

Abstract
Inflammatory cytokines such as tumor necrosis factor (TNF)-α and interleukin (IL)-1β stimulate glucuronosyl-transferase genes (S and P) in endothelial cells (ECs) and up-regulate sulfoglucuronosyl paragloboside (SGPG) expression, which serves as a ligand for T-cell adhesion. However, the mechanism of cytokine-mediated gene up-regulation has not been elucidated. To evaluate the precise mechanism of SGPG up-regulation, we have specifically inhibited the SGPG synthesis in the cerebromicrovascular EC line (SV-HCECs), a transformed brain ECs of human origin. SV-HCECs were transfected with small interfering RNA designed to mimic the human natural killer epitope-1 sulfotransferase (HNK-1ST), the ultimate enzyme that transfers the sulfate group to glucuronic acid for SGPG synthesis. An inhibition of SGPG expression along with a reduction of human CD4+-cell adhesion was observed in siRNA HNK-1ST (siHNK-1)-transfected cells after TNFα stimulation. A thorough screening of the signaling system confirmed that TNFα/IL-1β stimulation up-regulated nuclear factor κB (NFκB) signaling in SV-HCECs. siHNK-1 transfection interfered with the SGPG up-regulation after TNFα/IL-1β stimulation in transfected cells and reduced the T-cell adhesion. Hence, our study indicates that T-cell-SGPG adhesion in SV-HCECs may proceed through NFκB activation. In addition, siHNK-1 transfection reduced the NFκB activity compared with cells that were transfected with scrambled siRNA, before and after TNFα/IL-1β stimulation. This is the first report indicating that NFκB signaling is involved in SGPG gene expression in brain ECs by an unknown mechanism. Its down-regulation by inhibiting HNK-1ST expression may have a potential use in preventing the T-cell invasion and consequently nerve damage during inflammation.
Keywords: blood-brain/nerve barrier, endothelial cell, human natural killer antigen, NFκB signaling, glycosphingolipid
Glycosphingolipids (GSLs) are cell-surface molecules and play multifunctional roles. They may act as cell surface antigens; bind with microorganisms and with their toxins, hormones, and viruses; initiate or modulate signal transduction; and participate in cell adhesion (Bergelson, 1995; Karlsson, 1995; Riboni et al., 1997; Hakomori, 2000; Kolter and Sandhoff, 2006). Two GSLs, sulfoglucuronosyl paragloboside (SGPG) and its analogue sulfoglucuronosyl lactosaminylparagloboside (SGLPG; Chou et al., 1986; Ariga et al., 1987), are highly expressed during mouse embryonic brain development, but their concentration is reduced beyond the detection limit at later developmental stages (Schwarting et al., 1987), with an exception in murine cerebellum (Chou and Jungalwala, 1988). They have been characterized in peripheral nervous system (PNS) in an approximate ratio of SGPG:SGLPG of 10:1 (Kohriyama et al., 1987). SGPG has been identified as a minor component of total GSLs composition of bovine brain endothelial cells (ECs) and has been characterized in human cerebromicrovascular ECs (Kanda et al., 1994; Duvar et al., 2000), immortalized with SV40 T-antigen, and designated as SV-HCEC (Muruganandam et al., 1997). In vitro, SGPG concentration in ECs is elevated during inflammatory conditions with increasing T-cell adhesion (Kanda et al., 1995; Dasgupta et al., 2007). Hence, we postulated that SGPG participates in EC-leukocyte adhesion via recognition of cell adhesion molecules such as L-selectin (CD62L) that are abundantly expressed on the T-cell surface (Arbones et al., 1994; Rosen, 2004).
L-selectin is a carbohydrate-binding protein that binds with high endothelial venules (HEV) and promotes T-lymphocyte homing (Arbones et al., 1994). The interaction of these ligands is specific to carbohydrate configuration such as sialyl Lewis X (sLex) and its sulfoderivative, which contains a sulfate group at C6-GlcNAc moiety and bears the structure as NeuAcα2–3Galβ1–4[Fucα1–3(sulfo-6)]GlcNAc (Rosen, 2004). The carbohydrate ligand, 6-sulfo(GlcNAc)-sLex, of both O- and N-glycans reacts with L-selectin and mediates in vivo lymphocyte homing, and this has been demonstrated using mutant mice lacking both core 1 and core 2 extension enzymes of O-glycan synthesis (Mitoma et al., 2007). Studies indicate that the sulfation of sLex is carried out by two different GlcNAc-sulfotransferases, GlcNAc6ST-1 and GlcNAc6ST-2 (Mitoma et al., 2007). Mice that are deficient in both GlcNAc-sulfotransferases completely lack 6-sulfo(GlcNAc)-sLex and show impaired lymphocytes homing (Kawashima et al., 2005; Uchimura et al., 2005), indicating the importance of sulfation in L-selectin binding. Both SGPG and SGLPG contain a terminal sulfated carbohydrate acid (sulfated glucuronic acid) with a neolacto-(GlcNAc-Gal) tetrasaccharide backbone (Chou et al., 1986; Ariga et al., 1987), and we have previously provided evidence that SGPG can act as a ligand for L-selectin (Kanda et al., 1995; Dasgupta et al., 2007).
