

Abstract
Introduction
C-reactive protein (CRP) is a non-specific marker of inflammation linked to cardiovascular disease and possibly colon cancer. Polymorphisms in CRP have been associated with differential CRP concentrations among healthy adults, with some evidence for functional effects on CRP expression.
Methods
A linkage-disequilibrium-based tagSNP-selection algorithm identified six tagSNPs for Europeans (−821A>G, −390C>T/A 90A>T 838G>C 2043G>A and 4363C>A), defining 6 haplotypes >1% frequency. In a case-control study of adenomatous (n=491) or hyperplastic (n=184) polyps vs. polyp-free controls (n=583) we investigated these SNPs in relation to colorectal polyp risk.
Results
Individuals with 838 GC or CC genotypes had a modestly, although not statistically significantly, increased risk of adenomas (OR=1.4 95% CI 0.9-2.1) and a nearly 2-fold increased risk of concurrent adenomas and hyperplastic polyps (OR=2.0 95% CI 1.1-3.6). Increased risk for concurrent adenomas and hyperplastic polyps was also observed for haplotype ACACAC. No other main associations were detected. Risk of adenomas associated with 2043G>A differed with NSAID use. Among NSAID non-users, there was a suggestion that the GA or AA genotypes were associated with decreased risk of adenomas; this was not seen among NSAID users (p-interaction = 0.03). We also observed interactions between UGT1A1 [TA](7) promoter repeat polymorphism and CRP tagSNPs −390C>T/A and 90A>T, in which only the homozygous variant CRP genotype was associated with increased adenoma risk among those with the UGT1A1 6rpt/6rpt genotype (p-interaction= 0.02 and 0.04 for −390C>T/A and 90A>T, respectively).
Conclusions
These results provide limited support for associations between genetic variation in CRP and colorectal polyp risk. The observed interactions should be evaluated further.
Keywords: CRP, UGT, CYP2C9, colorectal cancer, colorectal polyps, NSAIDs, aspirin
Introduction
C-reactive protein (CRP) is a non-specific acute-phase protein secreted by the liver in response to pro-inflammatory cytokines such as IL-6. Elevated CRP levels have been associated with increased risk of colorectal cancer in four prospective studies [1–4], but not in two others [5, 6]. Genetic polymorphisms in CRP have been associated with changes in CRP serum or plasma concentrations [7–15]. However, several of these polymorphisms are in intronic or untranslated regions of the CRP gene and associations with CRP concentrations may be attributable to a linked, but not yet identified, polymorphism.
Non-steroidal anti-inflammatory drugs (NSAIDs), including COX-2 specific NSAIDs (coxibs), have been consistently associated with a reduced risk of colorectal neoplasia (reviewed in [16]) and have been shown in clinical trials to prevent polyp recurrence [17–21]. NSAIDs have also been shown in some, but not all, studies to reduce CRP concentrations (reviewed in [22]). Thus, the reduction of risk in colorectal neoplasia afforded by NSAIDs may be partly through or marked by their effects on CRP concentrations (see Figure).
Figure.
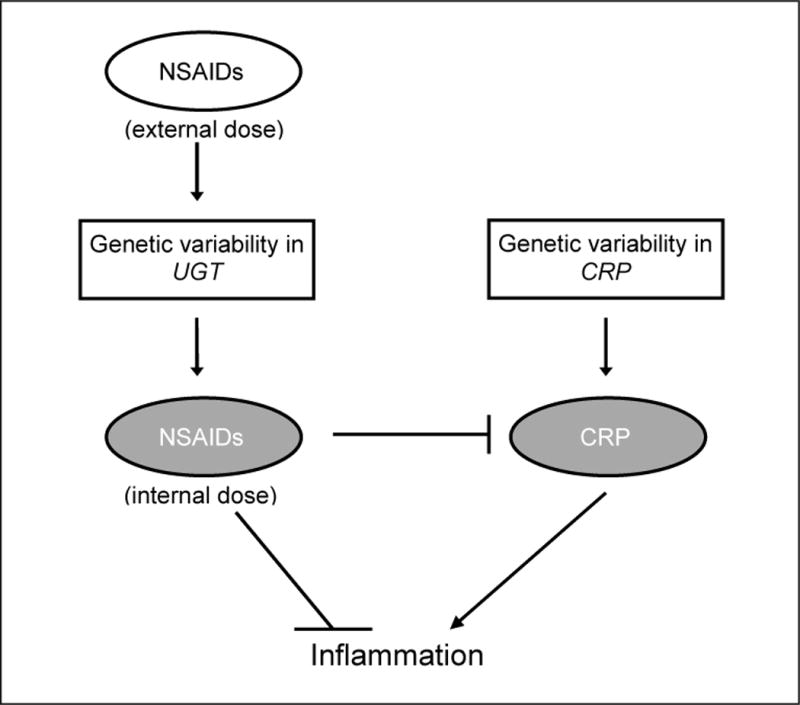
CRP and NSAID use in inflammation.
NSAIDs are primarily metabolized by oxidation or by glucuronidation. The former is achieved primarily by cytochrome P540 2C9 (CYP2C9) [23] and the latter by the UDP-glucoronosyltransferases (UGTs), such as UGT1A1, UGT1A6, UGT1A9, UGT2B4, and UGT2B7 [24]. Polymorphisms in several of these enzymes have been shown to alter drug metabolism [25–29]. Additionally, polymorphisms in CYP2C9 and UGT1A6 have been associated with risk of colorectal polyps or cancer and shown to interact with NSAID use in some, but not all, studies [30–33]. Because functional polymorphisms in NSAID-metabolism genes may alter the ability of NSAIDs to reduce CRP concentrations, we hypothesized that polymorphisms in NSAID metabolism may be effect modifiers of the relationship between genetic variation in CRP and risk of colorectal neoplasia (see Figure).
We selected tagSNPs to capture common CRP haplotypes (≥ 1% haplotype frequency); these haplotypes were analyzed in a colonoscopy-based case-control study of colorectal polyps. We also investigated whether CRP genotype or haplotype associations differed by NSAID use or by polymorphisms in genes encoding NSAID-metabolizing enzymes (UGTs and CYP2C9).
