

Abstract
Cationic Rh(II) complexes are able to catalyze the regioselective hydroamination of propargyl ureas in a 6-endo fashion. This transformation permits access to interesting substitution patterns of dihydropyrimidines which have found use as nucleotide exchange factor inhibitors.
Keywords: dihydropyrimidone, heterocycle, hydroamination, Rhodium catalysis, ARF 6
1. Introduction
Accesible through multicomponent reactions, the dihydropyrimidone scaffold has been prevalent in both pharmaceutical and academic screening campaigns for decades.1 Given the cornucopia of biological activities reported for this heterocyclic core,2 methodologies have developed advancing the vetted Biginelli condensation3 to include enantioselective variants and orthogonal substitution patterns.4,5 Recently Rovis and co-workers reported an enantioselective Rh(I) catalyzed [4 + 2] cycloaddition of α, β-unsaturated imines and isocyanates to generate dihydropyrimidones, highlighting the continuing need for methodologies to access this core.6
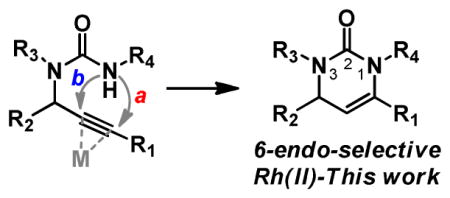
Our group has recently been interested in the cyclization of propargylguanidines, particlulary from the vantage of controlling the cyclization in either a 5-exo or 6-endo fashion.7 The application of this strategy to propargyl ureas introduces an added level of selectivity: that is not only are 5-exo and 6-endo modes of cyclization operable but they can occur through either the urea nitrogen or oxygen (Figure 1).
Figure 1.

Dihydropyrimidone strategy.
The cyclization of propargyl ureas and related propargycarbmates have been extensively studied, most frequently using salts of gold,8 silver,9 palladium,10 and copper.11 Kinetically favored, cyclization to form the 5-membered ring is most commonly observed, both with N- and O-connectivity. Recently Toste and co-workers described the isolation of the dihydrooxazinoneimine in significant quantities from the cationic Au(I)-catalyzed cyclization of N-Ts-propargyl ureas,8o but without fail the six-membered rings are the minor component of the cyclization event. To the best of our knowledge the cyclization of propargyl ureas, selectively in a 6-endo fashion through the urea nitrogen, has not been observed. Achieving this selectivity would greatly add to our ability to create new substitution patterns of a biomedically significant small molecule scaffold.
2. Results and Discussion
2.1. Cationic-Rh(II) cyclizations of propargyl ureas
Having noted the ability of dirhodium-(II)-carboxylates to selectively catalyze the 6-endo cyclization of propargylguanidines, in direct contrast to other π-Lewis acids, we were encouraged to explore the use of these catalysts in the cyclization of propargyl ureas.7b From our previous research AgOAc and Rh2(Oct)4 were preferred to selectively promote the 5-exo and 6-endo modes of cyclization, respectively.
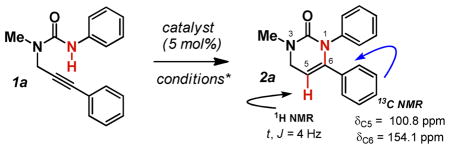
We initially examined the ability of πLewis acids to catalyze the cyclization of propargylurea 1a (Table 1). Surprisingly AgOAc failed to catalyze the cyclization at room temperature or 70 °C (entries 2 and 3) although Van der Eycken has recently reported that this transformation is possible with 20 mol% AgOTf at 110 °C, confirming that the propargyl ureas are much less nucleophilic than their di-Boc guanidine counterparts.9e More discouraging was the fact that Rh2(Oct)4 also failed to promote the cyclization (entries 4 and 5). The more Lewis acidic Rh2(TFA)4 was then examined. While unreactive at room temperature (entry 6), formation of a single cyclization product (2a) occurred when the temperature was increased to 70 °C, albeit in 50% isolated yield (entry 7). Encouragingly, the product did appear to have cyclized in a 6-endo fashion as evidenced by the coupling constant of the vinylic proton at C5 (3J = 4.0 Hz). Further the 13C chemical shifts of C5 and C6 were much more consistent with enamine connectivity versus that of an enol-ether. We anticipated that a suitable cationic Rh(II) complex, with more labile ligands, might lend the greater Lewis acidity and improve the reactivity. To test this assumption, we turned our attention to the cationic catalyst [Rh2(OAc)2(MeCN)6][BF4]2.12 To our delight, this provided 2a in 94% isolated yield with no trace of the other three possible cyclization products observable by 1H NMR.
Table 1.
Catalysts and solvents screen
| ||||
|---|---|---|---|---|
| entry | catalyst (5 mol %) | solvent | temp. (°C) | isolated yield (%) 2a |
| 1 | N/A | CH2Cl2 | 80 | NR |
| 2 | AgOAc | HOAc/CH2Cl2 | rt | NR |
| 3 | AgOAc | HOAc/CH2Cl2 | 70 | NR |
| 4 | Rh2(Oct)4 | CH2Cl2 | rt | NR |
| 5 | Rh2(Oct)4 | CH2Cl2 | 70 | NR |
| 6 | Rh2(TFA)4 | CH2Cl2 | rt | NR |
| 7 | Rh2(TFA)4 | CH2Cl2 | 70 | 50 |
| 8 | [Rh2(OAc)2(MeCN)]2[BF4]2 | CH2Cl2 | 80 | 94 |
All reactions were run at 0.1M in a Schlenk tube under an N2 atmosphere for 24h.