Our earlier study indicates that SGPG expression in bovine brain ECs is up-regulated by stimulation with interleukin (IL)-1β, and the up-regulation plays a significant role in T-cell adhesion (Kanda et al., 1995). More recently, we reported that inflammatory cytokines such as tumor necrosis factor (TNF)-α and IL-1β stimulate glucuronosyltransferase genes (GlcAT-P and GlcAT-S) and elevate SGPG concentrations in SV-HCECs, promoting T-cell adhesion (Dasgupta et al., 2007). Although our study indicated that SGPG played a significant role during inflammatory responses, the precise mechanism of SGPG elevation and its role as the molecular ligand for L-selectin demands further study. We have now elucidated the mechanism of T-cell adhesion to ECs by regulating the in situ SGPG expression and examining the cell signaling pathways. By blocking specifically the SGPG expression and assessing the T-cell binding, we have unequivocally established that SGPG is a ligand for T-cell adhesion. We have further demonstrated that the SGPG up-regulation is mediated through the nuclear factor κB (NFκB) signaling pathway. Thus, our study has revealed a novel mechanism regarding the signaling pathway for SGPG expression under inflammatory conditions.
MATERIALS AND METHODS
Cytokines and human TNFα and IL-1β were purchased from PeproTech (Rocky Hill, NJ), siRNA HNK-1 sulfotransferase (HNK-1ST) SMARTpool was purchased from Dharmacon Inc. (Thermo Fisher Scientific, Lafayette, CO), with the following primer sequences: duplex 5 sense: GCU GAU UGU UCU AAA UGG AUU, antisense: 5′-P UCC AUU UAG AAC AAU CAG CUU; duplex 6 sense: GUA AGA GAU CCC UUC GAA AUU, antisense: 5′-P UUU CGA AGG GAU CUC UUA CUU; duplex 7 sense: UGA CAA CCA UGC CGG AGG UUU, antisense: 5′-P ACU UCC GGC AUG GUU GUC AUU; duplex 8 sense: CUA GCA AGU UCA UCA CGU UUU, antisense: 5′-P AAC GUG AUG AAC UUG CUA GUU.
A scrambled siRNA SMARTpool was received to compare the transfection performance and validity of the results. The siRNA sample was dissolved in 1× siRNA buffer (0.5 ml = 20 μM) and preserved into 10 tubes (50 μl each). The dual luciferase reporter assay system was purchased from Promega (Madison, WI). Cell culture media and growth factors were purchased from Invitrogen (Carlsbad, CA). A human CD4+ (T) cell enrichment (positive selection) kit was purchased from Stemcell Technologies (Vancouver, British Columbia, Canada). Monoclonal antibody against SGPG (NGR 50, mouse IgG) was a generous gift from Dr. Toshio Ariga, and serum from a patient with peripheral neuropathy (IgM paraproteinemia, LT-serum, IgM) was also used as the source of SGPG antibody (Ariga et al., 1987; Kanda et al., 1995; Dasgupta et al., 2007). All reagents, buffers, and chemicals were of analytical grade or higher.
Cell Culture
An EC line of human cerebromicrovascular origin (SV-HCECs; Muruganandam et al., 1997) was grown in a 0.5% gelatin-coated dish in the medium (media 199; Invitrogen) containing insulin-transferrin-selenium (Invitrogen), heparin (Sigma, St. Louis, MO), and penicillin-streptomycin (Cellgro) as described previously (Muruganandam et al., 1997; Duvar et al., 2000). Jurkat cells culture was grown and maintained in RPMI 1640 (Cellgro) medium containing 10% FBS and penicillin-streptomycin (Dasgupta et al., 2007).