Materials and Methods
Participant recruitment has been previously described [30, 34, 35]. Briefly, adenoma and hyperplastic polyp cases and polyp-free controls were recruited through a large multiclinic gastroenterological practice in the Twin cities area of Minnesota from April 1991-April 1994. Eligibility criteria have been described elsewhere [34]; participants were aged 30-74 years, English-speaking residents of the Twin Cities metropolitan area with no known genetic syndrome associated with increased risk of colon neoplasia and no individual history of cancer (except non-melanoma skin cancer), prior colorectal polyps, or inflammatory bowel disease. Information on use of aspirin or other NSAIDs, diet, physical activity, anthropometrics, demographics, and medical history was obtained via questionnaire. The participation rate for all colonoscoped patients was 68%.
TagSNP selection
The CRP coding region and 2KB 5’ and 3’ of the gene was resequenced by the University of Washington-Fred Hutchinson Cancer Research Center Variation Discovery Resource (UW-FHCRC VDR) [36]. TagSNPs were selected using the LD Select algorithm developed by Carlson and colleagues [37] at the UW-FHCRC VDR. However, we used more stringent criteria specifically, a minor allele frequency of 4% (i.e. any variant that occurred twice in the resequencing effort) and an r2 value of 0.90. This resulted in the selection of six tagSNPs estimated by the Genome Variation Server (http://gvs.gs.washington.edu/GVS/index.jsp) to cover 85% of the variation in the CRP locus: −821A>G (rs2794521), −390C>T/A (rs3091244), 90A>T (rs1417938), 838G>C (L184L, rs1800947), 2043G>A (rs1205), and 4363C>A (rs3093075).
Genotyping
The selected CRP tagSNPs were detected by allelic discrimination using the 5’ nuclease assay on a 7900HT sequence detection system (Applied Biosystems, Foster City, CA). The 5’nuclease genotyping assays were validated by genotyping 92 individuals by both 5’ nuclease assay and RFLP or sequencing. There were no discrepancies between the two assays. The 20μl genotyping reactions contained 1x Taqman Core Reagents (Applied Biosystems), except for −821A>G for which the universal master mix was used, 0.5 units AmpliTaq DNA polymerase, 0.2 units AmpErase UNG, primers, probes, and 4ng genomic DNA. Primers, probes, Mg2+ concentrations, and cycling conditions are listed in Table 1. The triallelic polymorphism at −390 (−390C>T/A ) was run as a real-time assay and all the other polymorphisms as end-point assays. Positive controls for all the genotypes as well as four negative controls were included on each plate. For quality control purposes, genotyping for 94 randomly selected samples was repeated. There were no discrepancies.
Table 1.
PCR Conditions
| Polymorphism | PCR | Primers/Probes | [Mg2+] | [Primers, Probes] | Amplicon | Cycling |
|---|---|---|---|---|---|---|
| −821A>G | FP | 5'GGCCGTCATTTAGTGCCAA3' | 200nM | 50°C, 2min, 95°C, 10min, 40x 94°C, 30sec; 58°C, 45sec; 72°C, 1min 72°C, 5min |
||
| RP | 5'GTGCTGCACCCATTAACTCATC3' | 200nM | ||||
| A-allele | 5’6FAM-CACCGCATGTTCT-3’MGB | 2.5mM | 100nM | 250bp | ||
| G-allele | 5'VIC-CACCGCGTGTTCT-3’MGB | 100nM | ||||
|
| ||||||
| −390C>T/A | FP | 5'TCAGATTTCCTTTGTCAAACTCTATGA3' | 200nM |
50°C, 2min, 95°C, 10min, 40x 95°C, 15sec; 60°C, 90sec |
||
| RP | 5'TCCACTTTGGCTATCTATCCTGC3' | 200nM | ||||
| C-allele | 5’VIC-AACATATTAAACGAGTGGCCAT-3’MGB | 4mM | 100nM | 138bp | ||
| T-allele | 5’TET-CATATTAAACAAGTGGCCATC-3’MGB | 100nM | ||||
| A-allele | 5’6FAM-TAACATATTAAACTAGTGGCCATC-3’MGB | 100nM | ||||
|
| ||||||
| 90A>T | FP | 5'TGGCCAGACAGGTAAGGGC3' | 200nM | 50°C ,2min, 95°C, 10min, 40x 95°C, 15sec; 59°C, 90sec |
||
| RP | 5'ACCATGAAGGATGCTCCACTG3' | 200nM | ||||
| A-allele | 5’VIC-TCAGATCAAATCTCTCCCAT-3’MGB | 4mM | 100nM | 141bp | ||
| T-allele | 5’6FAM-TCAGATCAAAACTCTCCCATA-3’MGB | 100nM | ||||
|
| ||||||
| 838G>C | FP | 5'TGGGAGACATTGGAAATGTGAAC3' | 200nM | 50°C, 2min, 95°C, 10min, 40x 95°C, 15sec; 59°C, 1 min |
||
| RP | 5'CCGCCAAGATAGATGGTGTTAAT3' | 200nM | ||||
| G-allele | 5’VIC-TTGTGCTGTCACCAGA-3’MGB | 3mM | 100nM | 76bp | ||
| C-allele | 5’6FAM-TTTGTGCTCTCACCAGA-3’MGB | 100nM | ||||
|
| ||||||
| 2043G>A | FP | 5'GCCATCTTGTTTGCCACATG3' | 200nM | 50°C, 2min, 95°C, 10min, 40X 95°C, 15sec; 60°C, 1 min |
||
| RP | 5'CCCTTGGCTCCTCCACTTC3' | 200nM | ||||
| G-allele | 5'VIC-TGTCCTCACAGTCTC-3’MGB | 5mM | 100nM | 70bp | ||
| A-allele | 5'6-FAM-TGTCCTCATAGTCTCT-3’MGB | 150nM | ||||
|
| ||||||
| 4363C>A | FP | 5'AACCTAAAATCTCCCTGTGTCAGAA3' | 200nM | 50°C, 2min, 95°C, 10min, 40x 95°C, 15sec; 61°C, 2min 30sec |
||
| RP | 5'CTACTTACTTTGTCAGCTGGGACTCC3' | 200nM | ||||
| C-allele | 5’VIC-TTTCCATCAGGTCCCA-3’MGB | 4mM | 100nM | 263bp | ||
| A-allele | 5'6-FAM-TCCATCATGTCCCAGC-3’MGB | 100nM | ||||
Genotyping of CYP2C9, UGT1A1, and UGT1A6 has been described previously [27, 30]. Briefly, polymorphisms in CYP2C9 and UGT1A6 were genotyped by restriction fragment length polymorphism and oligonucleotide ligation assay. The UGT1A1 polymorphism was genotyped by PCR.
