three hydrogen bonds showing a charge complementarity to the wedge of xGKRP (Fig. 2A and Fig. S5). Glu238 of loop L4 in the large domain and Arg179 of α4 in the small domain of xGK form ion pairs with Arg300 and Asp412 of xGKRP, respectively (Fig. 2A and B). Ile462 of xGKRP makes backbone-to-backbone hydrogen bonds with Leu236 and Glu238 of loop L4 in xGK. Lys136 of L3 in xGK makes a hydrogen bond with a backbone carboxyl group of Leu463 in xGKRP (Fig. 2B). Mutation in Lys136 of xGK, which is equivalent to Lys143 in rat GK, was shown to decrease the inhibition exerted by GKRP in the study of rat GK (22). It is noteworthy that a single ion pair is the main interaction between xGKRP and the small domain of xGK. The mutations of Arg301 (Arg300 in xGKRP) or Asp413 (Asp412 in xGKRP) in hGKRP to Ala display comparable binding affinities to hGK as WT hGKRP does (Table 1). However, interestingly, hGKRPD413A showed a significantly decreased inhibitory effect on hGK compared with WT hGKRP (Fig. 2C). This result indicates that GKRP inhibits the GK activity mainly by a conformational restriction through its ion pair to the GK small domain.
Table 1.
Kd values of hGK for WT hGKRP and its mutants in the presence and absence of F6P (200 μM) or F1P (100 μM) by ITC
| Type | Kd, nM |
| WT hGKRP | 230 ± 26 |
| WT hGKRP + F6P | 95 ± 25 |
| WT hGKRP + F1P | ND |
| hGKRPL463A/F465A | ND |
| hGKRPD413A | 375 ± 97 |
| hGKRPR301A | 235 ± 34 |
| hGKRPE348A/H351P | 207 ± 27 |
| hGKRPE348A/H351P + F6P | 194 ± 21 |
| hGKRPE348A/H351P + F1P | 225 ± 50 |
ND, not determined.
Molecular Mechanism for Release of GK from GKRP.
Our structural analysis revealed that GKRP binds to the super-open conformation of GK. During catalysis, however, GK is released from GKRP upon glucose binding and undergoes a conformational change to an open conformation (9). To demonstrate how GK dissociates from GKRP in response to glucose, we superimposed the xGK/xGKRP complex to the open conformation of hGK (PDB ID code 1V4S). The superimposition revealed that the ion pair of GKRP to the small domain of GK is no longer available between GKRP and an open conformation of GK (Fig. 3A). Moreover, Tyr62 and Arg156 of hGK sterically clash with Gly441 and Ile462 of xGKRP (Fig. 3 A and B). These two residues are located at the edge of the wedge in xGKRP. Moreover, Ile462 (Leu463 in hGKRP) was shown to be particularly crucial for a hydrophobic interaction between GK and GKRP through the analysis of the hGKRPL463A/F465A mutant (Fig. S4). Consequently, a structural rearrangement of the GK small domain gives rise to a dissociation of the complex. It is therefore likely that GK will be released from GKRP in a sigmoidal manner in response to glucose concentration as its activity. It should also be noted that GKRP-bound GK cannot catalyze the reaction as a result of a conformational restriction of the small domain, and thus only free GK exhibits its activity. Also, GK activator (GKA), which is bound in an open conformation of hGK (9), is revealed to locate in a distant site from GKRP binding site (Fig. S6). This result indicates that GKA dissociates GKRP (7, 9, 17, 23) through a structural rearrangement of GK.
Fig. 3.
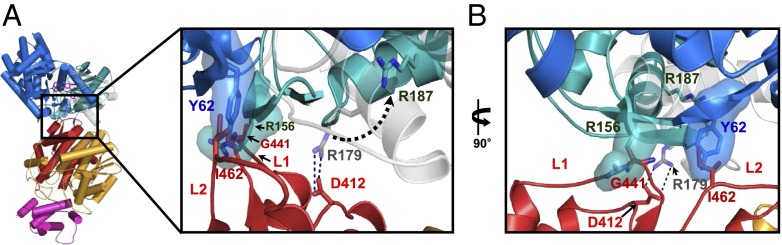
Structural changes of the interface in releasing GK from GKRP. (A) Close-up view of the xGK/xGKRP interface that superimposed onto open conformation of hGK. Inset represents the location of the interface. Superimposed open conformation of hGK (PDB ID code 1V4S) onto the xGK/xGKRP complex is shown in blue (large domain) and dark cyan (small domain). GKA and glucose are indicated in stick model (purple). Orientation of the xGK small domain is shown in gray. Dotted arrow indicates the movement of Arg-179 of xGK (Arg-187 in hGK) during a structural rearrangement of the GK small domain. (B) Rotated (90°) close-up view of the Inset in A shows the surface of Arg-156 and Tyr-62 in hGK. Oxygen and nitrogen atoms are presented in red and blue, respectively. The color notation in xGKRP domains are the same as in Fig. 1.