Human CD4+ Cells
Human CD4+ cells were prepared from human cord blood. After the policy and guidelines established by the Institutional Review Board of the Medical College of Georgia, cord blood samples were obtained from patients with full-term C-section delivery after obtaining written consent. The mononuclear fraction was isolated by standard centrifugation over Hespan (Hetastarch). Human cord blood CD4+ cells were purified by immunomagnetic selection by using a CD4+ positive selection kit (Stemcell Technology) according to the manufacturer’s instructions. CD4+ cells were plated for short-term expansion, and purified CD4+ cells were grown in RPMI medium containing 10% FBS before use for the adhesion assay.
Transfection of SV-HCECs With siRNA HNK-1ST
Cells were transfected in Amaxa Nucleofector equipment using the T20 program under aseptic conditions. Briefly, cells were grown in 3 × 100-mm dishes, taken out by trypsinization, counted, and divided into three equal parts (1.0 × 106 cells). Cells were then suspended in 0.1 ml of tranfecting media in presence of siRNA HNK-1ST (siHNK-1; 7.5 μl = 150 pmol). Two controls were prepared simultaneously using 1) scrambled siRNA mix (control 1) and 2) buffer only (minus siRNA) as mock transfection for comparison. The mixture containing the cell suspension was transferred into a sterile cuvette (2-mm gap) and zapped in the T20 program. Five hundred microliters of the prewarmed medium (complete) was added immediately, and the cells were aspirated carefully. The transfected cells were dispersed mostly in a 100-mm dish, and the rest were distributed in a sonic seal slide that had been precoated with 0.5% gelatin. The media were changed after 4–6 hr of transfection, and cells were incubated for 48 hr before the next experiment.
RT-PCR for HNK-1ST and Other Gene Expression
To verify the suppression of HNK-1ST expression in transfected cells, we purified RNA from PBS-washed collected cells using Trizol (Invitrogen) and prepared the RNA according to the manufacturer’s specification. The integrity of RNA was verified by using a 2% agarose gel. cDNA synthesis was carried out using with a script RT kit (Qiagen, Valencia, CA). With this cDNA as the template, PCR was performed with specific primers for three genes, HNK-1ST and two glucuronosyltransferases, GlcAT-S and GlcAT-P. The template volume for the reaction mixture was 1–2 μl. The synthesis of cDNA was subjected to PCR under settings described previously (Dasgupta et al., 2007). All products were run on a 2% agarose gel and compared with a 100-bp DNA ladder. All template volumes were normalized with β-actin. The following sequences of primers were used for this study. β-actin sense: 5′-TGACGGGG TCACCCACACTGTGCCCATCTA-3′, antisense: 5′-CTA-GAAGCATTTGCGGTGGACGATGGAGGG-3′; HNK-1ST sense: 5′-ATGTTCATGGTGGCCAGCAAG-3′, antisense: 5′-GCTCCATTTAGGAAGACGACTAGC-3′; GlcAT-P sense: 5′-GGGTEATGAGGAGCTGTGGG-3′, anti-sense: 5′-CT AGCCATGGAAGGTTGGGC-3′; GlcAT-S sense: 5′-ATG AAGTCCGCGCTGTGCAG-3′, antisense: 5′-ACATGCA GGTGCGTGTTGGG-3′.
Real-Time PCR for Quantification of HNK-1ST Gene Expression in Transfected Cells
SV-HCEC is an EC line of human origin. Hence the degree of HNK-1ST gene in transfected cells was measured by using primers designed from genes of human origin and running 40 cycles in a Bio-Rad iCycler. The efficacy of suppression of HNK-1ST expression was compared with the expression of siHNK-1-transfected cells with scrambled siRNA-transfected cells and/or with mock transfection. Real-time PCR was performed with the following settings: 94°C, 30 sec (denaturation), 56°C (30 sec annealing), and 72°C, 1 min (elongation), followed by 72°C, 2 min (final elongation), one cycle, with all samples normalized to β-actin (Liour et al., 2005).-Fold changes for the treated samples was measured by the formula-2ΔΔCt (Pfaff, 2001) and expressed relative to untreated samples such as regular control, scrambled-, or mock-transfected cells. The following primers for HNK-1ST were used: sense 5′-TGAGGAACTGAAGCCAACTGAGAA-3′, antisense 5′-AAACTTGGAGACAGGAGTGTGCGA-3′.
