

Abstract
Synthesis of 6-deoxycyclopropavir (10), a prodrug of cyclopropavir (1) and its in vitro and in vivo antiviral activity is described. 2-Amino-6-chloropurine methylenecyclopropane 13 was transformed to its 6-iodo derivative 14 which was reduced to prodrug 10. It is converted to cyclopropavir (1) by the action of xanthine oxidase and this reaction can also occur in vivo. Compound 10 lacked significant in vitro activity against human cytomegalovirus (HCMV), human herpes virus 1 and 2 (HSV-1 and HSV-2), human immunodeficiency virus type 1 (HIV-1), human hepatitis B virus (HBV), Epstein-Barr virus (EBV), vaccinia virus and cowpox virus. In contrast, prodrug 10 given orally was as active as cyclopropavir (1) reported previously [Kern, E. R.; Bidanset, D. J.; Hartline, C. B.; Yan, Z.; Zemlicka, J.; Quenelle, D. C. et al. Antimicrob. Agents Chemother. 2004, 48, 4745] against murine cytomegalovirus (MCMV) infection in mice and against HCMV in severe combined immunodeficient (SCID) mice.
Keywords: Methylenecyclopropanes, Cyclopropavir, 6-Deoxycyclopropavir, Antiviral agents, Xanthine oxidase, Murine cytomegalovirus, Human cytomegalovirus
1. Introduction
Methylenecyclopropane analogues of nucleosides are established antiviral agents effective against beta- and gamma-herpe-sviruses.1 The second generation guanine analogue, cyclopropavir (CPV, 1, Chart 1), is currently under preclinical development as a potential drug for human cytomegalovirus (HCMV) infections exceeding the efficacy of ganciclovir (GCV, 2).2–5 Previously, 2-ami-nopurine acyclic nucleoside analogues 3, 4a, 5 and 6 were investigated as prodrugs of ganciclovir (2),6 acyclovir (7),7,8 penciclovir (8)9 and A-5021 (9).10 The hallmark of 6-deoxypurines 3, 4a, 5 and 6 is a lack of in vitro antiviral activity but an improved in vivo potency owing to an increased oral absorption and bioavailability over the parent analogues 2, 7, 8 and 9. The key cellular enzymes for conversion of 2-aminopurine prodrugs to parent guanine analogues are xanthine and aldehyde oxidases.7,11 A double prodrug of penciclovir (8), famciclovir (Famvir)12 4b, became an approved drug against shingles.13
Chart 1.

6-Deoxypurine prodrugs of antiviral nucleosides.
Therefore, it was of interest to synthesize 6-deoxycyclopropavir (10), a potential prodrug of cyclopropavir (1) and investigate its in vitro and in vivo antiviral efficacy as well as substrate activity toward xanthine oxidase.
2. Results
2.1. Synthesis
Previously, 6-deoxysynguanol (11), a potential prodrug of synguanol (12), was obtained by desulfuration of 6-thiosynguanol by RaneyNiin31% overall yield.14 The lower yield was caused by a partial reduction of the methylenecyclopropane moiety. This complication could be avoided by using directly 2-amino-6-chloropurine methylenecyclopropane 13, an intermediate2 in the synthesis of cyclopropavir (1), and a suitable reducing agent such as tri-n-butyltin hydride (Bu3SnH)–2,2″-azobisisobutyronitrile15 (AIBN, Scheme 1). However, an attempted reduction of 13 under those conditions has not been successful. Therefore, compound 13 was transformed to transient 6-iodo derivative 14 using trimethylsilyl iodide (Me3SiI) in N,N-dimethylformamide (DMF). The crude product was smoothly reduced with Bu3SnH and AIBN to give 6-deoxycyclopropavir (10) in 63% overall yield.
Scheme 1.

Synthesis of 6-deoxycyclopropavir and its reaction with xanthine oxidase. Reagents and conditions: (a) Me3SiI, DMF, 0 °C; (b) Bu3SnH, AIBN, DMF, 65–75 °C; (c) xanthine oxidase, pH 7.5.