Modulation Mechanism of GK by Allosteric Effectors of GKRP.
It was reported that the allosteric effectors of GKRP such as F1P and F6P bind GKRP and alter the interaction between GK and GKRP and eventually the GK activity (11, 14, 16). As xGKRP has been shown to be insensitive to the binding of F6P and F1P (24), we used hGKRP to investigate the molecular mechanism by which effectors of GKRP modulate the GK activity. We first examined the activity of hGK at varying concentrations of F1P and F6P in the presence of hGKRP. As shown in Fig. 4A, F1P showed a more significant effect on the GK activity than F6P. Next, we performed molecular dynamics simulations by using the model structure of hGKRP in the presence and absence of F1P and F6P based on the xGKRP structure. Displacement of α-carbons in SIS domain 2 and C-terminal motif was shown to occur upon the binding of F6P and F1P in the time course of simulations (Fig. S7). The superimposition of F1P-bound hGKRP onto the xGK/xGKRP complex revealed that the C-terminal of α10 moves away from F1P as a result of a steric hindrance between His351 and the hydroxyl group of C6 in F1P (Fig. 4 B and C). Consequently, the helices α11 to α13 and a β-sheet, β6 to β10, of SIS domain 2 rotate outward from the core helix α14 (Fig. 4B) and displace the L2 toward GK, causing a steric hindrance to the GK binding with Leu463 of hGKRP (Fig. 4D). As shown in the hGKRPL463A/F465A mutant, Leu463 is the key residue of hydrophobic interaction in the GK/GKRP complex. Thus, F1P perturbs the binding of GK to GKRP and disrupts the complex by altering hydrophobic interaction between GK and GKRP. It is noteworthy that F1P is likely to be more efficient than glucose or GKA in releasing GK from GKRP. Although F1P, glucose, and GKA bind to distant sites from the interface of the GK/GKRP complex (9), F1P induces direct disruption of the interface to release GK from GKRP, whereas glucose and GKA induce slow and energetically unfavorable rearrangement of the GK small domain (9, 17). The superimposition of F6P-bound hGKRP onto apo-hGKRP revealed that the interaction between the hydroxyl group of C1 in F6P and Glu348 of α10 in SIS domain 2 shifts α10 downward (Fig. S8 A and B) and rotates the helices α11 to α13 and a β-sheet, β6 to β10, of SIS domain 2 toward F6P (Fig. S8A), thereby moving L1 and L2 of the wedge close to α14, enhancing the binding of GK compared with apo-hGKRP (Fig. S8C). Thus, His351 and Glu348 are likely to be the key residues in the allosteric regulation of hGKRP by its effectors. To verify the modulation mechanism described earlier, we mutated Glu348 and His351 in hGKRP to Asp and Pro, respectively. The structural consistency between hGKRPE348A/H351P and WT hGKRP were confirmed based on CD analysis and binding of F6P to hGKRPE348A/H351P by isothermal titration calorimetry (ITC; Fig. S9 and Table S2). The binding affinity of hGKRPE348A/H351P for hGK measured by ITC was almost the same as WT apo-hGKRP regardless of the presence of F1P or F6P (Table 1). The inhibitory effect of hGKRPE348A/H351P on the hGK activity was comparable to hGKRP in the absence of F1P and F6P, and remained unchanged regardless of the presence of either (Fig. 4E). This result confirms the modulation mechanism of allosteric GKRP by F1P and F6P and the key residues inducing the conformational changes in GKRP. Furthermore, our result provided some insight into why hGKRP responds to the occupation of its allosteric site by F1P and F6P whereas xGKRP does not. Sequence alignment of various GKRPs revealed that His351 of hGKRP is conserved in other species except xGKRP, which has Pro instead of His (Fig. S1). In addition, mutation of His351 to Pro resulted in no response of hGKRP to the binding of F1P and F6P as xGKRP. It is therefore likely that His351 of hGKRP has a key role in the interaction with GK upon the binding of F6P and F1P to hGKRP, acting as a pivotal residue inducing the structural change of hGKRP. On the contrary, His-351, which plays a critical role in the interaction with GK, is not conserved in xGKRP, and consequently xGKRP does not respond to the occupation of its allosteric site by F1P and F6P.