Immnunooverlay Analysis of the SGPG Concentration in Transfected Cells
Because SGPG is a minor component of the total GSLs in SV-HCEC (Duvar et al., 2000; Dasgupta et al., 2007), we cultured the control and transfected cells in separate 150-mm dishes to obtain sufficient cells for SGPG analysis by the immunooverlay method. Control, scrambled, siRNA-transfected cells and siHNK-1-transfected cells were grown for 48 hr as described. Cells were then treated with TNFα for 24 hr. Cells were then collected, washed with cold PBS, and preserved at −20°C. Lipids were extracted and the SGPG fraction was purified from the lipid extract using DEAE A25 (acetate form) as described previously (Dasgupta et al., 2007). Each purified fraction was dissolved in a defined volume of solvent (determined by the protein content) and equal volumes of the samples were applied via HPTLC with standard SGPG. The plate was developed in chloroform:methanol:0.25% CaCl2 (55:45:10; v/v) and was exposed to NGR 50 antibody, followed by a secondary antibody conjugated with peroxidase. After washing with PBS, a chemiluminescnce reagent was added to the plate, and the bands were identified after exposure to an X-ray film (Dasgupta et al., 2007). Bands in each lane were scanned in ImageJ and quantified.
Immunocytochemical Localization of SGPG
To verify the effect of siHNK-1 transfection on SGPG expression, we also performed immunocytochemical localization of SGPG in control and transfected cells (Dasgupta et al., 2007). Cells, cultured on the slides, were grown for 48 hr and treated with cytokines for 24 hr. Cells were then washed with 1× Hank’s balanced salt solution and fixed with 4% paraformaldehyde. The fixed cells were permeabilized and then treated with the Mab NGR 50 or LT serum followed by the secondary antibody (anti-mouse IgG or anti-human-IgM) conjugated with TRITC. Cells were further stained with Hoechst 33258 (nuclear stain) and visualized under a phase-contrast microscope.
T-Cell Adhesion
To establish the role of SGPG as a ligand for T-cell adhesion unequivocally, we examined adhesion properties of siHNK-1-transfected SV-HCECs with Jurkat cells that were tagged with Vybrant DiO (Dasgupta et al., 2007). Briefly, SV-HCECs were transfected with siHNK-1 as described earlier. Cells were then grown in 60-mm dishes for 48 hr and then incubated with TNFα overnight. The following sets of cells were prepared for adhesion study: set 1: control (untreated) SV-HCECs; set 2: control (untreated) SV-HCECs plus TNFα (100 ng/ml); set 3: SV-HCECs with scrambled siRNA transfection; set 4: SV-HCECs with scrambled siRNA transfection plus TNFα (100 ng/ml); set 5: SV-HCECs with siHNK-1 transfection; set 6: SV-HCECs with siHNK-1 transfection plus TNFα (100 ng/ml).
Each set was prepared in triplicate. Jurkat cells were washed with RPMI 1640 (without FBS) to remove the serum and incubated with Vybrant DiO at 37°C for 30 min. Tagged cells were washed and resuspended in RPMI 1640 to a concentration of 1 × 106 cells/ml. A defined amount of cells (1 ml) was added in each well and incubated at 37°C for 30 min. Unattached cells were removed by washing with PBS. Total cells were removed by incubating with trypsin, and the attached cells were counted in a flow cytometer. The number of adhered cells was determined in the Cell Quest program.
In addition, we purified human T cells (CD4+) from human cord blood using a CD4+ isolation kit (positive selection) according to the manufacturer’s protocol. These cells were grown for 48 hr in RPMI 1640 medium containing 10% FBS. Cells were counted and suspended in 0.5 ml of 2% FBS-PBS and then labeled with anti-CD4+ serum tagged with FITC as described. Initial blocking was performed with Fc blockers (1/40) for 5 min at 4°C (ice), followed by incubation with CD4+ antibody (FITC-conjugated; 1/200) for 15 min in ice. Cells were then washed with PBS/RPMI (without FBS) for adhesion assay. A portion of the cells (20 μl) was examined for labeling efficacy by using flow cytometry (85–90% positive).
Transfected SV-HCECs (siHNK-1 and scrambled siRNA) were dispensed in a 24-well culture plate (approximately 2.5 × 104 cells/well). Cells were grown for 48 hr and then treated with cytokines overnight and washed with cold PBS. A suspension of CD4+ cells (0.3 ml) in RPMI medium (approximately 5 × 105 cells/ml) was added and incubated at 37°C for 30 min. Unattached cells were removed by washing with PBS. Total cells were then removed by incubating with trypsin, and the number of attached cells was determined using a flow cytometer. For statistical evaluation, a group of three wells in which cells were grown and treated under same experimental conditions was considered as a single set. Six individual sets of experiment were prepared as described above. Experiments were repeated at least twice for confirmatory results.