2.2. Biological activity
2.2.1. Substrate activity for xanthine oxidase
6-Deoxycyclopropavir (10) was a substrate for xanthine oxidase (Scheme 1, Figs. 1 and 2) thereby establishing that this 6-deoxy-guanosine analogue could be a prodrug of cyclopropavir (1). The assay was performed under the conditions previously reported14 for 6-deoxysynguanol (11). The reaction of 10 was noticeably slower than that of 11 (T/1/2 400 vs 30 min).
Figure 1.
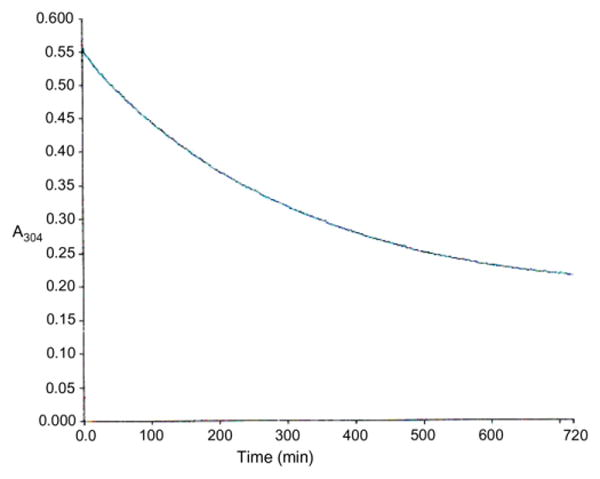
Time-course of oxidation of 6-deoxycyclopropavir (10) to cyclopropavir (1) with xanthine oxidase. Conditions: 6-deoxycyclopropavir (10, 0.07 mM) in 0.05 M Na2HPO4 (pH 7.5, 1.2 mL), xanthine oxidase from cow milk (20 units/mL, 20 μL, 0.4 units), room temperature, reaction was continuously monitored at 304 nm (Fig. 1). UV spectra at T = 0 and T = 720 min are shown in Figure 2.
Figure 2.
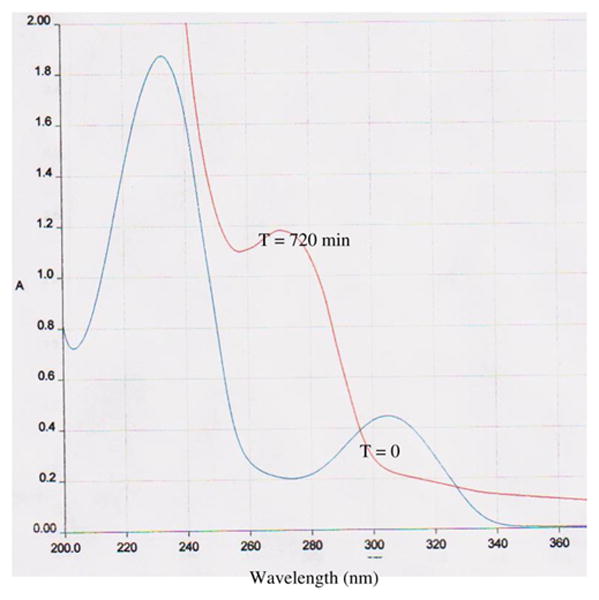
UV spectra of 6-deoxycyclopropavir (10) and cyclopropavir (1). UV spectrum before addition of enzyme (T = 0) and after incubation with enzyme (T = 720 min). The spectra at T = 0 and T = 720 min corresponded to those of authentic 6-deoxycyclopropavir (10) and cyclopropavir (1), respectively.
2.2.2. In vitro antiviral activity
As expected, no antiviral activity 6-deoxycyclopropavir (10) was found in vitro against human cytomegalovirus (HCMV, Towne strain, plaque reduction) in human foreskin fibroblasts (HFF) with EC50 and CC50 both greater than 100 μM. Similarly, no antiviral activity was observed against HCMV AD169 strain, HSV-1, HSV-2 and VZV (cytopathic effect assay), EC50 and CC50 were greater than 300 μM. Likewise, no activity was noted against vaccinia virus and cowpox virus at 100 μm. Some antiviral activity accompanied by cytotoxicity was found against Epstein–Barr virus (EBV) in Akata cells (DNA hybridization), with EC50 and CC50 values of 27 and 71 μM, respectively, but it was not well separated from cytotoxicity, thus its significance is doubtful.