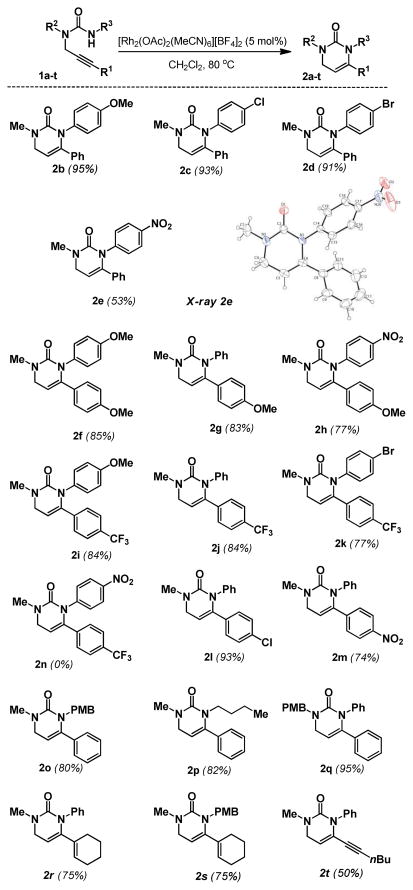
As mentioned, the competency of the more Lewis-acidic Rh(II) catalysts suggests that the propargyl ureas are inherently less nucleophilic than the related guanidines. Therefore it is likely that the nucleophilicity of the urea nitrogen and the electrophilicity of the metal-alkyne complex, might be critically coupled to the success of this reaction. Defining the limits of reactivity at these positions became the emphasis of our study on substrate scope (Table 2). With the phenyl substituted alkyne (R1 = Ph), cyclization is productive with urea substituents that span the electronic spectrum from p-MeOPh to p-NO2Ph (e.g. 2a–e). The p-NO2-Ph substituted urea 2e proved to be a highly crystalline solid, from which we were able to unambiguously prove the connectivity of these dihydropyrimidones via X-ray crystallography. Electron rich alkynes (e.g. p-MeOPh substituted) also reacted well with electron rich and poor ureas, 2f–h. If the alkyne is electron poor (e.g. p-CF3Ph substituted) it reacts well with most ureas (2i–k) but fails to react with an electron poor p-NO2Ph substituted urea (e.g. 2n). Other electron poor alkynes can also react as long as the urea is not extremely electron poor (2l–m). The reaction is also successful if the substrate is N1, N3-dialkyl substituted (2o,p) or N1, N3-alkyl/aryl substituted (2q). Alkyl substituted alkynes, while reactive gave complex mixtures that were intractable on a preparative scale. However, ene-ynes and yne-ynes react cleanly to give (2r–t) which are logical precursors to alkyl substitution patterns. Substrates bearing an N3-Tosyl or N3-Boc group are also not reactive, further defining that highly electron-withdrawing groups shut down the reaction. It is noteworthy that all of the successful cyclizations resulted in exclusive formation of the dihydropyrimidone.
Table 2.
Reaction Scope
|
When contemplating this unique reactivity, it is quite remarkable that Rh(II) catalyzes this bond forming event with two-fold thermodynamic selectivity: first to generate the 6-endo product and secondly to generate the C-N bond. One potential explanation for this is the Markovnikov-selective hydration of the alkyne followed by condensation of the urea on the resultant ketone. However, the addition of water (up to 1 equiv.) or desiccants did not affect the efficiency of the reaction. Further, Rh(II) does not appear to be a competent Lewis acid for the condensation of a preformed urea-ketone.13 Examples of alkyne activation by dirhodium(II) complex are quite rare.14 Thus further investigations are warranted to understand this unique selectivity, and the potential reversibility of the initial amino- or oxo-rhodation.
2.2. Application of the resultant dihydropyrimidones
A testament to the value of these scaffolds, we identified compound 2k capable of inhibiting proliferation of the LN-229 glioblastoma cell line (IC50 = 25 μM) (Fig. 2A). The EGF-dependent proliferation of glioblastoma cells has been directly linked to the activation of Adenosine diphosphate-ribosylation factor 6 (ARF6).15 Known inhibitors of this enzyme, e.g. secin-44, are comprised of a 1,2-disubstituted triazole which shares the diaryl-orienation as delivered by the methodology described above.16 One drawback with the triazole inhibitors is their poor solubility. We reasoned that replacement of the triazole backbone with a dihydropyrimidine should reduce the amount of unsaturation, increase polarity and thus increase solubility. To test this we prepared the analog 4 bearing the secin sidechains, via reduction of the nitro group in 2h with Ni2B followed EDCI mediated coupling to 5-bromofuroic acid. Indeed compound 4 inhibits activation of ARF6. As shown in Fig. 2B, activation of ARF6 occurs only in the presence of ARNO (ARF nucleotide-binding site opener) (lanes 1 and 2).17 Addition of 4 inhibits this activation by nearly 50% at 50 μM (lane 3), suggesting that this small molecule might be useful for inhibiting cellular behaviors that are controlled by activated ARF6. We further investigated whether compound 4 can inhibit cell proliferation, and it does so in a dose dependent manner with an IC50 of 17 μM (Fig. 2C, all measurements after 24 hr).18 This activity is similar to secin-44 which has an IC50 of 15 μM and contrasts with an inactive variant secin-44neg (in which an acetyl group replaces the bromofuranoic acid appendage). Moreover, 4 had improved solubility (soluble up to 100 μM) compared to secin-44 which will enable us to better evaluate the inhibition of ARF6 activation as a therapeutic target in vivo.
Figure 2.

Manipulation of the secin scaffold for increased solubility.
3. Conclusions
In conclusion, we have demonstrated that cationic dirhodium(II) is capable of catalyzing the chemo- and regioselective hydroamination of propargyl ureas. The ability of Rh(II) to selectively catalyze the 6-endo selective cyclization through the urea nitrogen further illustrates its unique ability to activate alkynes for nucleophilic addition. This methodology delivers dihydropyrimidones with as of yet unexplored substitution patterns, in high yield and as single regioisomers. These new structures have found use in cascade reactions and as nucleotide exchange factor inhibitors.
4. Experimental Section
4.1. General
All reactions requiring anhydrous condition were conducted in flame-dried glassware under a positive pressure of nitrogen. Commercially available reagents were used as received; otherwise, materials were purified according to Purification of Laboratory Chemicals.19 Dichloromethane (CH2Cl2), tetrahydrofuran (THF), were degassed with nitrogen and passed through a solvent purifaction system (Innovative Technologies Pure Soly). Triethylamine (Et3N) was distilled from CaH2 immediately prior to use. Reactions were monitored by TLC and visualized by a dual short wave/long wave UV lamp and stained with KMnO4. Flash chromatography was performed on Merk silica gel Kieselgel 60 (230–400 mesh) from EM science with the indicated HPLC grade solvents.