Haplotypes
Haplotypes were inferred using proc haplotype in SAS Genetics v.9 (SAS Institute, Cary, NC). All haplotypes predicted to occur with more than 1% frequency were analyzed separately and the haplotypes with lower frequency were grouped together for analyses. The most common haplotype among the controls was used as the referent group.
Statistical Methods
Three cases groups were defined: adenomas (n=477, hyperplastic polyps (n=177, and those with concurrent adenomas and hyperplastic polyps (n=112). Cases with concurrent adenomas and hyperplastic polyps were included in the adenoma and hyperplastic analyses.
Unconditional logistic regression was used to estimate odds ratios (ORs) and corresponding 95% confidence intervals (CI) for the associations between CRP genotypes and haplotypes and polyp risk. For SNPs with a minor allele frequency <10%, we grouped the homozygous variant genotypes with the heterozygous genotypes. A logistic regression model using GEE was used to analyze haplotype effects, using the haplotype probabilities as weights and clustering the haplotypes for each individual [38]. Global tests of haplotype associations were calculated using score tests in the genmod procedure. Covariates considered in our models included age, sex, body mass index, dietary intakes of fiber, alcohol, and energy, postmenopausal hormone use, and smoking. Effect modification by NSAID use was evaluated by the inclusion of multiplicative interaction terms in logistic regression models. Because use of NSAIDs may be associated with other known risk factors for colorectal neoplasia, we adjusted our NSAID-interaction analyses for the variables listed above. All statistical analyses were carried out using SAS v.9.
Results
A total of 1719 subjects were recruited into this study, of which 1217 had DNA available for genotyping. Characteristics of the study population have been described previously [30, 34, 35]. Briefly, the study population was mostly Caucasian (97.2%) and tagSNPs were selected within that group; adenoma cases tended to be older than hyperplastic polyp cases and controls. Both sets of cases were more likely to be male than controls. Genotype frequencies for all polymorphisms were in Hardy-Weinberg Equilibrium among the controls.
Risk of colon polyps associated with CRP genotypes and haplotypes are presented in Table 2 and in Supplemental Table S1 online. There were no differences in risk of adenomas or hyperplastic polyps for any of the genotypes, except possibly 838G>C, for which there was a non-significant increase in risk of adenoma (OR 1.4, 95%CI 0.9-2.1) and a marginally significant increased risk of concurrent adenomatous and hyperplastic polyps associated with the C allele (OR 2.0, 95%CI 1.1-3.6). This was no longer significant when Bonferroni correction for multiple testing was applied. Similarly, global tests of the haplotype associations were statistically non-significant (see Tables 2 and S1). There were no associations with adenoma risk observed for any of the individual imputed haplotypes; however the ACACAC haplotype and the grouping of all rare haplotypes were both associated with increased risk of concurrent adenomas and hyperplastic polyps. Multivariate adjustment for BMI, fiber intake, total energy intake, alcohol, hormone use (women), and smoking did not alter odds ratio estimates; thus results adjusted for age and sex are presented. Tests for heterogeneity of odds ratios, using the contrast statement in multinomial regression in SAS (the logistic procedure with the glogit link specified) in adenoma models vs. concurrent adenomas and hyperplastic polyps were statistically non-significant (p=0.86).
Table 2.
Risk of colorectal polyps associated with CRP genotypes1
| Controls (N=562) |
Adenomas (N=477) |
Concurrent adenomas and hyperplastic polyps (N=112) |
|||
|---|---|---|---|---|---|
| N | N | OR (95% CI) | N | OR (95% CI) | |
| CRPGenotype | |||||
| −821A>G rs2794521 | |||||
| AA | 294 | 245 | 1.0 (ref.) | 61 | 1.0 (ref.) |
| AG | 217 | 180 | 1.0 (0.7-1.3) | 41 | 0.8 (0.5-1.3) |
| GG | 51 | 53 | 1.4 (0.9-2.1) | 11 | 1.2 (0.5-2.5) |
| −390C>T/A rs3091244 (all genotypes) | |||||
| CC | 221 | 196 | 1.0 (ref.) | 43 | 1.0 (ref.) |
| CT | 215 | 181 | 1.0 (0.7-1.3) | 43 | 1.1 (0.6-1.8) |
| CA | 56 | 37 | 0.9 (0.6-1.5) | 12 | 1.5 (0.7-3.2) |
| TT | 42 | 42 | 1.1 (0.7-1.8) | 10 | 1.1 (0.5-2.4) |
| AA | 4 | 1 | 0.2 (0.1-1.4) | 0 | 0 (--) |
| TA | 25 | 20 | 0.9 (0.4-1.6) | 4 | 0.8 (0.3-2.7) |
| −390C>T/A rs3091244 (grouped by putative phenotype as in [15]) | |||||
| CC | 221 | 196 | 1.0 (ref.) | 43 | 1.0 (ref.) |
| CT or CA | 271 | 218 | 1.0 (0.7-1.3) | 55 | 1.2 (0.7-1.7) |
| TT, AA, or TA | 71 | 63 | 1.0 (0.6-1.4) | 14 | 0.9 (0.4-1.8) |
| 90A>T rs1417938 | |||||
| AA | 281 | 236 | 1.0 (ref.) | 57 | 1.0 (ref.) |
| AT | 239 | 200 | 1.0 (0.8-1.3) | 46 | 1.0 (0.6-1.5) |
| TT | 43 | 42 | 1.1 (0.6-1.8) | 10 | 1.0 (0.4-2.1) |
| 838G>C rs1800947 (L184L) | |||||
| GG | 504 | 414 | 1.00(ref.) | 94 | 1.0 (ref.) |
| GC or CC | 59 | 64 | 1.4 (0.9-2.1) | 19 | 2.0 (1.1-3.6) |
| 2043G>A rs1205 | |||||
| GG | 241 | 218 | 1.0 (ref.) | 50 | 1.0 (ref.) |
| GA | 262 | 210 | 0.9 (0.7-1.2) | 51 | 1.0 (0.6-1.7) |
| AA | 60 | 50 | 0.9 (0.6-1.4) | 12 | 1.1 (0.5-2.2) |
| 4363C>A rs3093075 | |||||
| CC | 477 | 419 | 1.0 (ref.) | 96 | 1.0 (ref.) |
| CA or AA | 86 | 57 | 0.9 (0.6-1.3) | 16 | 1.1 (0.6-2.0) |
| CRPhaplotype2(−821A>G; −390C>T/A; 90A>T; 838G>C; 2043G>A; 4363C>A) | |||||
| GCAGGC | 28.6 | 29.3 | 1.0 (ref.) | 26.9 | 1.0 (ref.) |
| ATTGGC | 28.4 | 29.7 | 1.0 (0.9-1.2) | 30.0 | 1.1 (0.8-1.4) |
| ACAGGC | 1.3 | 1.9 | 1.2 (0.8-1. 8) | 2.6 | 1.5 (0.8-2.8) |
| ACAGAC | 28.1 | 25.1 | 0.9 (0.8-1.1) | 24.3 | 1.0 (0.7-1.3) |
| ACACAC | 5.2 | 7.1 | 1.2 (0.9-1.6) | 7.8 | 1.6 (1.0-2.5) |
| AAAGGA | 8.1 | 6.3 | 0.9 (0.7-1.2) | 7.0 | 1.1 (0.7-1.78) |
| all rare haplotypes | 0.4 | 0.8 | 1.5 (0.7-3.1) | 1.3 | 2.0 (1.0-3.8) |
|
| |||||
| global p=0.63 | global p=0.49 | ||||
Age and sex adjusted.