Fig. 4.

Modulation in the binding of GKRP to GK by allosteric effectors. (A) The activity of hGK measured at different concentrations of F1P and F6P in the presence of an equal molar ratio of hGKRP to hGK and 10 mM glucose. The activity indicates average and SDs from triplicate experiments. (B) Superimposition of F1P-bound hGKRP onto the xGK/xGKRP complex. The helices α11 to α13 and a β-sheet, β6 to β10, of SIS domain 2 of hGKRP (green) and xGKRP (orange) are shown. xGK is represented in cyan. (C) Close-up view of the binding site for F1P and F6P. Stick representations of F1P (green) and F6P (orange) are shown. Residues from hGKRP (green) and xGKRP (orange) are also represented. (D) Rotated (90°) close-up view of the binding interface between GK and GKRP. L1 and L2 of hGKRP (green) and xGKRP (orange) are shown. The surface of Leu463 of hGKRP is shown. (E) Inhibition of hGK activity by hGKRPE348A/H351P. The hGK activity was determined with WT hGKRP or hGKRPE348A/H351P in the presence and absence of 0.1 mM F1P and 0.2 mM F6P. The activity represents average and SDs from triplicate experiments.
Implications for Physiological Role of GKRP.
GKRP is known to be located mainly in hepatocytes with an excess ratio to GK, regulating the allosteric GK in response to glucose concentration (12, 14, 15). To get some insight into the physiological implications for the role of GKRP, we attempted to investigate the regulatory features of GKRP. To this end, we first examined the dissociation of GK from GKRP in response to glucose, and determined the binding affinity of hGKRP and hGKRPD413A for hGK at varying glucose concentrations by using ITC (Fig. 5A and Fig. S10). As shown in Fig. 5A, the Kd of hGKRP for hGK displays a sigmoidal curve with respect to glucose concentration as presumed, showing an inflection point at approximately 20 mM glucose. On the contrary, the binding affinity of hGKRPD413A for hGK greatly decreased even at 5 mM glucose. This result indicates that the ion pair between GKRP and the GK small domain is a key for generating a sigmoidal-type release of GK from GKRP in response to glucose concentration. To investigate the effect of GKRP on the kinetic behavior of GK, we measured the hGK activity with increasing ratios of hGKRP to hGK (Fig. 5B). The activity curve of hGK for a fourfold excess of hGKRP became more sigmoidal, resulting in a higher Hill coefficient (2.8) than with hGK alone (1.8; Table S3). The increased sigmoidicity of the GK activity curve is coincident with previous in vivo reports showing the free GK activity or detritiation of [2-3H]glucose of hepatocytes in response to glucose (25). Thus, the regulation of GK by GKRP in a sigmoidal pattern also seems to be valid in vivo. Interestingly, the hGK activity curve in the presence of hGKRPD413A or WT hGKRP and 25 mM KCl showed a decreased sigmoidicity compared with that with only WT hGKRP (Fig. 5C and Table S3). This result demonstrates that the increased sigmoidicity of the GK activity curve in the presence of GKRP is attributed to the ion pair between GKRP and the GK small domain. KCl seems to perturb the ion pair between GKRP and the GK small domain. Thus, previous studies describing GKRP as a classical competitive inhibitor (11, 26) are likely to be misled by addition of KCl. Our results imply that the release of GK from GKRP in a sigmoidal manner in response to glucose concentration is crucial for blood glucose control by the liver.
Fig. 5.

Dissociation and kinetic behavior of GK in the presence of GKRP and glucose. (A) Effect of glucose on the dissociation of hGK from hGKRP. The Kd values of hGK for WT hGKRP and hGKRPD413A were determined by ITC at different glucose concentrations. (B) Activity of hGK determined with increasing the ratio of hGKRP to hGK in response to glucose. (C) Activity of hGK determined at a fourfold excess of hGKRP and hGKRPD413A to hGK with respect to glucose. The hGK activity in the presence of 25 mM KCl and a fourfold excess of hGKRP is also shown (□). The activity indicates average and SDs from triplicate experiments.