1HNMR spectra were recorded on Varian Unity-300 MHz, Inova-400 MHz, or VXR-500 MHz as indicated. The chemical shifts (δ) of proton resonances are reported relative to CHCl3 using the following format: chemical shift in ppm [multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, app = apparent), coupling constant(s) (J in Hz), integral]. 13C NMR spectra were recorded at 75, 100, or 125 MHz. The chemical shifts of carbon resonances are reported relative to the deuterated solvent peak.20, 21 Infrared spectra were obtained using a Thermo Nicolet 380-FT IR spectrometer fitted with a SmartOrbit sample system. All absorptions are reported in cm−1 relative to polystyrene. Mass spectra were determined by ESI/APCI-TOF for HRMS in the University of Utah mass spectrometry facility.
4.2. Substrate preparation: Synthesis of Propargyl ureas
Given the focus of this manuscript, details for the preparation and spectral characterization of the substrate propargyl ureas corresponding to 1a–t are given in the supporting data section.
4.3. Cationic Rh(II)-catalyzed hydroamination of propargyl ureas
4.3.1. Representative procedure for the synthesis of 2a–t: 3-Methyl-1,6-diphenyl-3,4-dihydropyrimidin-2(1H)-one (2a)
[Rh2(OAc)2(CH3CN)6][BF4]2 (16.6 mg, 0.022 mmol, 5 mol%), 1-methyl-3-phenyl-1-(3-phenylprop-2-ynyl)urea (1a, 116 mg, 0.44 mmol, 1.0 equiv), and CH2Cl2 (4.5 mL) was mixed in an flame-dried Schlenk tube under N2. The reaction mixture was stirred at 80 °C overnight. The resulting solution was purified through the flash column chromatography (silica gel, Hexane/EtOAc = 6:1 to 1:1) to give the product 2a (109 mg, 94%). 1H NMR (CDCl3, 500 MHz) δ 7.14-7.00 (m, 10H), 5.13 (t, J = 4.0 Hz, 1H), 4.09 (d, J = 4.0 Hz, 2H), 3.00 (s, 3H); 13C NMR (CDCl3, 100 MHz) δ 154.6, 141.7, 139.4, 136.0, 128.9, 127.8, 127.8, 127.7, 127.6, 125.9, 100.9, 48.0, 35.3; IR (neat) 3853, 3744, 3648, 3060, 2923, 2360, 2338, 1700, 1676, 1489, 1280, 908, 727, 693, 623 cm−1; HRMS (ESI) Calculated for C17H16N2ONa m/z (M+Na) 287.1160 Obsd 287.1159.
4.3.2. 1-(4-Methoxyphenyl)-3-methyl-6-phenyl-3,4-dihydropyrimidin-2(1H)-one (2b)
1H NMR (CDCl3, 500 MHz) δ 7.10-7.02 (m, 7H), 6.65 (dd, J = 2.0, 6.0 Hz, 2H), 5.06 (t, J = 4.0 Hz, 1H), 4.08 (d, J = 4.0 Hz, 2H), 3.67 (s, 3H), 2.99 (s, 3H); 13C NMR (CDCl3, 100 MHz) δ 157.4, 154.9, 141.9, 136.2, 132.4, 130.1, 128.0, 127.7, 127.6, 113.2, 100.3, 55.2, 48.1, 35.4; IR (neat) 2932, 2835, 2359, 2241, 1675, 1654, 1607, 1509, 1493, 1239, 724 cm−1; HRMS (ESI) Calculated for C18H19N2O2 m/z (M+H) 295.1447 Obsd 295.1447.
4.3.3. 1-(4-Chlorophenyl)-3-methyl-6-phenyl-3,4-dihydropyrimidin-2(1H)-one (2c)
1H NMR (CDCl3, 500 MHz) δ 7.14-7.06 (m, 9H), 5.15 (t, J = 4.0 Hz, 1H), 4.09 (d, J = 4.0 Hz, 1H), 3.00 (s, 3H); 13C NMR (CDCl3, 100 MHz) δ 154.4, 141.4, 138.1, 135.7, 131.4, 130.1, 128.0, 127.9, 127.8, 101.6, 18.0, 35.4; IR (neat) 3853, 3743, 3688, 3058, 2926, 2360, 2340, 1680, 1576, 1489, 1282, 1091, 746 cm−1; HRMS (ESI) Calculated for C17H16N2OCl m/z (M+H) 299.0951 Obsd 299.0958.
4.3.4. 1-(4-Bromophenyl)-6-phenyl-3-methyl-3,4-dihydropyrimidin-2(1H)-one (2d)
1H NMR (CDCl3, 500 MHz) δ 7.25-7.00 (m, 9H), 5.16 (t, J = 4.0 Hz, 1H), 4.08 (d, J = 4.0 Hz, 1H), 3.00 (s, 3H); 13C NMR (CDCl3, 100 MHz) δ 154.3, 141.3, 138.6, 135.7, 130.9, 130.4, 128.0, 127.9, 127.8, 119.4, 101.7, 47.9, 35.4; IR (neat) 2923, 2361, 2338, 1677, 1664, 1484, 1249, 915, 868, 822, 744 cm−1; HRMS (ESI) Calculated for C17H15N2ONaBr m/z (M+Na) 365.0265 Obsd 365.0265.
4.3.5. 3-Methyl-1-(4-nitrophenyl)-6-phenyl-3,4-dihydropyrimidin-2(1H)-one (2e)
1H NMR (CDCl3, 500 MHz) δ 7.99-7.97 (m, 2H), 7.35-7.33 (m, 2H); 7.14-7.07 (m, 5H), 5.31 (t, J = 4.0 Hz, 1H), 4.10 (d, J = 4.0 Hz, 1H), 3.3 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 153.4, 145.6, 144.7, 140.8, 135.2, 128.6, 128.3, 128.2, 127.4, 123.1, 103.9, 47.7, 35.5; IR (neat) 2927, 2245, 1946, 1682, 1592, 1516, 1493, 1403, 1342, 1247, 1112, 854, 754, 590 cm−1; HRMS (ESI) Calculated for C17H16N3O3 m/z (M+Na) 310.1192 Obsd 310.1193.