2.2.3. In vivo antiviral activity
2.2.3.1. Effect of oral treatment with 6-deoxycyclopropavir (10) or GCV(2) in MCMV infections of mice
In contrast, 6-deoxy-cyclopropavir (10) was active in vivo although some minimal toxicity was associated with the 50 mg/kg dose of 10 to uninfected mice. There was an approximate 17% weight loss compared to vehicle treated or untreated mice although no clinical signs, such as ruffled coat appearance, were seen (Fig. 3). This is also possibly reflected by a higher mortality in MCMV infected mice given 50 mg/kg at both the 24 and 48 h times (Table 1). At the 16.7 or 5.6 mg/kg dose, less weight loss was observed in uninfected mice and, in MCMV-infected mice, 6-deoxycyclopropavir (10) significantly reduced final mortality rates (P ≤0.001) when treatment was initiated at 24–48 h post infection. GCV (2), the positive control, also reduced mortalities at the two higher doses of 50 and 16.7 mg/kg at both times of initiation of treatment (P ≤0.05).
Figure 3.

Mean body weight in grams for five mice/group is shown over the experimental dosing period of day 1–5. Day 6 is a post-dosing phase of recovery. None of the uninfected mice were ruffled coated or behaviorally affected by any dosage level of 6-deoxycyclopropavir (10).
Table 1.
Effect of oral treatment with 6-deoxycyclopropavir (10) on the mortality of BALB/c mice inoculated intraperitoneally with MCMV
| Treatmenta | Mortality Number Percent | P-value | MDD ± STDEVb | P-value | |
|---|---|---|---|---|---|
| Placebo + 24 h | |||||
| 0.4% CMC | 10/15 | 67 | – | 4.9 ± 0.7 | – |
| GCV + 24 h | |||||
| 50 mg/kg | 1/15 | 7 | <0.01 | 6.0 ± 0 | NSc |
| 16.7 mg/kg | 3/15 | 20 | <0.05 | 6.3 ± 0.6 | – |
| 5.6 mg/kg | 13/15 | 87 | NS | 6.8 ± 0.8 | <0.001 |
| 6-Deoxycyclopropavir + 24 h | |||||
| 50 mg/kg | 7/15 | 47 | NS | 8.3 ± 4.9 | 0.01 |
| 16.7 mg/kg | 0/15 | 0 | <0.001 | – | – |
| 5.6 mg/kg | 1/15 | 7 | <0.001 | 6.0 ± 0 | NS |
| Placebo + 48 h | |||||
| 0.4% CMC | 15/15 | 100 | – | 5.1 ± 0.9 | – |
| GCV + 48 h | |||||
| 50 mg/kg | 3/15 | 20 | <0.001 | 5.7 ± 0.6 | NS |
| 16.7 mg/kg | 8/15 | 53 | <0.01 | 7.3 ± 1.5 | 0.001 |
| 5.6 mg/kg | 14/15 | 93 | NS | 6.4 ± 0.9 | 0.001 |
| 6-Deoxycyclopropavir + 48 h | |||||
| 50 mg/kg | 10/15 | 67 | <0.05 | 11.9 ± 6.1 | <0.001 |
| 16.7 mg/kg | 1/15 | 7 | <0.001 | 7.0 ± 0 | 0.09 |
| 5.6 mg/kg | 0/15 | 0 | <0.001 | – | – |
6-deoxycyclopropavir (10) was suspended in 0.4% CMC and given p.o. in 0.2 mL doses. GCV (2) was prepared in sterile saline and given p.o. in 0.2 mL doses. Animals were treated once daily for five days beginning 24 or 48 h after viral inoculation.
MDD = mean day of death. STDEV = standard deviation.