Discussion
We have demonstrated the molecular mechanism for the allosteric regulation of GK by GKRP and effectors based on the structure of the GK/GKRP complex. Our structural analysis revealed that GKRP binds to a super-open conformation of GK mainly through hydrophobic interaction, inhibiting the GK activity by locking a small domain of GK. We also showed how allosteric effectors of GKRP such as F1P and F6P indirectly modulate the GK activity in negative and positive way, respectively. More importantly, GKRP was revealed to release GK in a sigmoidal manner in response to glucose concentration by restricting a structural rearrangement of the GK small domain via a single ion pair. This study elucidates the molecular mechanism by which an allosteric protein regulates a monomeric allosteric enzyme. The GK/GKRP system exemplifies a unique case showing a cascade regulation mechanism of an allosteric enzyme by an allosteric regulatory protein and its effectors. Our results demonstrate the role for GKRP as an allosteric switch (27) that turns GK “on” and “off” in response to glucose concentration, providing crucial insight into the understanding of blood glucose homeostasis and the development of new antidiabetes drugs.
The sigmoidal nature driven by GKRP in regulating GK can be characterized into two distinct features. One is the release of GK from GKRP in a sigmoidal manner with respect to glucose concentration, leading to an amplification of the GK activity at high glucose levels. The other is an increased sigmoidicity of the GK activity curve, resulting in a turning-off of GK at low glucose concentration. This sigmoidal feature results from the binding of GKRP to a super-open conformation of GK and consequent restriction of a structural rearrangement of GK through a single ion pair. Those two features in regulation of GK by GKRP seem to be essential for two major functions of the liver, glucose clearance and glucose production in blood glucose homeostasis (3, 10). As GK itself displays a sigmoidal activity curve with respect to glucose concentration (1, 3), a release of GK from GKRP in a sigmoidal manner would significantly amplify the glucose clearance rate by the liver at high glucose levels. In addition, as free GK in cytosol can be degraded by proteasome after ubiquitination (28, 29), a sigmoidal-type release of GK by GKRP can minimize the loss of GK within the low glucose levels. At low glucose concentration, GK remains turned off as a result of an increased sigmoidicity of the GK activity curve by GKRP. Therefore, the production of glucose will be efficient because of a blocking of fertile cycling of endogenous glucose to G6P (10) and maintenance of the cytosolic concentration of glucose at a higher level than in blood stream (5 mM) for facilitated diffusion of endogenous glucose by liver glucose transporter 2 (30). Thus, our results indicate that GKRP acts as the allosteric switch that turns GK on and off in response to glucose concentration in blood glucose control by the liver.
The activity of hGK in the presence of hGKRP at high glucose concentration is lower than expected, and this seems to be mainly caused by the difference in the conditions between in vitro and in vivo (Fig. S11). In the case of in vitro system, GK released from GKRP by glucose can rebind to GKRP, which results in a decreased GK activity. On the contrary, in vivo, released GK is translocated into the cytosol by its own nuclear exporting signal (31) and separated from GKRP by nuclear membrane. Accordingly, translocated free GK exhibits its full activity in the cytosol, which is crucial for fast glucose clearance.
GK remains almost turned off in the range of 5 to 10 mM of glucose by the action of GKRP (Fig. 5B). Thus, the negative effector such as F1P is likely to be essential for glucose clearance in this range of glucose. Accordingly, precursors of F1P, such as fructose or sorbitol (16), are likely to be important for glucose clearance by liver after a meal. Indeed, supply of fructose together with glucose was shown to increase glycogen synthesis by liver in the rat and dog (32, 33). Although fructose can be efficient to relieve hyperglycemia in diabetes, the use of fructose as a substituent for glucose for diabetes patients has raised the controversy (34, 35). F1P is effective for releasing GK from GKRP, but a high amount or long-term supply of fructose might result in unexpected loss of GK as a result of proteasomal degradation of free GK in cytosol (29), which can cause the hyperglycemia to be more serious.