4.3.6. 1,6-Bis(4-methoxyphenyl)-3-methyl-3,4-dihydropyrimidin-2(1H)-one (2f)
1H NMR (CDCl3, 500 MHz) δ 7.02-7.00 (m, 4H), 6.66-6.61 (m, 4H), 5.00 (t, J = 4.0 Hz, 1H), 4.04 (d, J = 4.0 Hz, 2H), 3.68 (s, 3H), 3.67 (s, 3H), 2.97 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 158.7, 157.3, 155.0, 141.7, 132.4, 130.0, 129.2, 128.6, 113.1, 113.0, 99.4, 55.1, 54.9, 48.0, 35.3; IR (neat) 2933, 2836, 2361, 2338, 2241, 1675, 1652, 1608, 1509, 1437, 1360, 1290, 1241, 1174, 1109, 1031, 907, 827, 723, 592 cm−1; HRMS (ESI) Calculated for C19H21N2O3 m/z (M+H) 325.1552 Obsd 325.1558.
4.3.7. 6-(4-Methoxyphenyl)-3-methyl-1-phenyl-3,4-dihydropyrimidin-2(1H)-one (2g)
1H NMR (CDCl3, 500 MHz) δ 7.14-6.61 (m, 9H), 5.07 (t, J = 4.0 Hz, 1H), 4.06 (d, J = 4.0 Hz, 2H), 3.68 (s, 3H), 2.99 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 158.8, 154.7, 131.4, 139.5, 129.1, 129.0, 128.5, 127.8, 125.8, 113.1, 100.2, 55.0, 48.0, 35.4; IR (neat) 2932, 2836, 2360, 2242, 1658, 1608, 1511, 1487, 1431, 1401, 1356, 1289, 1243, 1174, 1028, 908, 865, 761, 724, 696, 592 cm−1; HRMS (ESI) Calculated for C18H19N2O2 m/z (M+H) 295.1447 Obsd 295.1448.
4.3.8. 6-(4-Methoxyphenyl)-3-methyl-1-(4-nitrophenyl)-3,4-dihydropyrimidin-2(1H)-one (2h)
1H NMR (CDCl3, 300 MHz) δ 8.00 (dd, J = 2.4, 4.2 Hz, 2H), 7.34 (dd, J = 2.4, 7.2 Hz, 2H), 7.00 (dd, J = 2.4, 6.9 Hz, 2H), 6.66 (dd, J = 2.1, 6.6 Hz, 2H), 5.27 (t, J = 4.0 Hz, 1H), 4.08 (d, J = 4.0 Hz, 2H), 3.71 (s, 3H), 3.02 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 159.3, 154.0, 145.7, 144.7, 140.6, 128.75, 128.73, 127.6, 123.2, 102.8, 55.1, 47.6, 35.5; IR (neat) 2926, 2838, 1680, 1606, 1593, 1511, 1488, 1434, 1403, 1338, 1292, 1246, 1176, 1111, 1070, 1030, 877, 843, 757, 696, 589 cm−1; HRMS (ESI) Calculated for C18H18N3O4 m/z (M+H) 340.1297 Obsd 340.1298.
4.3.9. 1-(4-Methoxyphenyl)-3-methyl-6-(4-(trifluoromethyl)phenyl)-3,4-dihydropyrimidin-2(1H)-one (2i)
1H NMR (CDCl3, 500 MHz) δ 7.39 (d, J = 8.0 Hz, 2H), 7.24 (d, J = 7.5 Hz, 2H), 7.05 (dd, J = 1.5, 7.0 Hz, 2H), 6.69 (dd, J = 6.5, 7.0 Hz, 2H), 5.15 (t, J = 4.0 Hz, 1H), 4.12 (J = 4.0 Hz, 2H), 3.70 (s, 3H), 3.01 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 157.6, 154.7, 140.8, 139.7, 132.0, 129.8, 128.1, 124.8, 124.77, 124.74, 124.71, 113.4, 101.8, 55.2, 48.0, 35.4; IR (neat) 2936, 2839, 1712, 1671, 1608, 1510, 1441, 1410, 1322, 1246, 1165, 1110, 1066, 1018, 830, 737, 556 cm−1; HRMS (ESI) Calculated for C19H17N2O2F3Na m/z (M+Na) 385.1140 Obsd 385.1152.
4.3.10. 3-Methyl-1-phenyl-6-(4-(trifluoromethyl)phenyl)-3,4-dihydropyrimidin-2(1H)-one (2j)
1H NMR (CDCl3, 500 MHz) δ 7.36 (d, J = 8.0 Hz, 2H), 7.22 (d, J = 8.5 Hz, 2H), 7.21-7.12 (m, 4H), 7.05-7.03 (m, 1H), 5.20 (t, J = 4.0 Hz, 1H), 4.11 (d, J = 4.5 Hz, 2H), 3.00 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 154.4, 140.7, 139.6, 139.1, 128.8, 128.6, 128.3, 128.1, 128.0, 126.2, 124.83, 124.80, 124.78, 124.74, 102.6, 48.0, 35.4; IR (neat) 2929, 2348, 2246, 1676, 1488, 1404, 1365, 1320, 1281, 1248, 1163, 1107, 1066, 1017, 908, 845, 763, 695, 601, 552 cm−1; HRMS (ESI) Calculated for C18H16N2OF3 m/z (M+H) 333.1215 Obsd 333.1223.
4.3.11. 1-(4-Bromophenyl)-3-methyl-6-(4-(trifluoromethyl)phenyl)-3,4-dihydropyrimidin-2(1H)-one (2k)
1H NMR (CDCl3, 500 MHz) δ 7.40 (d, J = 7.5 Hz, 2H), 7.30-7.21 (m, 4H), 7.02 (dd, J = 1.5, 8.0 Hz, 2H), 5.23 (t, J = 4.0 Hz, 1H), 4.10 (d, J = 4.0 Hz, 2H), 3.00 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 154.1, 139.3, 138.3, 131.3, 130.2, 127.9, 125.1, 124.8, 119.6. 103.5, 47.9, 35.4; IR (neat) 2931, 2360, 2339, 1679, 1485, 1403, 1364, 1322, 1248, 1164, 1122, 1067, 1013, 846, 823, 785, 603, 551 cm−1; HRMS (ESI) Calculated for C18H14N2OF3NaBr m/z (M+Na) 433.0139 Obsd 433.0146.