NS = Not significant when compared to the placebo control.
2.2.3.2. Effect of treatment with 6-deoxycyclopropavir (10) or cidofovir (CDV) on HCMV replication in SCID-human thymus/liver tissue implants in mice
The SCID mice implanted with human fetal thymus or liver tissue under the kidney capsule were used to determine the efficacy of the 6-deoxycyclopropavir (10) against HCMV replication in a visceral organ. In this model, the implanted thymus and liver tissue have been shown to grow and become vascularized. The thy/liv implants were infected with 9000 PFU of the Toledo strain of HCMV. Starting 24 h after infection, mice were treated with vehicle, 20 mg/kg CDV or 12.5 or 6.25 mg/kg of 6-deoxycyclopropavir (10) once daily for 28 days. At 14, 21, 28 and 35 days after infection, implants were biopsied and HCMV titers quantified by real time PCR. The results summarized in Table 2 indicated that 6-deoxycyclopropavir (10) was efficacious in this model achieving statistical significance at day 28 (P <0.01) at the 12.5 mg/kg dose. Although not statistically significant, trends towards lowered viral genome equivalent numbers were seen on days 14 and 21 for 12.5 mg/kg and also on day 28 at the 6.25 mg/kg dose. CDV, the positive control, significantly inhibited HCMV replication on days 14, 28 and 35 (P ≤0.05), as expected. In MCMV infections of BALB/c mice and in HCMV infections in the SCID-human model, 6-deoxycyclopropavir (10) was highly active to active, respectively, when given orally.
Table 2.
The effect of once daily oral treatment with 6-deoxycyclopropavir (10) or CDV given intraperitoneally on the replication of HCMV in SCID human thymus/liver tissue implants
| Treatmenta | Parameter | Days post infection | |||
|---|---|---|---|---|---|
|
| |||||
| 14 | 21 | 28 | 35 | ||
| Vehicle | Log10geb | 3.50 ± 0.878 | 3.82 ± 1.24 | 3.73 ± 0.784 | 3.28 ± 1.422 |
| 0.4% CMC | % Positive | 92 (11/12) | 92 (11/12) | 92 (11/12) | 67 (8/12) |
| CDV | Log10geb | 2.47 ±0.786e | 2.83 ± 1.081c | 2.08 ± 0.649e | 1.59 ±0.0d |
| 20 mg/kg | % Positive | 82 (9/11) | 100 (10/10) | 50 (6/12) | 0 (0/5) |
| 6-Deoxycyclopropavir | Log10geb | 3.03 ± 0.710c | 2.94 ± 1.022c | 2.76 ± 0.768e | 2.69 ± 0.69 |
| 12.5 mg/kg | % Positive | 100 (12/12) | 82 (9/11) | 50 (6/12) | 83 (10/12) |
| 6-Deoxycyclopropavir | Log10geb | 3.90 ± 1.119 | 3.14 ± 1.165 | 2.81 ± 1.198c | 3.32 ± 0.935 |
| 6.25 mg/kg | % Positive | 92 (11/12) | 92 (11/12) | 58 (7/12) | 100 (11/11) |
Treatment administered once daily for 28 days.
Values are the mean log10 genome equivalents/animal ± standard deviation.
Indicates P ≤60.1.
Indicates P ≤60.05.
Indicates P ≤60.01 by Mann–Whitney non-parametric statistical evaluation in comparison to vehicle control.