Allosteric activators of GK (i.e., GKAs) have been of great significance as antidiabetes drugs, and a number of diverse GKAs have been reported (5, 7–9). However, GKAs may cause serious side effects, as they change a sigmoidal activity curve of GK into a hyperbolic one (8, 9), and consequently alter the sensing of blood glucose level by GK in pancreatic β-cells, resulting in hyperinsulinemic hypoglycemia. As an approach to avoid the side effect, moderate or hepatocyte-specific GKAs are under development (36–38). With the same rationale and based on our result, GKRP can be a target for the development of new antidiabetes drugs that activate GK in hepatocytes. As GKRP is predominantly located in hepatocytes, the GKRP-targeting drugs can activate GK specifically in hepatocytes, thus preventing possible hypoglycemia. The binding site of allosteric effectors in GKRP and the interface of the GK/GKRP complex can be most promising drug targets.
Materials and Methods
The genes encoding GK and GKRP from X. laevis and humans were expressed in Escherichia coli and purified by Ni affinity, anion exchange, and gel-filtration chromatography. The crystals of Xenopus GK/GKRP complex were obtained by the hanging-drop vapor diffusion method, and the structure was solved by using the MR method. The binding affinities of GK for GKRP mutants were measured by ITC. The activities of GK in the presence and absence of GKRP and allosteric effectors were determined by a G6P dehydrogenase-coupled assay. Detailed materials and methods are described in SI Materials and Methods.
Supplementary Material
Acknowledgments
We thank J. J. Song and W. D. Jones for helpful discussion and S. H. Kim (LG Life Science) for providing the human GK and GKRP genes. This research was supported by the Pioneer Research Center Program (2008-2000218); the Advanced Biomass R&D Center (2011-0031363); the Intelligent Synthetic Biology Center (2011-0031950) through the National Research Foundation of Korea funded by the Ministry of Science, Information and Communication Technology and Future Planning; and the World-Class University (WCU) program and the Brain Korea 21 of the Ministry of Education (Korea).
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 3W0L).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1300457110/-/DCSupplemental.
References
- 1.Cornish-Bowden A, Cardenas ML. Glucokinase: A monomeric enzyme with positive cooperativity. In: Matschinsky FM, Magnuson MA, editors. Frontiers in Diabetes. Vol 16. Basel: Karger; 2004. pp. 125–134. [Google Scholar]
- 2.Iynedjian PB, Möbius G, Seitz HJ, Wollheim CB, Renold AE. Tissue-specific expression of glucokinase: Identification of the gene product in liver and pancreatic islets. Proc Natl Acad Sci USA. 1986;83(7):1998–2001. doi: 10.1073/pnas.83.7.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Agius L. Glucokinase and molecular aspects of liver glycogen metabolism. Biochem J. 2008;414(1):1–18. doi: 10.1042/BJ20080595. [DOI] [PubMed] [Google Scholar]
- 4.Matschinsky FM, et al. Glucokinase as pancreatic β cell glucose sensor and diabetes gene. J Clin Invest. 1993;92(5):2092–2098. doi: 10.1172/JCI116809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Matschinsky FM. Assessing the potential of glucokinase activators in diabetes therapy. Nat Rev Drug Discov. 2009;8(5):399–416. doi: 10.1038/nrd2850. [DOI] [PubMed] [Google Scholar]
- 6.Vionnet N, et al. Nonsense mutation in the glucokinase gene causes early-onset non-insulin-dependent diabetes mellitus. Nature. 1992;356(6371):721–722. doi: 10.1038/356721a0. [DOI] [PubMed] [Google Scholar]
- 7.Grimsby J, et al. Allosteric activators of glucokinase: Potential role in diabetes therapy. Science. 2003;301(5631):370–373. doi: 10.1126/science.1084073. [DOI] [PubMed] [Google Scholar]
- 8.Matschinsky FM, et al. Handbook of Experimental Pharmacology. 2011. Research and development of glucokinase activators for diabetes therapy: Theoretical and practical aspects. eds Schwanstecher M (Springer, Berlin), Vol 203, pp 357–401. [DOI] [PubMed] [Google Scholar]