4.3.12. 6-(4-Chlorophenyl)-3-methyl-1-phenyl-3,4-dihydropyrimidin-2(1H)-one (2l)
1H NMR (CDCl3, 500 MHz) δ 7.17-7.02 (m, 9H), 5.13 (t, J = 4.0 Hz, 1H), 4.09 (d, J = 4.0 Hz, 2H), 3.00 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 154.5, 140.8, 139.2, 134,6, 133.4, 129.1, 128.9, 128.0, 126.2, 101.5, 48.0, 35.4; IR (neat) 3234, 3050, 2922, 2049, 1677, 1660, 1591, 1488, 1432, 1398, 1278, 1244, 1105, 1088, 1013, 909, 828, 773, 754, 744, 697, 564 cm−1; HRMS (ESI) Calculated for C17H16N2OCl m/z (M+H) 299.0951 Obsd 299.0949.
4.3.13. 3-Methyl-6-(4-nitrophenyl)-1-phenyl-3,4-dihydropyrimidin-2(1H)-one (2m)
1H NMR (CDCl3, 500 MHz) δ 7.97 (d, J = 9.5 Hz, 2H), 7.29 (d, J = 6.5 Hz, 1H), 7.17-7.23 (m, 4H), 7.07-7.06 (m, 1H), 5.28 (t, J = 4.0 Hz, 1H), 4.14 (d, J = 4.0 Hz, 2H), 3.02 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 154.2, 146.9, 142.5, 140.2, 139.0, 128.6, 128.4, 128.3, 126.5, 123.2, 103.8, 48.0, 35.5; IR (neat) 3076, 2924, 2853, 2244, 1674, 1597, 1515, 1489, 1434, 1402, 1342, 1314, 1281, 1249, 1108, 1013, 910, 852, 767, 729, 699, 561 cm−1; HRMS (ESI) Calculated for C17H15N3O3 m/z (M+H) 332.1011 Obsd 332.1015.
4.3.14. 1-(4-Methoxybenzyl)-3-methyl-6-phenyl-3,4-dihydropyrimidin-2(1H)-one (2o)
1H NMR (CDCl3, 300 MHz) δ 7.29-7.27 (m, 2H); 7.15-7.12 (m, 2H), 6.87-6.84 (m, 2H), 6.71-6.68 (m, 2H), 4.82 (t, J = 3.9 Hz, 1H), 4.56 (s, 2H), 4.92 (d, J = 3.9 Hz, 1H), 3.73 (s, 3H), 2.95 (s, 3H); 13C NMR (CDCl3, 75 MHz) δ 158.3, 155.8, 141.8, 135.7, 131.4, 128.4, 128.2, 128.1, 113.5, 100.2, 55.1, 47.8, 46.7, 35.4; IR (neat) 2932, 2834, 2360, 2339, 1652, 1611, 1510, 1494, 1442, 1242, 1174, 1031, 815, 727, 700, 645, 568 cm−1; HRMS (ESI) Calculated for C19H20N2O2Na m/z (M+Na) 331.1422 Obsd 331.1431.
4.3.15. 1-Butyl-3-methyl-6-phenyl-3,4-dihydropyrimidin-2(1H)-one (2p)
1H NMR (CDCl3, 500 MHz) δ 7.35-7.24 (m, 5H), 4.82 (t, J = 4.0 Hz, 1H), 4.10 (d, J = 4.0 Hz, 1H), 3.45-3.42 (m, 2H), 2.94 (s, 3H), 1.31-1.29 (m, 2H), 1.10-1.06 (m, 2H), 0.71 (t, J = 7.5 Hz, 1H); 13C NMR (CDCl3, 125 MHz) δ 155.9, 141.9, 135.9, 128.1, 128.0, 99.9, 99.8, 47.7, 43.7, 35.2, 31.5, 19.5, 13.6; IR (neat) 2955, 2928, 2870, 2360, 2338, 1652, 1493, 1431, 1400, 1275, 1083, 765, 700 cm−1; HRMS (ESI) Calculated for C15H21N2O m/z (M+H) 245.1654 Obsd 245.1652.
4.3.16. 3-(4-Methoxybenzyl)-1,6-diphenyl-3,4-dihydropyrimidin-2(1H)-one (2q)
1H NMR (CDCl3, 500 MHz) δ 7.50 (dd, J = 2.5, 7.0 Hz, 2H), 7.43-7.19 (m, 10H), 7.04 (dd, J = 2.5, 7.0 Hz, 2H), 5.26 (t, J = 4.0 Hz, 1H), 4.74 (s, 2H), 4.14 (d, J = 4.0 Hz, 2H), 3.98 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 159.0, 154.5, 141.4, 139.5, 135.9, 129.7, 129.3, 128.9, 128.7, 128.6, 128.5, 127.8, 127.6, 127.5, 126.6, 125.9, 113.9, 113.8, 101.3, 55.1, 50.8, 45.0; IR (neat) 2928, 2834, 2173, 2065, 1709, 1660, 1512, 1445, 1283, 1246, 1174, 1110, 1032, 816, 757, 696, 614, 566 cm−1; HRMS (ESI) Calculated for C24H23N2O2 m/z (M+H) 371.1760 Obsd 371.1763.
4.3.17. 6-Cyclohexenyl-3-methyl-1-phenyl-3,4-dihydropyrimidin-2(1H)-one (2r)
1H NMR (CDCl3, 500 MHz) δ 7.27-7.14 (m, 5H), 5.73 (d, J = 4.0 Hz, 2H), 4.89 (t, J = 4.0 Hz, 1H), 3.94 (d, J = 4.0 Hz, 1H), 2.93 (s, 3H), 2.03-1.86 (m, 2H), 1.59-1.57 (m, 2H), 1.32-1.25 (m, 2H), 1.21-1.16 (m, 2H); 13C NMR (CDCl3, 125 MHz) δ 154.6, 144.5, 139.4, 134.4, 129.4, 129.1, 128.9, 127.5, 126.0, 97.7, 47.9, 35.3, 27.4, 25.1, 22.1, 21.6; IR (neat) 3330, 3058, 2929, 2857, 1708, 1649, 1596, 1489, 1435, 1396, 1339, 1265, 1104, 1072, 920, 833, 802, 755, 731, 695, 641, 576 cm−1; HRMS (ESI) Calculated for C17H21N2O m/z (M+H) 269.1654 Obsd 269.1654.