TNFα and IL-1β Stimulate NFκB Signaling in SV-HCECs
To investigate further the mechanism of cytokine-mediated up-regulation of GlcAT gene expression, we examined the role of cell signaling in SV-HCECs after TNFα and IL-1β stimulation. Cultured cells were collected (2.88 × 106 cells) and divided into seven groups (three wells in each group) each containing 1 × 105 cells/well: group 1, negative control (background): 3 × 105 cells using 0.8 μg of empty plasmid vector plus 0.08 μg of pRL-CMV (Renilla luciferase) internal control plasmid and 2.5 μl of siRNA scramble in 100 μl of media and divided into three wells after electroporation; group 2, control: 3 × 105 cells using 0.4 μg of pNFκB-luc, a multimerized κB-luciferase reporter gene plasmid, plus 0.08 μg of pRL-CMV (Renilla luciferase) internal control plasmid and 2.5 μl of siRNA scramble in 100 μl of media, with cells divided into three wells after electroporation; group 3: control (group 2) + TNFα: cells were divided into three wells after electroporation; group 4, control (group 2) + IL-1β: cells were divided into three wells after electroporation; group 5, siHNK-1 (~50 nM): 3 × 105 cells using 0.4 μg of pNFκB-luc, a multimerized κB-luciferase reporter gene plasmid, 0.08 μg of pRL-CMV (Renilla luciferase) internal control plasmid and 2.5 μl of si HNK-1 (~50 nM) in 100 μl of media and divided into three wells after electroporation; group 6, siHNK-1 (~50 nM, group 5) + TNFα: cells were divided into three wells after electroporation; group 7, siHNK-1 (~50 nM, group 5) + IL-1β: cells were divided into three wells after electroporation.
Cells were incubated for 24 hr, and cells in groups 3 and 6 were treated with TNFα (100 ng/ml), and the cells in groups 4 and 7 were treated with IL-1β at a concentration of 10 ng/ml. Cell extracts were prepared, and the levels of luciferase activity were determined by using the dual luciferase reporter assay system in accordance with the manufacturer’s (Promega, Madison, WI) instructions. Briefly, firefly (NFκB) and Renilla (internal control) luciferase were read with a microplate luminometer (PE Applied Biosystems), and background was subtracted. The NFκB luciferase was normalized to the internal control luciferase, and the NFκB activity of the control experiment was designated as 100%. Experiments were performed in triplicate and repeated at least twice to obtain a standard deviation.
RESULTS
Expression of HNK-1 Sulfotransferase (HNK-1ST) and Glucuronosyltransferase (GlcAT) Genes After siRNA HNK-1ST (siHNK-1) Transfection
We have extended our studies by examining the precise role of SGPG in inflammation and cell adhesion. To determine the specific expression of SGPG in cell adhesion, we silenced the SGPG expression by transfecting the SV-HCECs with siRNA HNK-1ST (siHNK-1) and scrambled siRNA (control). The degree of HNK-1ST inhibition was quantitatively measured by using real-time PCR and was on the order of 2−2 to 2−3 (Liour et al., 2005; Pfaffl, 2001). In other words, approximately 80% (or more) inhibition of HNK-1ST expression was observed in the siHNK-1-transfected cells. In addition, we examined and compared the expression of three genes HNK-1ST, GlcAT-P, and GlcAT-S. Our result indicated that there was a reduction in HNK-1ST expression as well as the expression of GlcAT-P and GlcAT-S after cytokine stimulation (Fig. 1). Although GAT-P expression is visible in controls only after cytokine stimulation (Fig. 1), our pervious study using the same cell lines indicated a 20-and 10-fold stimulation of GAT-S and GAT-P, respectively, after cells were exposed to cytokine (Dasgupta et al., 2007). SGPG expression was further confirmed by immunooverlay assay of purified SGPG fractions from the transfected cells and by immunocytochemical detection.
Fig. 1.

Expression of GlcAT-P, GlcAT-S, and HNK-1ST genes in control (scrambled) and siHNK-1-transfected cells.