Percents rather than total N are reported for haplotypes since they are inferred rather than determined.
We detected a significant interaction between regular aspirin or other NSAID use and the 2043G>A polymorphism (Table 3). Among those with the common allele for 2043G>A (i.e. GG), NSAID use was associated with a decreased risk of adenoma (OR 0.4, 95% CI 0.3-0.7), whereas among those with at least one A allele, NSAID use was not associated with a further decrease in adenoma risk (AG: OR 0.6, 95% CI 0.4-0.9; AA: OR 0.5, 95% CI 0.4-1.2; p-interaction = 0.03). However, when Bonferroni correction is applied for multiple testing, the p-value for this interaction is 0.18. No other NSAID interactions were observed for CRP tagSNPs (see Table S2 online). For CRP haplotypes, no interactions with NSAID use were observed (p-interaction=0.63 for adenomas). Because BMI is an important predictor of CRP levels, we also investigated potential interactions between BMI and CRP genotypes. No statistically significant interactions were observed (data not shown).
Table 3.
Association between CRP 2043G>A and risk of adenoma, stratified by NSAID use*
| Common Referent Group
|
Separate Referent Groups
|
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Aspirin or other NSAID use
|
Aspirin or other NSAID use
|
||||||||||||
| No
|
Yes
|
No
|
Yes
|
||||||||||
| Controls | Cases | Controls | Cases | Controls | Cases | Controls | Cases | ||||||
| (N) | (N) | OR (95% CI) | (N) | (N) | OR (95% CI) | (N) | (N) | OR (95% CI) | (N) | (N) | OR (95% CI) | ||
|
|
|
||||||||||||
| 2043G>A rs1205 | |||||||||||||
| GG | 124 | 145 | 1.0 (ref.) | 117 | 73 | 0.4 (0.3-0.7) | 124 | 145 | 1.0 (ref.) | 117 | 73 | 1.0 (ref.) | |
| GA | 154 | 125 | 0.6 (0.4-0.9) | 108 | 85 | 0.6 (0.4-0.9) | 154 | 125 | 0.6 (0.4-0.9) | 108 | 85 | 1.4 (0.9-2.3) | |
| AA | 34 | 33 | 0.7 (0.4-1.4) | 26 | 17 | 0.5 (0.2-1.1) | 34 | 33 | 0.7 (0.4-1.4-) | 26 | 17 | 1.2 (0.5-2.6) | |
|
| |||||||||||||
| p-interaction = 0.03 | |||||||||||||
Adjusted for age, sex, BMI, intakes of total calories, alcohol, and fiber, smoking (pack-years) and hormone use (females).
Statistically significant interactions were observed between two CRP polymorphisms (−390C>T/A and 90A>T) and the UGT1A1 promoter TA repeat polymorphism. Among those who were homozygous variant for either CRP genotype, the most frequent UGT1A1 genotype (6rpt/6rpt) was associated with increased risk of adenoma, whereas having an increased number of UGT1A1 repeats was associated with decreased risk (Table 4). No other interactions between CRP and UGT or CYP2C9 polymorphisms were observed (see Tables S3–S5 online).
Table 4.
CRP genotype, UGT1A1 genotype, and risk of colorectal adenoma1
| Common Referent Group
|
Separate Referent Groups
|
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| UGT1A1 genotype
|
UGT1A1 genotype
|
||||||||||||
| 6/6 (wildtype)
|
6/7, 6/8, 7/7, 7/8
|
6/6 (wildtype)
|
6/7, 6/8, 7/7, 7/8
|
||||||||||
| Controls | Cases | Controls | Cases | Controls | Cases | Controls | Cases | ||||||
| CRP Genotype | (N) | (N) | OR (95% CI) | (N) | (N) | OR (95% CI) | (N) | (N) | OR (95% CI) | (N) | (N) | OR (95% CI) | |
|
|
|
|
|||||||||||
| −390C>T/A rs3091244 (grouped by putative phenotype) | |||||||||||||
| CC | 101 | 92 | 1.0 (ref.) | 117 | 103 | 1.1 (0.7-1.7) | 101 | 92 | 1.0 (ref.) | 117 | 103 | 1.0 (ref.) | |
| CT or CA | 114 | 91 | 1.0 (0.7-1.6) | 154 | 126 | 1.1 (0.7-1.6) | 114 | 91 | 1.0 (0.7-1.6) | 154 | 126 | 1.0 (0.7-1.4) | |
| TT, AA, or TA | 25 | 32 | 2.1 (1.0-4.2) | 45 | 30 | 0.7 (0.4-1.2) | 25 | 32 | 2.1 (1.0-4.2) | 45 | 30 | 0.6 (0.3-1.1) | |
| p-interaction = 0.02 | p-trend=0.08 | p-trend=0.19 | |||||||||||
| 90A>T rs1417938 | |||||||||||||
| AA | 120 | 105 | 1.0 (ref.) | 156 | 130 | 1.1 (0.7-1.6) | 120 | 105 | 1.0 (ref.) | 156 | 130 | 1.0 (ref.) | |
| AT | 104 | 86 | 1.1 (0.7-1.7) | 133 | 112 | 1.0 (0.7-1.6) | 104 | 86 | 1.1 (0.7-1.7) | 133 | 112 | 1.0 (0.7-1.4) | |
| TT | 16 | 25 | 2.6 (1.2-5.6) | 27 | 17 | 0.7 (0.3-1.4) | 16 | 25 | 2.6 (1.2-5.6) | 27 | 17 | 0.6 (0.3-1.2) | |
| p-interaction = 0.04 | p-trend=0.07 | p-trend=0.31 | |||||||||||
Adjusted for age, sex, BMI, intakes of total calories, alcohol, and fiber, current NSAID use, smoking (pack-years) and hormone use (females).