4.3.18. 6-Cyclohexenyl-1-(4-methoxybenzyl)-3-methyl-3,4-dihydropyrimidin-2(1H)-one (2s)
1H NMR (CDCl3, 500 MHz) δ 7.13 (d, J = 9.0 Hz, 2H), 6.80 (dd, J = 2.0, 7.0 Hz, 2H), 5.65-5.64 (m, 1H), 4.68 (t, J = 4.0 Hz, 1H), 4.57 (s, 2H), 3.78 (d, J = 4.0 Hz, 2H), 3.76 (s, 3H), 2.87 (s, 3H), 2.04-2.02 (m, 2H), 1.92-1.91 (m, 2H), 1.56-1.55 (m, 4H); 13C NMR (CDCl3, 100 MHz) δ 158.3, 155.9, 144.0, 133.6, 132.0, 128.8, 128.3, 113.5, 97.1, 55.1, 47.5, 46.6, 35.1, 28.4, 25.0, 22.3, 21.7; IR (neat) 2931, 2835, 2243, 1646, 1511, 1440, 1403, 1379, 1289, 1244, 1174, 1035, 905, 818, 724, 645, 570 cm−1; HRMS (ESI) Calculated for C19H25N2O2 m/z (M+H) 313.1916 Obsd 313.1911.
4.3.19. 6-(Hex-1-ynyl)-3-methyl-1-phenyl-3,4-dihydropyrimidin-2(1H)-one (2t)
1H NMR (CDCl3, 400 MHz) δ 7.35-7.32 (m, 2H), 7.28-7.24 (m, 1H), 7.25-7.22 (m, 2H), 5.19 (t, J = 4.0 Hz, 1H), 4.02 (d, J = 4.0 Hz, 2H), 2.93 (s, 3H), 2.00 (t, J = 6.8 Hz, 1H), 1.14-1.09 (m, 2H), 1.05-0.99 (m, 2H), 0.74 (t, J = 7.4 Hz, 1H); 13C NMR (CDCl3, 125 MHz) δ 153.6, 139.4, 129.6, 128.1, 127.2, 125.1, 103.3, 94.4, 74.4, 48.6, 35.1, 29.8, 21.4, 18.5, 13.4; IR (neat) 2955, 2928, 2870, 2360, 2229, 1668, 1597, 1488, 1433, 1325, 1284, 1250, 1101, 1070, 1023, 913, 760, 695, 567 cm−1; HRMS (ESI) Calculated for C17H21N2O m/z (M+H) 269.1654 Obsd 269.1652.
4.4. Synthesis of the secin-44 analogue (4)
N2H4·H2O (101.0 mg) was added to a reflux mixture of nickel boride7 (12.8 mg), 6-(4-methoxyphenyl)-3-methyl-1-(4-nitrophenyl)-3,4-dihydropyrimidin-2(1H)-one (2h) (16.1 mg, 0.05 mmol) in absolute EtOH (4.0 mL) and stirred at reflux for 30 minutes. Then, the solution was cooled to room temperature, filtered through a short pad of celite. The filtrate was concentrated to give the aniline which was used for the coupling reaction without further purification.
Under N2, DCM (2.0 mL) was added to a mixture of 5-bromofuran-2-carboxylic acid (21.0 mg, 0.11 mmol), EDCI (96 mg, 0.5 mmol), DMAP (15.3 mg, 0.125 mmol) and 1-(4-aminophenyl)-6-(4-methoxyphenyl)-3-methyl-3,4-dihydropyrimidin-2(1H)-one (16.0 mg, 0.05 mmol). The mixture was stirred at room temperature overnight until the aniline had been consumed as judged by TLC analysis. The mixture was diluted with EtOAc (5 mL), washed with NaHCO3 (aq.), HCl (1M), NaHCO3 (aq.), brine and dried over MgSO4. After filtration, the solution was concentrated and the residue was purified through preparative TLC (silica gel, EtOAc/Hexane = 3:1) to give the coupling product (4, 13.0 mg, 50%). 1H NMR (CDCl3, 500 MHz) δ 7.91 (s, 1H), 7.44-7.42 (m, 2H), 7.12-7.10 (m, 2H), 7.02-7.00 (m, 2H), 6.64-6.62 (m, 2H), 6.46 (d, J = 4.0 Hz, 1H), 5.06 (t, J = 4.0 Hz, 1H), 4.06 (d, J = 4.0 Hz, 2H), 3.69 (s, 3H), 2.99 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 159.0, 154.8, 154.6, 149.3, 141.4, 136.0, 134.9, 129.6, 129.2, 128.4, 124.7, 119.2, 117.4, 114.6, 113.2, 100.2, 55.0, 48.0, 35.4; IR (neat) 3303, 2930, 2359, 2341, 1654; 1602, 1511, 1463, 1405, 1357, 1295, 1248, 1174, 1144, 1108, 1012, 952, 925, 854, 833, 798, 735, 668 cm−1; HRMS (ESI) Calculated for C23H20N3O4NaBr m/z (M+Na) 504.0535 Obsd 504.0553.
4.5. Arf6 pull-down experiments
Plasmid DNA construction
The ARF6 and ARNO coding sequences were amplified by PCR and then ligate into pcDNA3.1/myc-His A (Invitrogen) in fusion with Myc and a C-terminal His Tag.
Active ARF6 pull-down
The plasmid DNA was transfected into HEK293T cell using Lipofectamine 2000 (Invitrogen) when cell density is around 90%. The medium was changed to DMEM+1% FBS at 24 hrs post transfection and incubated overnight. The compound was diluted with DMEM+1% FBS according to 1:200 to get final concentration of 50 μM or 100 μM in 0.5% DMSO. Transfected cell was treated with compound or DMSO only for 4 hr before pull-down. The pull-down was performed using active Arf6 pull-down and detection kit from Thermo Scientific (cat#: 26186).