3. Discussion
In previous studies, cyclopropavir (1) was determined to be highly efficacious in four different animal models of cytomegalovirus infections.4 First, a lethal inoculum of murine cytomegalovirus administered intraperitoneally to BALB/c mice was effectively treated with oral doses ranging from 50 to 1 mg/kg CPV (1) when treatment was initiated 24 or 48 h post viral inoculation resulting in fewer viral induced mortalities (P <0.01). Even when treatment was started 72 h post viral inoculation with oral doses of CPV (1) of 10 and 3 mg/kg, survival was significantly improved (P <0.05). Tissue samples from SCID mice infected with MCMV revealed that mice treated with 10 mg/kg CPV (1) had decreased virus titers in liver, lung, spleen and kidney samples by 2 to 5 log10 compared to vehicle treated mice. Also, two models of human cytomegalovirus using surgically implanted human fetal tissues in SCID mice revealed that titers of HCMV were significantly reduced by oral CPV (1) at doses of 45 and 15 in the SCID-human retinal or SCID-human thymus/liver models (P ≤0.002). Lower doses of 10 or 3 mg/kg CPV (1) were also evaluated in both models. Neither dosage level showed significant efficacy in the ocular model, but viral reduction was statistically significant using 10 mg/kg in the thymus/liver model of HCMV.
Several other active antiviral compounds have been chemically modified for increased oral bioavailability and improved efficacy. The most noteworthy of those was cidofovir which was coupled to a lipid side chain to create hexadecyloxypropyl-cidofovir16 or CMX001. Not only was the antiviral compound orally bioavailable, but the systemic distribution, as well as, cellular uptake was dramatically improved.17,18 By improving uptake and distribution, reduction in the quantities of active component administered to patients should therefore reflect fewer toxic side effects while maintaining effective antiviral properties. These studies using the prodrug 6-deoxycyclopropavir (10) support that hypothesis. The lowest orally administered dose of 5.6 mg/kg of 6-deoxycyclopropavir (10) completely protected BALB/c mice from mortality due to MCMV even when treatment was initiated 48 h post viral inoculation. Further studies with this compound may be warranted using additional lower doses to determine the lowest effective dose in the MCMV model. Also, 12.5 mg/kg of 6-deoxycyclopropavir (10) was effective in the SCID-human thymus/liver model of HCMV which is a lower dose than the previously reported4 higher doses using 45 or 15 mg/kg of CPV (1) in the SCID-human thymus/liver model and ocular model. However, the lowest effective dose of 10 mg/kg dose of CPV (1) was effective in the thymus/liver model of HCMV, as previously reported,4 which appears to show that CPV (1) is only marginally more active in vivo than 6-deoxycyclopropavir (10).
As HCMV is still considered one of the most likely infectious agents involved in the failure of solid organ transplantation, the need is critical in the development of antiviral therapies for use in this important human disease.19 6-Deoxycyclopropavir (10) may potentially be useful in the treatment of HCMV infections alone as a novel therapy or in combination with other approved medications.
4. Conclusion
6-Deoxycyclopropavir (10), a prodrug of cyclopropavir (1), was synthesized and evaluated for in vitro and in vivo antiviral activity. It had little antiviral activity in culture but it was effective against MCMV infections in mice and against HCMV infections in SCID mice.
Compound 10 was a substrate for xanthine oxidase.
5. Experimental
5.1. General methods
The NMR spectra were determined using Mercury 400 spectrometer in CD3SOCD3 at 400 (1H) and 100 (13C) MHz, respectively. UV spectra were measured in ethanol. Mass spectra were performed on MICROMASS QUATTRO LC-MS instrument in an electro-spray ionization mode (ESI-MS) using aqueous MeOH and NaCl. In vitro antiviral2,20 and xanthine oxidase14 assays were performed as described before. For a description of xanthine oxidase catalyzed oxidation of 6-deoxycyclopropavir (10) see Figures 1 and 2.