Because CYP2C9 and UGT1A6 are the major biotransformation enzymes involved in the metabolism of NSAIDs and polymorphisms in these genes have been previously found to alter associations of NSAID use with colorectal neoplasia [30–33], we investigated whether non-synonymous SNPs in CYP2C9 (*2 and *3 alleles) and UGT1A6 (T181A + R184S or R184S alone) combined with NSAID use interacted with CRP SNPs. For the UGT1A6 polymorphism, the variant alleles are associated with slower drug metabolism [39], thus regular NSAID users with one or more variant alleles are likely to have the highest NSAID concentrations and, perhaps, the lowest risk of colorectal polyps [30]. Based on this hypothesis, we defined a three-level variable based on putative NSAID exposure: those who were wildtype and who didn’t use NSAIDs (=high risk) were in one group, those with any variant allele and who were regular NSAID users (=low risk) in a second group and a third group contained all other combinations (=intermediate). A statistically significant interaction was observed for UGT1A6/NSAID use and CRP −390C>T/A. Among those who were wildtype for UGT1A6 and did not use NSAIDs, there was no association with the CRP −390C>T/A polymorphism. However, among those with at least one variant UGT1A6 allele who regularly used NSAIDs, the lowest risk of adenoma was among those with the CRP homozygous variant alleles (Table 5; p-interaction = 0.03, Bonferroni correction p=0.18). Similar results were observed for hyperplastic polyps, although the interaction was not statistically significant. We followed a similar procedure for CYP2C9; however no interactions were observed (see Tables S6 and S7 online).
Table 5a.
CRP genotype, UGT1A6 genotype and NSAID use and risk of colorectal adenoma (common referent group)1
|
UGT1A6 genotype/regular NSAID use
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| wt and no NSAIDs
|
All others | 181A + 184S or 184S and NSAID use | |||||||
| Controls | Cases | Controls | Cases | Controls | Cases | ||||
| (N) | (N) | OR (95% CI) | (N) | (N) | OR (95% CI) | (N) | (N) | OR (95% CI) | |
|
|
|||||||||
| CRPgenotype | |||||||||
| −390C>T/A rs3091244 (grouped by putative phenotype) | |||||||||
| CC | 61 | 56 | 1.0 (ref.) | 111 | 98 | 1.0 (0.6-1.7) | 49 | 43 | 1.0 (0.5-1.8) |
| CT or CA | 68 | 55 | 1.0 (0.6-1.8) | 132 | 118 | 1.1 (0.7-1.8) | 70 | 46 | 0.8 (0.5-1.5) |
| TT, AA, or TA | 12 | 17 | 1.9 (0.7-5.0) | 32 | 37 | 1.5 (0.7-2.9) | 27 | 9 | 0.3 (0.1-0.7) |
| p-interaction = 0.03 | |||||||||
Adjusted for age, sex, BMI, intakes of total calories, alcohol, and fiber, smoking (pack-years) and hormone use (females).
Discussion
There is growing interest in the role of CRP in colorectal neoplasia. Studies of plasma or serum CRP measures and colorectal neoplasia risk generally suggest that increasing CRP concentrations are associated with increased risk of colorectal cancer [1–4], although these results are not entirely consistent [5, 6]. Our findings suggest that genetic variability in CRP is unlikely to play a role in colorectal neoplasia risk, but that polymorphisms in CRP may be relevant to the pharmacogenetics of NSAIDs. Out of six tagSNPs tested for an association with risk of colorectal polyps, only one, 838G>C, was associated with increased risk of concurrent adenomatous and hyperplastic polyps. This SNP is a synonymous polymorphism in exon 2 of the CRP gene (L184L) and the G allele has been previously, although not consistently, associated with higher plasma CRP levels [8, 12, 13, 40, 41].
To our knowledge only one previous study has examined polymorphisms in CRP and risk of colorectal neoplasia [4]. In that study, no association was observed between three tagSNPs (rs1130864, rs1205, and rs3093068) and colorectal cancer risk. One of these SNPs, rs1205, was included in our study; we also found no association. The other two SNPs in that study are in LD (r2 ≥ 90%) with two SNPs that we included in our study (rs1130864 is in LD with 90A>T and rs3093068 is in LD with 4363C>A); we also found no associations with these SNPs. No previous studies have examined potential interactions between variation in CRP and regular NSAID use. However, given the importance of aspirin and other NSAIDs for chemoprevention of cardiovascular disease and colorectal neoplasia, and the increasing use of CRP as a biomarker for inflammation, we suggest that potential interactions between NSAID use and CRP genetic variation should be considered in future etiologic studies of the associations between CRP and colorectal cancer risk.
In this exploratory study, we observed several interactions with NSAID use, NSAID-metabolizing enzymes, or the combination, suggesting that research into the role of CRP in cancer risk should take NSAID use into account. NSAID use has been reported to reduce CRP levels (reviewed in [22]). For the NSAID interaction, the OR among the homozygous variant NSAID users was higher than would be expected in NSAID use and CRP genotype were independent risk factors (expected: 0.7*0.4=0.28 observed: 0.5, p-0.05). This observed interaction suggests that NSAID use may not be beneficial to all, but rather may be more effective among people with higher underlying inflammation. We have previously reported on NSAID-gene interactions in genes relevant to prostaglandin synthesis [42–44] and functional polymorphisms in NSAID-metabolizing enzymes UGT1A6 and CYP2C9 have also been reported to alter the NSAID-colorectal neoplasia association [30–33]. Although our findings require confirmation, the current results add to the evidence that pharmacogenetic studies are necessary to truly understand which patients are most likely to benefit from NSAID chemoprevention and which are most likely to experience side effects.