4.6. Solubility determinations
Solubility determinations were made by adding a 10mM solution of secin-44 or compound 4 in DMSO to pull down medium (DMEM+1% FBS) to final concentrations of 1, 10, 50, 100 and 200 μM. After standing overnight the solutions were observed under a microscope and the concentration at which the compound began crystallizing was noted as its maximum solubility.
Supplementary Material
Acknowledgments
We are thankful to Dr. Jim Muller for help with mass spectral analysis. REL is grateful to the National Institutes of Health, General Medical Sciences (R01-GM090082 and P41-GM08915) for financial support of this research. DYL is supported by grants from the National Institutes of Health, NCI (R01HL066277) and the Department of Defense (W81XWH0910260).
Footnotes
Analytical data for all new compounds including 1H and 13C NMR spectra. Supplementary data associated with this article can be found in the online version, at http://for doi.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.(a) Kappe CO. Eur J Med Chem. 2000;35:1043. doi: 10.1016/s0223-5234(00)01189-2. [DOI] [PubMed] [Google Scholar]; (b) Singh K, Arora D, Singh K, Singh S. Mini-Rev Med Chem. 2009;9:95. doi: 10.2174/138955709787001686. [DOI] [PubMed] [Google Scholar]; (c) Kappe CO. Tetrahedron. 1993;49:6937. [Google Scholar]
- 2.(a) Mayer TU, Kapoor TM, Haggarty SJ, King RW, Schreiber SL, Mitchison TJ. Science. 1999;286:971. doi: 10.1126/science.286.5441.971. [DOI] [PubMed] [Google Scholar]; (b) Atwal KS, Swanson BN, Unger SE, Floyd DM, Moreland S, Hedberg A, O’Reilly BC. J Med Chem. 1991;34:806. doi: 10.1021/jm00106a048. [DOI] [PubMed] [Google Scholar]
- 3.Biginelli P. Gazz Chim Ital. 1893;23:360. [Google Scholar]
- 4.For excellent reviews see: Kappe CO. J Org Chem. 1997;62:7201. doi: 10.1021/jo971010u.Kappe CO. Acc Chem Res. 2000;33:879. doi: 10.1021/ar000048h.Wan JP, Liu Y. Synthesis. 2010;23:3943.
- 5.For enantioselective variants see: Li N, Chen XH, Song J, Luo S-W, Fan W, Gong L-Z. J Am Chem Soc. 2009;131:15301. doi: 10.1021/ja905320q.Gong LZ, Chen XH, Xu XY. Chem Eur J. 2007;13:8920. doi: 10.1002/chem.200700840.Huang Y, Yang F, Zhu C. J Am Chem Soc. 2005;127:16386. doi: 10.1021/ja056092f.Lou S, Taoka BM, Ting A, Schaus SE. J Am Chem Soc. 2005;127:11256. doi: 10.1021/ja0537373.Goss JM, Schaus SE. J Org Chem. 2008;73:7651. doi: 10.1021/jo801463j.Chen XH, Xu XY, Liu H, Cun LF, Gong LZ. J Am Chem Soc. 2006;128:14802. doi: 10.1021/ja065267y.
- 6.Oberg KM, Rovis T. J Am Chem Soc. 2011;133:4785. doi: 10.1021/ja200766k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Giles RL, Sullivan JD, Steiner AM, Looper RE. Angew Chem Int Ed. 2009;48:3116. doi: 10.1002/anie.200900160. [DOI] [PubMed] [Google Scholar]; (b) Gainer MJ, Bennett NR, Takahashi Y, Looper RE. Angew Chem Int Ed. 2011;50:684. doi: 10.1002/anie.201006087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gold catalyzed intramolecular annulation of substituted aminoalkynes: Verniest G, Padwa A. Org Lett. 2008;10:4379. doi: 10.1021/ol801847j.Hashmi ASK. Chem Rev. 2007;107:3180. doi: 10.1021/cr000436x.Istrate FM, Buzas AK, Jurberg ID, Odabachian Y, Gagosz F. Org Lett. 2008;10:925. doi: 10.1021/ol703077g.Hirano K, Inaba Y, Watanabe T, Oishi S, Fujii N, Ohno H. Adv Synth Catal. 2010;352:368.Perl NR, Ide ND, Prajapati S, Perfect HH, Duron SG, Gin DY. J Am Chem Soc. 2010;132:1802. doi: 10.1021/ja910831k.Ye D, Zhang X, Zhou Y, Zhang D, Zhang L, Wang H, Jiang H, Liu H. Adv Synth Catal. 2009;351:2770.Enomoto T, Girard AL, Yasui Y, Takemoto Y. J Org Chem. 2009;74:9158. doi: 10.1021/jo901906b.Enomoto T, Obika S, Yasui Y, Takemoto Y. Synlett. 2008:1647.Hashmi ASK, Rudolph M, Schymura S, Visus J, Frey W. Eur J Org Chem. 2006:4905.Kang JE, Kim HB, Lee JW, Shin S. Org Lett. 2006;8:3537. doi: 10.1021/ol061307r.Hashmi ASK, Weyrauch JP, Frey W, Bats JW. Org Lett. 2004;6:4391. doi: 10.1021/ol0480067.Milton MD, Inada Y, Nishibayashi Y, Uemura S. Chem Commun. 2004:2712. doi: 10.1039/b411180j.Ritter S, Horino Y, Lex J, Schmalz HG. Synlett. 2006:3309.Verniest G, England D, De Kimpe N, Padwa A. Tetrahedron. 2010;66:1496.Campbell MJ, Toste FD. Chem Sci. 2011;2:1369. doi: 10.1039/C1SC00160D.