5.2. 6-Deoxycyclopropavir (10)
Iodotrimethylsilane (10 mL, 71 mmol) was added to a solution of (Z)-2-amino-6-chloro-9-{[2,2-bis(hydroxymethyl)cyclopropylidene]methyl}purine2 (13, 2.0 g, 7.1 mmol) in DMF (80 mL) at 0 °C with stirring which was continued for 3 h. The DMF was evaporated in vacuo and a liquid residue was added into saturated solution of NaHCO3 (50 mL) at 0 °C. The mixture was lyophilized and the residue was put on a silica gel column as a suspension in chromatographic solvent. The elution was performed using CH2Cl2-MeOH (50:1 to 10:1) to give the crude (Z)-2-amino-6-iodo-9-{[2,2-bis(hydroxymethyl)cyclopropylidene]methyl}purine (14). A mixture of this intermediate, AIBN (0.25 g, 1.4 mmol) and Bu3SnH (20 mL, 71 mmol) in DMF (60 mL) was stirred at 65-75 °C for 30 min. The solvent was evaporated in vacuo, the residue consisting of two liquid phases (the upper layer was colorless and the lower one was red) was extracted with hexanes (2 × 60 mL). Hexane layer was decanted and the red oily material was chromatographed on silica gel using CH2Cl2–MeOH (20:1 to 10:1). The resultant solid was recrystallized from methanol to give 6-deoxycyclopropavir (10, 1.10 g, 63%) as a pale yellow solid, mp 222–224 °C. UV max (ethanol) 310 nm (ε 7200), 236 (ε 30,000). 1H NMR δ 8.75 (s, 1H, H6), 8.59 (s, 1H, H8), 7.23 (s, 1H, H1′), 6.60 (s, 2H, NH2), 5.00 (t, 2H, J =5.4 Hz, OH), 3.49, 3.67 and 3.50, 3.66 (2AB, 4H, J =10.6 Hz, H5′), 1.32 (d, 2H, J =1.6 Hz, H3). 13C NMR 161.5 (C2), 151.9 (C4), 150.0 (C6), 140.1 (C8), 127.3 (C5), 118.1 (C2′), 110.4 (C1′), 62.9 (C5′), 31.4 (C4′), 11.6 (C3′); ESI-MS (MeOH+ NaCl) 248 (88.9, M+H), 270 (89.4, M+Na), 517 (100.0, 2M+Na). Anal. Calcd for C11H13N5O20.2H2O: C, 52.66; H, 5.34; N, 27.92. Found: C, 52.65; H, 5.30; N, 27.77.
5.3. Animal models
5.3.1. Antiviral compounds
6-Deoxycyclopropavir (10) was synthesized as described in this communication. Ganciclovir (GCV, 2) was purchased from the University of Alabama Hospital Pharmacy and used as a positive control in MCMV experiments. GCV (2) was diluted in sterile saline and 6-deoxycyclopropavir in 0.4% carboxymethylcellulose (CMC) at various dosages and administered to mice in 0.2 mL orally. Cidofovir (CDV, Gilead Pharmaceuticals, Foster City, CA) was diluted in saline for ip administration in a 0.1 mL volume providing 20 mg/kg as a positive control in HCMV experiments.
5.3.2. Tissue, cells and viruses
Human foreskin fibroblast (HFF) cells were used as primary cultures for HCMV. These cells were propagated in MEM containing 10% FBS, L-glutamine with or without antibiotics.22 Human fetal tissue was obtained from Advanced Biosciences Resources, Alameda, CA The Smith strain of MCMV was obtained originally from Dr. June Osborne, University of Wisconsin, and the Toledo strain of HCMV from Dr. Edward Mocarski, Palo Alto, CA.
5.3.3. Mice
Female BALB/c mice, 3 weeks of age were obtained from Charles River Laboratories, Raleigh, NC. Male SCID mice, 5–8 weeks of age were obtained from Frederick Cancer Research Laboratories, MD. Mice were group housed in microisolator cages and utilized at a quantity of 15–24 mice per treatment group for statistical analysis. Mice were obtained, housed, utilized and euthanized according to USDA and AAALAC regulatory policies. All animal procedures were approved by The University of Alabama at Birmingham, Institutional Animal Care and Use Committee prior to initiation of studies.
5.3.4. Experimental MCMV infections
MCMV infections were initiated by ip inoculation of BALB/c mice using an approximate LD90 and observed daily for 21 days. GCV (2) or 6-deoxycyclopropavir (10) was administered orally (p.o.) once daily for 5 days. GCV (2) was diluted in sterile saline and administered once daily at 50, 16.7 and 5.6 mg/kg for 5 consecutive days beginning either 24 or 48 h post viral inoculation. 6-Deoxycyclopropavir (10) was suspended in 0.4% carboxymethylcellulose (CMC) and administered once daily at 50, 16.7 and 5.6 mg/kg for 5 consecutive days beginning either 24 or 48 h post viral inoculation. Uninfected mice were treated with 6-deoxycyclopropavir (10) in groups of 5 for evaluation of toxicity by clinical signs and body weight.