- 9.Kamata K, Mitsuya M, Nishimura T, Eiki J, Nagata Y. Structural basis for allosteric regulation of the monomeric allosteric enzyme human glucokinase. Structure. 2004;12(3):429–438. doi: 10.1016/j.str.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 10.Nordlie RC, Foster JD, Lange AJ. Regulation of glucose production by the liver. Annu Rev Nutr. 1999;19:379–406. doi: 10.1146/annurev.nutr.19.1.379. [DOI] [PubMed] [Google Scholar]
- 11.Van Schaftingen E. A protein from rat liver confers to glucokinase the property of being antagonistically regulated by fructose 6-phosphate and fructose 1-phosphate. Eur J Biochem. 1989;179(1):179–184. doi: 10.1111/j.1432-1033.1989.tb14538.x. [DOI] [PubMed] [Google Scholar]
- 12.Vandercammen A, Van Schaftingen E. Species and tissue distribution of the regulatory protein of glucokinase. Biochem J. 1993;294(pt 2):551–556. doi: 10.1042/bj2940551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brown KS, Kalinowski SS, Megill JR, Durham SK, Mookhtiar KA. Glucokinase regulatory protein may interact with glucokinase in the hepatocyte nucleus. Diabetes. 1997;46(2):179–186. doi: 10.2337/diab.46.2.179. [DOI] [PubMed] [Google Scholar]
- 14.de la Iglesia N, Veiga-da-Cunha M, Van Schaftingen E, Guinovart JJ, Ferrer JC. Glucokinase regulatory protein is essential for the proper subcellular localisation of liver glucokinase. FEBS Lett. 1999;456(2):332–338. doi: 10.1016/s0014-5793(99)00971-0. [DOI] [PubMed] [Google Scholar]
- 15.Toyoda Y, et al. Evidence for glucokinase translocation by glucose in rat hepatocytes. Biochem Biophys Res Commun. 1994;204(1):252–256. doi: 10.1006/bbrc.1994.2452. [DOI] [PubMed] [Google Scholar]
- 16.Detheux M, Vandercammen A, Van Schaftingen E. Effectors of the regulatory protein acting on liver glucokinase: A kinetic investigation. Eur J Biochem. 1991;200(2):553–561. doi: 10.1111/j.1432-1033.1991.tb16218.x. [DOI] [PubMed] [Google Scholar]
- 17.Futamura M, et al. An allosteric activator of glucokinase impairs the interaction of glucokinase and glucokinase regulatory protein and regulates glucose metabolism. J Biol Chem. 2006;281(49):37668–37674. doi: 10.1074/jbc.M605186200. [DOI] [PubMed] [Google Scholar]
- 18.Anderka O, et al. Biophysical characterization of the interaction between hepatic glucokinase and its regulatory protein: Impact of physiological and pharmacological effectors. J Biol Chem. 2008;283(46):31333–31340. doi: 10.1074/jbc.M805434200. [DOI] [PubMed] [Google Scholar]
- 19.Teplyakov A, Obmolova G, Badet-Denisot MA, Badet B, Polikarpov I. Involvement of the C terminus in intramolecular nitrogen channeling in glucosamine 6-phosphate synthase: Evidence from a 1.6 A crystal structure of the isomerase domain. Structure. 1998;6(8):1047–1055. doi: 10.1016/s0969-2126(98)00105-1. [DOI] [PubMed] [Google Scholar]
- 20.Veiga-da-Cunha M, Sokolova T, Opperdoes FR, Van Schaftingen E. Evolution of vertebrate glucokinase regulatory protein from a bacterial N-acetylmuramate 6-phosphate etherase. Biochem J. 2009;423(3):323–332. doi: 10.1042/BJ20090986. [DOI] [PubMed] [Google Scholar]
- 21.Veiga-da-Cunha M, Van Schaftingen E. Identification of fructose 6-phosphate- and fructose 1-phosphate-binding residues in the regulatory protein of glucokinase. J Biol Chem. 2002;277(10):8466–8473. doi: 10.1074/jbc.M105984200. [DOI] [PubMed] [Google Scholar]
- 22.Veiga-da-Cunha M, Courtois S, Michel A, Gosselain E, Van Schaftingen E. Amino acid conservation in animal glucokinases. Identification of residues implicated in the interaction with the regulatory protein. J Biol Chem. 1996;271(11):6292–6297. doi: 10.1074/jbc.271.11.6292. [DOI] [PubMed] [Google Scholar]
- 23.Zorn JA, Wells JA. Turning enzymes ON with small molecules. Nat Chem Biol. 2010;6(3):179–188. doi: 10.1038/nchembio.318. [DOI] [PubMed] [Google Scholar]