Because we chose tagSNPs rather than candidate functional SNPs for this study, we had no prior hypotheses as to the association between CRP polymorphisms and colorectal polyp risk. However, several of these tagSNPs have been previously associated with plasma or serum CRP levels, indicating that at least one of them has functional effects. Most consistently, the T and A alleles of −390C>T/A have been previously associated with increased plasma CRP concentrations [13–15, 45, 46]. Our results do not support a role for this polymorphism in polyp risk, but we did observe an interaction between −390C>T/A and the combination of UGT1A6 genotype and regular NSAID use. In this interaction, those with two variant -390 alleles (i.e. AA, TT or AT) had a decreased risk in combination with having UGT1A6 variant alleles, which are associated with slower drug metabolism, and NSAID use. If the CRP alleles are associated with increased CRP production, then this is the group in which NSAID use, particularly among slow metabolizers, would be expected to be the most effective. This and other reported interactions require confirmation in other studies.
This was an initial exploratory study of the association between CRP SNPs and colorectal neoplasia. We performed 48 tests for interaction; at α=0.05, by chance, we would expect at least 2 to be significant. We recognize that our sample size was relatively small and that our findings may be false positives. However, because this study was meant to inform further investigations of CRP variability and neoplasia risk, we decided to report all significant and near-significant findings.
In summary, we found limited evidence for an association between genetic variability in CRP and colorectal polyp risk, yet these polymorphisms may interact with NSAID use or NSAID-metabolizing enzymes. Further research is required to confirm these findings in larger studies of colorectal neoplasia. A comprehensive investigation of the genetics of biologic pathways relevant to inflammation, including prostaglandin synthesis and pro- and anti-inflammatory cytokines will further our understanding of the role of inherited susceptibility in colorectal cancer risk. Clearly such studies require information on use of NSAIDs.
Supplementary Material
Table 5b.
CRP genotype, UGT1A6 genotype and NSAID use and risk of colorectal adenoma (separately by strata of UGT1A6 genotype and NSAID use)1
|
UGT1A6 genotype/regular NSAID use
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| wt and no NSAIDs
|
All others | 181A + 184S or 184S and NSAID use | |||||||
| Controls | Cases | Controls | Cases | Controls | Cases | ||||
| (N) | (N) | OR (95% CI) | (N) | (N) | OR (95% CI) | (N) | (N) | OR (95% CI) | |
|
|
|||||||||
| CRPgenotype | |||||||||
| −390C>T/A rs3091244 (grouped by putative phenotype) | |||||||||
| CC | 61 | 56 | 1.0 (ref.) | 111 | 98 | 1.0 (ref.) | 49 | 43 | 1.0 (ref.) |
| CT or CA | 68 | 55 | 1.0 (0.6-1.8) | 132 | 118 | 1.0 (0.7-1.6)) | 70 | 46 | 0.9 (0.5-1.7) |
| TT, AA, or TA | 12 | 17 | 1.9 (0.7-5.0) | 32 | 37 | 1.4 (0.8-2.7) | 27 | 9 | 0.3 (0.1-0.7) |
| p-trend=0.28 | p-trend=0.32 | p-trend=0.02 | |||||||
Adjusted for age, sex, BMI, intakes of total calories, alcohol, and fiber, smoking (pack- years) and hormone use (females).
Acknowledgments
The authors would like to thank Dr. Roberd Bostick and Lisa Fosdick for their contributions to the initial establishment of this study. We would also like to thank Dr. Chris Carlson for advice regarding tagSNP selection and functional relevance of genetic variants in CRP.
References
- 1.Erlinger TP, Platz EA, Rifai N, Helzlsouer KJ. C-reactive protein and the risk of incident colorectal cancer. JAMA. 2004;291:585–90. doi: 10.1001/jama.291.5.585. [DOI] [PubMed] [Google Scholar]
- 2.Otani T, Iwasaki M, Sasazuki S, Inoue M, Tsugane S. Plasma C-reactive protein and risk of colorectal cancer in a nested case-control study: Japan Public Health Center-based prospective study. Cancer Epidemiol Biomarkers Prev. 2006;15:690–5. doi: 10.1158/1055-9965.EPI-05-0708. [DOI] [PubMed] [Google Scholar]
- 3.Gunter MJ, Stolzenberg-Solomon R, Cross AJ, Leitzmann MF, Weinstein S, Wood RJ, et al. A prospective study of serum C-reactive protein and colorectal cancer risk in men. Cancer Res. 2006;66:2483–7. doi: 10.1158/0008-5472.CAN-05-3631. [DOI] [PubMed] [Google Scholar]
- 4.Siemes C, Visser LE, Coebergh JW, Splinter TA, Witteman JC, Uitterlinden AG, et al. C-reactive protein levels, variation in the C-reactive protein gene, and cancer risk: the Rotterdam Study. J Clin Oncol. 2006;24:5216–22. doi: 10.1200/JCO.2006.07.1381. [DOI] [PubMed] [Google Scholar]
- 5.Zhang SM, Buring JE, Lee IM, Cook NR, Ridker PM. C-reactive protein levels are not associated with increased risk for colorectal cancer in women. Ann Intern Med. 2005;142:425–32. doi: 10.7326/0003-4819-142-6-200503150-00008. [DOI] [PubMed] [Google Scholar]
- 6.Ito Y, Suzuki K, Tamakoshi K, Wakai K, Kojima M, Ozasa K, et al. Colorectal cancer and serum C-reactive protein levels: a case-control study nested in the JACC Study. J Epidemiol. 2005;15(Suppl 2):S185–9. doi: 10.2188/jea.15.S185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Szalai AJ, McCrory MA, Cooper GS, Wu J, Kimberly RP. Association between baseline levels of C-reactive protein (CRP) and a dinucleotide repeat polymorphism in the intron of the CRP gene. Genes Immun. 2002;3:14–9. doi: 10.1038/sj.gene.6363820. [DOI] [PubMed] [Google Scholar]