5.3.5. SCID-human mouse model for HCMV infection in thymus/liver tissue
For the HCMV infection, 4- to 6-week old male SCID mice were anesthetized, and fragments of human fetal thymus and liver (thy/liv) were implanted under the capsule of one kidney using an 18-gauge trocar using techniques described previously.21,22 Following an implant growth period of 12–14 weeks, the grafts were inoculated with 9000 pfu of HCMV. Beginning 24 h after viral inoculation, the mice were treated once daily for 28 days. 6-Deoxycyclopropavir (10) was administered orally once daily at 12.5 or 6.25 mg/kg and CDV was injected intraperitoneally using 20 mg/kg. On days 14, 21, 28 and 35, five to 12 implants were biopsied (approximately 50% of the graft size), homogenized, and frozen at −70 °C until assayed for HCMV. Biopsies were also obtained one week after treatment ended (day 35) to determine if viral replication rebounded after cessation of treatment.
5.3.6. Viral replication in implant tissue
Biopsies were removed, weighed and homogenized using a Kontes tissue grinder (Vineland, NJ) in MEM containing 10% FBS, mM L-glutamine, 200 U/mL penicillin, 50 μg/mL gentamicin and 3 μg/mL amphotericin B. The homogenate was centrifuged at 1500 rpm for 15 min at 4 °C, the supernatant removed and frozen at -70 °C until assayed for HCMV using viral quantitation by PCR. A 200 μL aliquot was taken from each sample and total DNA was prepared using Wizard® SV 96 Genomic DNA Purification System (Promega, Madison WI). Viral load was quantified by real time PCR using primers 5″-AGG TCT TCA AGG AAC TCA GCA AGA-3″ and 5″-CGG CAA TCG GTT TGT TGT AAA-3″, with the probe 5″-(6FAM) CCG TCA GCC ATT CTC TCG GC TAMRA-3″, and the plasmid pMP217 to provide absolute quantification. Titers were expressed as log10 genome equivalent/sample (ge).
5.3.7. Evaluation of efficacy: statistics
In order to determine therapeutic efficacy in these models, animals treated with the 6-deoxycyclopropavir (10) or GCV (2) were compared to vehicle-treated animals and differences in mortality rates were evaluated using a Fisher's Exact Test. Difference in mean day of death (MDD) and virus titers in tissues were determined by a Mann-Whitney U Rank Sum Test. A p value of 0.05 or less was considered significant.
Acknowledgments
We thank L.M. Hrihorczuk from the Central Instrumentation Facility, Department of Chemistry, Wayne State University for mass spectra. The authors gratefully acknowledge the excellent technical assistance provided by Caroll Hartline, Deborah J. Collins and Terri L Rice in performance of the animal studies, also Julie Breitenbach and Kathy Borysko for some of the in vitro studies. This work was supported by SBIR grant 5 R44 AI054135, contracts N01-AI-15439 and HHSN272201000027I from the National Institute of Allergy and Infectious Diseases.