- 24.Veiga-Da-Cunha M, Detheux M, Watelet N, Van Schaftingen E. Cloning and expression of a Xenopus liver cDNA encoding a fructose-phosphate-insensitive regulatory protein of glucokinase. Eur J Biochem. 1994;225(1):43–51. doi: 10.1111/j.1432-1033.1994.00043.x. [DOI] [PubMed] [Google Scholar]
- 25.Agius L. The physiological role of glucokinase binding and translocation in hepatocytes. Adv Enzyme Regul. 1998;38:303–331. doi: 10.1016/s0065-2571(97)00001-0. [DOI] [PubMed] [Google Scholar]
- 26.Vandercammen A, Van Schaftingen E. Competitive inhibition of liver glucokinase by its regulatory protein. Eur J Biochem. 1991;200(2):545–551. doi: 10.1111/j.1432-1033.1991.tb16217.x. [DOI] [PubMed] [Google Scholar]
- 27.Goodey NM, Benkovic SJ. Allosteric regulation and catalysis emerge via a common route. Nat Chem Biol. 2008;4(8):474–482. doi: 10.1038/nchembio.98. [DOI] [PubMed] [Google Scholar]
- 28.Farrelly D, et al. Mice mutant for glucokinase regulatory protein exhibit decreased liver glucokinase: A sequestration mechanism in metabolic regulation. Proc Natl Acad Sci USA. 1999;96(25):14511–14516. doi: 10.1073/pnas.96.25.14511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bjørkhaug L, Molnes J, Søvik O, Njølstad PR, Flatmark T. Allosteric activation of human glucokinase by free polyubiquitin chains and its ubiquitin-dependent cotranslational proteasomal degradation. J Biol Chem. 2007;282(31):22757–22764. doi: 10.1074/jbc.M700517200. [DOI] [PubMed] [Google Scholar]
- 30.Olson AL, Pessin JE. Structure, function, and regulation of the mammalian facilitative glucose transporter gene family. Annu Rev Nutr. 1996;16:235–256. doi: 10.1146/annurev.nu.16.070196.001315. [DOI] [PubMed] [Google Scholar]
- 31.Shiota C, Coffey J, Grimsby J, Grippo JF, Magnuson MA. Nuclear import of hepatic glucokinase depends upon glucokinase regulatory protein, whereas export is due to a nuclear export signal sequence in glucokinase. J Biol Chem. 1999;274(52):37125–37130. doi: 10.1074/jbc.274.52.37125. [DOI] [PubMed] [Google Scholar]
- 32.Boyd ME, Albright EB, Foster DW, McGarry JD. In vitro reversal of the fasting state of liver metabolism in the rat. Reevaluation of the roles of insulin and glucose. J Clin Invest. 1981;68(1):142–152. doi: 10.1172/JCI110230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shiota M, et al. Inclusion of low amounts of fructose with an intraduodenal glucose load markedly reduces postprandial hyperglycemia and hyperinsulinemia in the conscious dog. Diabetes. 2002;51(2):469–478. doi: 10.2337/diabetes.51.2.469. [DOI] [PubMed] [Google Scholar]
- 34.Hallfrisch J, et al. Effects of dietary fructose on plasma glucose and hormone responses in normal and hyperinsulinemic men. J Nutr. 1983;113(9):1819–1826. doi: 10.1093/jn/113.9.1819. [DOI] [PubMed] [Google Scholar]
- 35.Hawkins M, et al. Fructose improves the ability of hyperglycemia per se to regulate glucose production in type 2 diabetes. Diabetes. 2002;51(3):606–614. doi: 10.2337/diabetes.51.3.606. [DOI] [PubMed] [Google Scholar]
- 36.Pfefferkorn JA, et al. Designing glucokinase activators with reduced hypoglycemia risk: discovery of N, N-dimethyl-5-(2-methyl-6-((5-methylpyrazin-2-yl)-carbamoyl) benzofuran-4-yloxy) pyrimidine-2-carboxamide as a clinical candidate for the treatment of type 2 diabetes mellitus. Med Chem Commun. 2011;2:828–839. [Google Scholar]
- 37.Bebernitz GR, et al. Investigation of functionally liver selective glucokinase activators for the treatment of type 2 diabetes. J Med Chem. 2009;52(19):6142–6152. doi: 10.1021/jm900839k. [DOI] [PubMed] [Google Scholar]
- 38.Pfefferkorn JA, et al. Discovery of (S)-6-(3-cyclopentyl-2-(4-(trifluoromethyl)-1H-imidazol-1-yl)propanamido)nicotinic acid as a hepatoselective glucokinase activator clinical candidate for treating type 2 diabetes mellitus. J Med Chem. 2012;55(3):1318–1333. doi: 10.1021/jm2014887. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.