- 8.Zee RY, Ridker PM. Polymorphism in the human C-reactive protein (CRP) gene, plasma concentrations of CRP, and the risk of future arterial thrombosis. Atherosclerosis. 2002;162:217–9. doi: 10.1016/s0021-9150(01)00703-1. [DOI] [PubMed] [Google Scholar]
- 9.Brull DJ, Serrano N, Zito F, Jones L, Montgomery HE, Rumley A, et al. Human CRP gene polymorphism influences CRP levels: implications for the prediction and pathogenesis of coronary heart disease.[see comment] Arterioscler Thromb Vasc Biol. 2003;23:2063–9. doi: 10.1161/01.ATV.0000084640.21712.9C. [DOI] [PubMed] [Google Scholar]
- 10.Russell AI, Cunninghame Graham DS, Shepherd C, Roberton CA, Whittaker J, Meeks J, et al. Polymorphism at the C-reactive protein locus influences gene expression and predisposes to systemic lupus erythematosus. Hum Mol Genet. 2004;13:137–147. doi: 10.1093/hmg/ddh021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Obisesan TO, Leeuwenburgh C, Phillips T, Ferrell RE, Phares DA, Prior SJ, et al. C-reactive protein genotypes affect baseline, but not exercise training-induced changes, in C-reactive protein levels. Arterioscler Thromb Vasc Biol. 2004;24:1874–9. doi: 10.1161/01.ATV.0000140060.13203.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Suk HJ, Ridker PM, Cook NR, Zee RY. Relation of polymorphism within the C-reactive protein gene and plasma CRP levels. Atherosclerosis. 2005;178:139–45. doi: 10.1016/j.atherosclerosis.2004.07.033. [DOI] [PubMed] [Google Scholar]
- 13.Kovacs A, Green F, Hansson LO, Lundman P, Samnegard A, Boquist S, et al. A novel common single nucleotide polymorphism in the promoter region of the C-reactive protein gene associated with the plasma concentration of C-reactive protein. Atherosclerosis. 2005;178:193–8. doi: 10.1016/j.atherosclerosis.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 14.Szalai AJ, Wu J, Lange EM, McCrory MA, Langefeld CD, Williams A, et al. Single-nucleotide polymorphisms in the C-reactive protein (CRP) gene promoter that affect transcription factor binding, alter transcriptional activity, and associate with differences in baseline serum CRP level. J Mol Med. 2005;83:440–7. doi: 10.1007/s00109-005-0658-0. [DOI] [PubMed] [Google Scholar]
- 15.Carlson CS, Aldred SF, Lee PK, Tracy RP, Schwartz SM, Rieder M, et al. Polymorphisms within the C-reactive protein (CRP) promoter region are associated with plasma CRP levels. Am J Hum Genet. 2005;77:64–77. doi: 10.1086/431366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bosetti C, Gallus S, La Vecchia C. Aspirin and cancer risk: an updated quantitative review to 2005. Cancer Causes Contr. 2006;17:871–88. doi: 10.1007/s10552-006-0033-7. [DOI] [PubMed] [Google Scholar]
- 17.Baron JA, Cole BF, Sandler RS, Haile RW, Ahnen D, Bresalier R, et al. A randomized trial of aspirin to prevent colorectal adenomas. New Engl J Med. 2003;348:891–9. doi: 10.1056/NEJMoa021735. [DOI] [PubMed] [Google Scholar]
- 18.Sandler RS, Halabi S, Baron JA, Budinger S, Paskett E, Keresztes R, et al. A randomized trial of aspirin to prevent colorectal adenomas in patients with previous colorectal cancer. New Engl J Med. 2003;348:883–90. doi: 10.1056/NEJMoa021633. [DOI] [PubMed] [Google Scholar]
- 19.Baron JA, Sandler RS, Bresalier RS, Quan H, Riddell R, Lanas A, et al. A randomized trial of rofecoxib for the chemoprevention of colorectal adenomas. Gastroenterology. 2006;131:1674–82. doi: 10.1053/j.gastro.2006.08.079. [DOI] [PubMed] [Google Scholar]
- 20.Arber N, Eagle CJ, Spicak J, Racz I, Dite P, Hajer J, et al. Celecoxib for the prevention of colorectal adenomatous polyps. New Engl J Med. 2006;355:885–895. doi: 10.1056/NEJMoa061652. [DOI] [PubMed] [Google Scholar]
- 21.Bertagnolli MM, Eagle CJ, Zauber AG, Redston M, Solomon DH, Kim K, et al. Celecoxib for the prevention of sporadic colorectal adenomas. New Engl J Med. 2006;355:873–874. doi: 10.1056/NEJMoa061355. [DOI] [PubMed] [Google Scholar]
- 22.Prasad K. C-reactive protein (CRP)-lowering agents. Cardiovasc Drug Rev. 2006;24:33–50. doi: 10.1111/j.1527-3466.2006.00033.x. [DOI] [PubMed] [Google Scholar]
- 23.Miners JO, Birkett DJ. Cytochrome P4502C9: an enzyme of major importance in human drug metabolism. Br J Clin Pharmacol. 1998;45:525–38. doi: 10.1046/j.1365-2125.1998.00721.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kuehl GE, Lampe JW, Potter JD, Bigler J. Glucuronidation of nonsteroidal anti-inflammatory drugs (NSAIDs): identifying the enzymes responsible in human liver microsomes. Drug Metabol Dispos. 2005;33:1027–35. doi: 10.1124/dmd.104.002527. [DOI] [PubMed] [Google Scholar]
- 25.Takahashi H, Kashima T, Nomoto S, Iwade K, Tainaka H, Shimizu T, et al. Comparisons between in-vitro and in-vivo metabolism of (S)-warfarin: catalytic activities of cDNA-expressed CYP2C9, its Leu359 variant and their mixture versus unbound clearance in patients with the corresponding CYP2C9 genotypes. Pharmacogenetics. 1998;8:365–73. doi: 10.1097/00008571-199810000-00001. [DOI] [PubMed] [Google Scholar]
- 26.Rettie AE, Wienkers LC, Gonzalez FJ, Trager WF, Korzekwa KR. Impaired (S)-warfarin metabolism catalysed by the R144C allelic variant of CYP2C9. Pharmacogenetics. 1994;4:39–42. doi: 10.1097/00008571-199402000-00005. [DOI] [PubMed] [Google Scholar]