References
- 1.Zemlicka J. In: Advances in Antiviral Drug Research. De Clercq E, editor. Vol. 5. The Netherlands: Elsevier; Amsterdam: 2007. pp. 113–165. [Google Scholar]
- 2.Zhou S, Breitenbach JM, Borysko KZ, Drach JC, Kern ER, Gullen E, Cheng YC, Zemlicka J. J Med Chem. 2004;47:566. doi: 10.1021/jm030316s. [DOI] [PubMed] [Google Scholar]
- 3.Kern ER, Kushner NL, Hartline CB, Williams-Azziz SL, Harden EA, Zhou S, Zemlicka J, Prichard MN. Antimicrob Agents Chemother. 2005;49:1039. doi: 10.1128/AAC.49.3.1039-1045.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kern ER, Bidanset DJ, Hartline CB, Yan Z, Zemlicka J, Quenelle DC. Antimicrob Agents Chemother. 2004;48:4745. doi: 10.1128/AAC.48.12.4745-4753.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bowlin TL, Brooks JL, Zemlicka J. Preclinical, toxicokinetic and toxicology results for cyclopropavir, a promising new agent for the treatment of beta- and gamma-herpesviruses. Antiviral Res; 22nd International Conference on Antiviral Research; May 3–7, 2009; Miami Beach, Florida, USA. 2009. p. A47. [Google Scholar]
- 6.Krasny HC, Beauchamp LM, Krenitsky TA, De Miranda P. Drug Metab Dispos. 1995;23:1242. [PubMed] [Google Scholar]
- 7.Krenitsky TA, Hall WW, De Miranda P, Beauchamp LM, Shaeffer HJ, Whiteman PD. Proc Natl Acad Sci USA. 1984;81:3209. doi: 10.1073/pnas.81.10.3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rees PJ, Selby P, Prentice HG, Whiteman PD, Grant DM. J Antimicrob Chemother. 1986;18:215. doi: 10.1093/jac/18.supplement_b.215. [DOI] [PubMed] [Google Scholar]
- 9.Harnden MR, Jarvest RL, Boyd MR, Sutton D. J Med Chem. 1989;32:1738. doi: 10.1021/jm00128a012. [DOI] [PubMed] [Google Scholar]
- 10.Iwayama S, Suzuki K, Nakagawa T, Sekiyama T, Ohnishi T, Ohmura Y, Ono N, Nakagawa H, Ikemura O, Tsuji T, Okunishi M. Antiviral Res. 1998;37:A70. [Google Scholar]
- 11.Hall WW, Krenitsky TA. Arch Biochem Biophys. 1986;251:36. doi: 10.1016/0003-9861(86)90048-2. [DOI] [PubMed] [Google Scholar]
- 12.Vere Hodge RA. Antiviral Chem Chemother. 1993;4:67. doi: 10.1177/2040206618783924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ann. Rep. Med. Chem., Vol. 30, Cheng, X.-M., Chapter 31. To Market, To Market - 1994, 1995; pp 295–317.
- 14.Wang R, Chen X, Drach JC, Kern ER, Zemlicka J. Nucleosides, Nucleotides Nucleic Acids. 2003;22:135. doi: 10.1081/NCN-120019502. [DOI] [PubMed] [Google Scholar]
- 15.Neumann WP. Synthesis. 1987:665. [Google Scholar]
- 16.Beadle JR, Hartline C, Aldern KA, Rodriguez N, Harden E, Kern ER, Hostetler KY. Antimicrob Agents Chemother. 2002;46:2381. doi: 10.1128/AAC.46.8.2381-2386.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Quenelle DC, Lampert B, Collins DJ, Rice TL, Painter GR, Kern ER. J Infect Dis. 2010;202:1492. doi: 10.1086/656717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aldern KA, Ciesla SL, Winegarden KL, Hostetler KY. Mol Pharmacol. 2003;63:678. doi: 10.1124/mol.63.3.678. [DOI] [PubMed] [Google Scholar]
- 19.Eid AJ, Razonable RR. Drugs. 2010;70:965. doi: 10.2165/10898540-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 20.Li C, Prichard MN, Korba BE, Drach JC, Zemlicka J. Bioorg Med Chem. 2008;16:2148. doi: 10.1016/j.bmc.2007.11.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bidanset DJ, Beadle JR, Wan WB, Hostetler KY, Kern ER. J Infect Dis. 2004;190:499. doi: 10.1086/421912. [DOI] [PubMed] [Google Scholar]
- 22.Kemble GW, Duke GM, Mocarski ES. Human Cytomegalovirus Infection of the SCID-hu (Thy/Liv) Mouse. In: Zak O, Sande MA, editors. Handbook of Animal Models of Infection. Academic Press; London, UK: 1999. p. 951. [Google Scholar]