- 9.Silver: Ermolat’ev DS, Mehta VP, Van der Eycken EV. Synlett. 2007;20:3117.Yamada W, Sugawara Y, Cheng HM, Ikeno T, Yamada T. Eur J Org Chem. 2007:2604.Patil NT, Pahadi NK, Yamamoto Y. J Org Chem. 2005;70:10096. doi: 10.1021/jo051524q.Peng AY, Ding YX. Org Lett. 2005;7:3299. doi: 10.1021/ol051126+.Ag(I)-catalyzed N-cyclization: Peshkov VA, Pereshivko OP, Sharma S, Meganathan T, Parmar VS, Ermolat’ev DS, Van der Eycken EV. J Org Chem. 2011;76:5867. doi: 10.1021/jo200789t.Huang Q, Hunter JA, Larock RC. Org Lett. 2001;3:2973. doi: 10.1021/ol010136h.Asao N, Yudha S, Nogami T, Yamamoto Y. Angew Chem, Int Ed. 2005;44:5526. doi: 10.1002/anie.200500795.Su S, Porco JA., Jr J Am Chem Soc. 2007;129:7744. doi: 10.1021/ja072737v.Yu X, Yang X, Wu J. Org Biomol Chem. 2009;7:4526. doi: 10.1039/b913409c.Ghavtadze N, Fröhlich R, Würthwein EU. Eur J Org Chem. 2010:1787. doi: 10.1021/jo900270e.Chen Z, Yu X, Wu J. Chem Commun. 2010;46:6356. doi: 10.1039/c0cc01207f.Chen Z, Wu J. Org Lett. 2010;12:4856. doi: 10.1021/ol101988q.
- 10.Palladium: Majumdar KC, De Nirupam Roy B. Synthesis. 2010:4207.Narsireddy M, Yamamoto Y. J Org Chem. 2008;73:9698. doi: 10.1021/jo801785r.Patil NT, Wu H, Yamamoto Y. J Org Chem. 2007;72:6577. doi: 10.1021/jo0708137.Patil NT, Lutete LM, Wu H, Pahadi NK, Gridnev ID, Yamamoto Y. J Org Chem. 2006;71:4270. doi: 10.1021/jo0603835.Bajracharya GB, Huo Z, Yamamoto Y. J Org Chem. 2005;70:4883. doi: 10.1021/jo050412w.Lutete LM, Kadota I, Yamamoto Y. J Am Chem Soc. 2004;126:1622. doi: 10.1021/ja039774g.Saito A, Iimura K, Hanzawa Y. Tetrahedron Lett. 2010;51:1471.Bacchi A, Chiusoli GP, Costa M, Sani C, Gabriele B, Salerno G. J Organomet Chem. 1998;562:35.Bacchi A, Costa M, Gabriele B, Pelizzi G, Salerno G. J Org Chem. 2002;67:4450. doi: 10.1021/jo0200217.Gabriele B, Plastina P, Salerno G, Raffaella Mancuso R, Costa M. Org Lett. 2007;9:3319. doi: 10.1021/ol071332c.
- 11.Copper: Krasnova LB, Hein JE, Fokin VV. J Org Chem. 2010;75:8662. doi: 10.1021/jo1016376.Kimura MM, Kure S, Yoshida Z, Tanaka S, Fugami K, Tamaru Y. Tetrahedron Lett. 1990;31:4887.Ohe K, Ishihara T, Chatani N, Kawasaki Y, Murai S. J Org Chem. 1991;56:2267.
- 12.Rempel GA, Legzdins P, Smith H, Wilkinson G. Inorg Synth. 1972;13:87. [Google Scholar]; Crawford CA, Matonic JH, Streib WE, Huffman JC, Dunbar KR, Christou G. Inorg Chem. 1993;32:3125. doi: 10.1021/ic9610288. [DOI] [PubMed] [Google Scholar]
-
13.We prepared the urea-ketone which failed to cyclize under the reaction conditions. Similar substrates are known to give moderate to low yields of dihydropyrimidones in the presence of mineral acid catalysts, see: Xue N, Lu X, Hu YJ. Heterocyclic Chem. 2008;45:1095.
- 14.(a) Ota K, Chatani N. Chem Commun. 2008:2906. doi: 10.1039/b805100c. [DOI] [PubMed] [Google Scholar]; (b) Ota K, Lee SI, Tang JM, Takachi M, Nakai H, Morimoto T, Sakurai H, Kataoka K, Chatani N. J Am Chem Soc. 2009;131:15203. doi: 10.1021/ja9047637. [DOI] [PubMed] [Google Scholar]; (c) Lee SI, Chatani N. Chem Commun. 2009:371. doi: 10.1039/b812466c. [DOI] [PubMed] [Google Scholar]; (d) Shikanai D, Murase H, Hata T, Urabe H. J Am Chem Soc. 2009;131:3166. doi: 10.1021/ja809826a. [DOI] [PubMed] [Google Scholar]; (e) Miki K, Washitake Y, Ohe K, Uemura S. Angew Chem Int Ed. 2004;43:1857. doi: 10.1002/anie.200352949. [DOI] [PubMed] [Google Scholar]; (f) Moniz GA, Wood JL. J Am Chem Soc. 2001;123:5095. doi: 10.1021/ja015727h. [DOI] [PubMed] [Google Scholar]
- 15.Li M, Wang J, Ng SSM, Chan C, He M, Yu F, Lai L, Shi C, Chem Y, Yew T, Kung H, Lin MC. Cancer. 2009;115:4959. doi: 10.1002/cncr.24550. [DOI] [PubMed] [Google Scholar]
- 16.Stumpfe D, Bill A, Novak N, Loch G, Blockus H, Geppert H, Becker T, Schmitz A, Hoch M, Kolanus W, Famulok M, Bajorath J. ACS Chem Biol. 2010;5:839. doi: 10.1021/cb100171c. [DOI] [PubMed] [Google Scholar]
- 17.For leading references see: Donaldson JG, Klausner RD. Curr Opin Cell Biol. 1994;6:527. doi: 10.1016/0955-0674(94)90072-8.Frank S, Upender S, Hansen SH, Cassanova JE. J Biol Chem. 1998;273:23. doi: 10.1074/jbc.273.1.23.
- 18.See Supporting information for more detailed protocols and statistical analysis.
- 19.Armarego WLF, Chai CLL. Purification of Laboratory Chemicals. 5 2003. [Google Scholar]
- 20.Gottlieb HE, Kotlyar V, Nudelman A. J Org Chem. 1997;62:7512–7515. doi: 10.1021/jo971176v. [DOI] [PubMed] [Google Scholar]
- 21.Hoye TR, Hansen PR, Vyvyan JR. J Org Chem. 1994;59:4096–4103. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.