- 27.Lampe JW, Bigler J, Horner NK, Potter JD. UDP-glucuronosyltransferase (UGT1A1*28 and UGT1A6*2) polymorphisms in Caucasians and Asians: relationships to serum bilirubin concentrations. Pharmacogenetics. 1999;9:341–9. doi: 10.1097/00008571-199906000-00009. [DOI] [PubMed] [Google Scholar]
- 28.Iyer L, Das S, Janisch L, Wen M, Ramirez J, Karrison T, et al. UGT1A1*28 polymorphism as a determinant of irinotecan disposition and toxicity. Pharmacogenomics Journal. 2002;2:43–7. doi: 10.1038/sj.tpj.6500072. [DOI] [PubMed] [Google Scholar]
- 29.Sparks R, Ulrich CM, Bigler J, Tworoger SS, Yasui Y, Rajan KB, et al. UDP-glucuronosyltransferase and sulfotransferase polymorphisms, sex hormone concentrations, and tumor receptor status in breast cancer patients. Breast Cancer Res. 2004;6:R488–98. doi: 10.1186/bcr818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bigler J, Whitton J, Lampe JW, Fosdick L, Bostick RM, Potter JD. CYP2C9 and UGT1A6 genotypes modulate the protective effect of aspirin on colon adenoma risk. Cancer Res. 2001;61:3566–9. [PubMed] [Google Scholar]
- 31.Chan AT, Tranah GJ, Giovannucci EL, Hunter DJ, Fuchs CS. Genetic variants in the UGT1A6 enzyme, aspirin use, and the risk of colorectal adenoma. J Natl Cancer Inst. 2005;97:457–60. doi: 10.1093/jnci/dji066. [DOI] [PubMed] [Google Scholar]
- 32.Samowitz WS, Wolff RK, Curtin K, Sweeney C, Ma KN, Andersen K, et al. Interactions between CYP2C9 and UGT1A6 polymorphisms and nonsteroidal anti-inflammatory drugs in colorectal cancer prevention. Clin Gastroenterol Hepatol. 2006;4:894–901. doi: 10.1016/j.cgh.2006.04.021. [DOI] [PubMed] [Google Scholar]
- 33.Hubner RA, Muir KR, Liu JF, Logan RF, Grainge M, Armitage N, et al. Genetic variants of UGT1A6 influence risk of colorectal adenoma recurrence. Clin Cancer Res. 2006;12:6585–9. doi: 10.1158/1078-0432.CCR-06-0903. [DOI] [PubMed] [Google Scholar]
- 34.Potter JD, Bostick RM, Grandits GA, Fosdick L, Elmer P, Wood J, et al. Hormone replacement therapy is associated with lower risk of adenomatous polyps of the large bowel: the Minnesota Cancer Prevention Research Unit Case-Control Study. Cancer Epidemiol Biomarkers Prev. 1996;5:779–84. [PubMed] [Google Scholar]
- 35.Morimoto LM, Newcomb PA, Ulrich CM, Bostick RM, Lais CJ, Potter JD. Risk factors for hyperplastic and adenomatous polyps: evidence for malignant potential? Cancer Epidemiol Biomarkers Prev. 2002;11:1012–8. [PubMed] [Google Scholar]
- 36.UW-FHCRC Variation Discovery Resource. http://pga.gs.washington.edu/
- 37.Carlson CS, Eberle MA, Rieder MJ, Yi Q, Kruglyak L, Nickerson DA. Selecting a maximally informative set of single-nucleotide polymorphisms for association analyses using linkage disequilibrium. Am J Hum Genet. 2004;74:106–20. doi: 10.1086/381000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liang K-Y, Zeger S. Longitudinal data analysis using generalized linear models. Biometrika. 1986;73:13–22. [Google Scholar]
- 39.Ciotti M, Marrone A, Potter C, Owens IS. Genetic polymorphism in the human UGT1A6 (planar phenol) UDP-glucuronosyltransferase: pharmacological implications. Pharmacogenetics. 1997;7:485–95. doi: 10.1097/00008571-199712000-00007. [DOI] [PubMed] [Google Scholar]
- 40.Eklund C, Lehtimaki T, Hurme M. Epistatic effect of C-reactive protein (CRP) single nucleotide polymorphism (SNP) +1059 and interleukin-1B SNP +3954 on CRP concentration in healthy male blood donors. Int J Immunogenet. 2005;32:229–32. doi: 10.1111/j.1744-313X.2005.00515.x. [DOI] [PubMed] [Google Scholar]
- 41.Davey Smith G, Lawlor DA, Harbord R, Timpson N, Rumley A, Lowe GD, et al. Association of C-reactive protein with blood pressure and hypertension: life course confounding and mendelian randomization tests of causality. Arterioscler Thromb Vasc Biol. 2005;25:1051–6. doi: 10.1161/01.ATV.0000160351.95181.d0. [DOI] [PubMed] [Google Scholar]
- 42.Ulrich CM, Bigler J, Sparks R, Whitton J, Sibert JG, Goode EL, et al. Polymorphisms in PTGS1 (=COX-1) and risk of colorectal polyps. Cancer Epidemiol Biomarkers Prev. 2004;13:889–893. [PubMed] [Google Scholar]
- 43.Ulrich CM, Whitton J, Yu JH, Sibert J, Sparks R, Potter JD, et al. PTGS2 (COX-2) −765G > C promoter variant reduces risk of colorectal adenoma among nonusers of nonsteroidal anti-inflammatory drugs. Cancer Epidemiol Biomarkers Prev. 2005;14:616–9. doi: 10.1158/1055-9965.EPI-04-0510. [DOI] [PubMed] [Google Scholar]
- 44.Poole E, Bigler J, Whitton J, Potter J, Sibert J, Ulrich C. Prostacyclin synthase and arachidonate 5-lipoxygenase polymorphisms and risk of colorectal polyps. Cancer Epidemiol Biomarkers Prev. 2006;15:502–508. doi: 10.1158/1055-9965.EPI-05-0804. [DOI] [PubMed] [Google Scholar]
- 45.Kathiresan S, Larson MG, Vasan RS, Guo CY, Gona P, Keaney JF, Jr, et al. Contribution of clinical correlates and 13 C-reactive protein gene polymorphisms to interindividual variability in serum C-reactive protein level. Circulation. 2006;113:1415–23. doi: 10.1161/CIRCULATIONAHA.105.591271. [DOI] [PubMed] [Google Scholar]
- 46.Crawford DC, Sanders CL, Qin X, Smith JD, Shephard C, Wong M, et al. Genetic variation is associated with C-reactive protein levels in the Third National Health and Nutrition Examination Survey. Circulation. 2006;114:2458–65. doi: 10.1161/CIRCULATIONAHA.106.615740. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.