TLC-Immunooverlay of SGPG and Immunocytochemistry
The purified SGPG fractions were examined by immunooverlay assay. siHNK-1-transfected cells showed a clear reduction of SGPG concentration even after stimulation with TNFα (Fig. 2, lane 6) compared with control stimulation (lanes 2 and 4). The image was scanned and quantified in ImageJ. Although a minor increase (1.75-fold) in SGPG expression in lane 6 was observed after TNFα stimulation compared with the untreated transfected cells (lane 5), a 3.5–4.0-fold increase was observed in both control cells when stimulated with cytokines (lanes 2 and 4). This was comparable to the degree of inhibition of the HNK-1ST gene expression after siHNK-1 transfection (Fig. 1). The level of SGPG expression in siHNK-1-tranfected cells after TNFα stimulation was almost equivalent to that of siRNA-transfected control (Fig. 2, lanes 1 and 3). Our observation was further supported by immunocytochemical localization of SGPG in the tranfected cells. An up--regulation of SGPG expression was observed in controls (Fig. 3B), whereas TNFα-stimulated siHNK-1 transfected cells (Fig. 3C) showed a very similar SGPG expression compared with the nontransfected control (Fig. 3A). This further confirms the success of the transfection.
Fig. 2.

Thin-layer chromatography-immunooverlay of purified SGPG fractions from transfected SV-HCECs. Lane 1: Mock transfection. Lane 2: Mock transfection + TNFα. Lane 3: Control (scrambled siRNA transfection). Lane 4: Control + TNFα. Lane 5: siHNK-1 transfection. Lane 6: siHNK-1 + TNFα. Lanes 7–9: Standard SGPG at different concentrations. The amount of SGPG in each lane was quantified in ImageJ. The quantity of SGPG in lanes 5 and 6 was relevant to suppression of HNK-1ST by siHNK-1 trnasfection. The method is described in detail in the text.
Fig. 3.
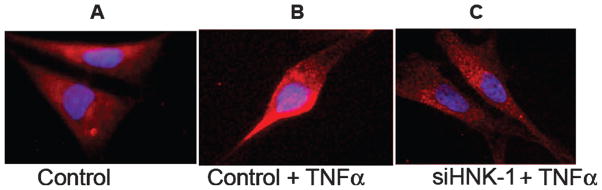
A–C: Immunocytochemical detection of SGPG in control and siHNK-1-transfected cells after TNFα stimulation. The method is described in detail in the text.
Cell Adhesion Assay
Because SGPG acts as a ligand for L-selectin and promotes T-cell adhesion (Kanda et al., 1995; Dasgupta et al., 2007), we tested the efficacy of T-cell (Jurkat and human CD4+ cells) adhesion in siHNK-1-transfected cells after TNFα stimulation. Previously, using Jurkat cells (as T cells), we tested our hypothesis that SGPG binds with T cells via L-selectin. More importantly, a similar experiment was carried out with human CD4+ cells from cord blood to confirm our previous results as well as to validate our present data. Our results indicate that SGPG is a ligand for both Jurkat (Fig. 4A) and CD4+ (Fig. 4B) cells, insofar as we observed a significant decrease (approximately 40%) in cell adhesion in SV-HCECs transfected with siHNK-1, which specifically prevented the SGPG up-regulation after TNFα stimulation.
Fig. 4.

A, B Quantitative assay of Jurkat cell (A) and CD4+ cell (B) adhesion with transfected SV-HCECs after TNFα stimulation. A reduction of Jurkat- and CD4+-cell adhesion was observed with siHNK-1-transfected SV-HCECs after TNFα stimulation. **Significant variation at P < 0.01, †significant variation at P < 0.05 as measured by t-test.
NFκB Signaling in SGPG Expression
To define the mechanism of TNFα/IL-1β-mediated GlcAT gene expression, we have examined several signaling pathways that might be involved in regulating SGPG expression. An increase in NFκB signaling after cytokine stimulation was observed in SV-HCECs, and this is consistent with previous studies suggesting that TNFα stimulates NFκB signaling in ECs (Hazra et al., 2000; Meiler et al., 2002). We also determined whether siHNK-1 transfection regulated NFκB signaling, because HNK-1ST is the final step for SGPG synthesis. A down-regulation of NFκB activity (0.5-fold) was observed in siHNK-1-transfected cells compared with scrambled transfection (Fig. 5). Although siHNK-1 inhibited NFκB activity in transfected cells, the degree of inhibition was comparatively less (0.4-fold) when the transfected cells were exposed to cytokines, TNFα and IL-1β (Fig. 5).
Fig. 5.

NFκB signaling in transfected SV-HCECs after TNFα and IL-1β stimulation. siHNK-1 transfection reduced the NFκB activity in SV-HCECs. An inhibition of NFκB activity was also observed in siHNK-1-transfected cells after cytokine stimulation. *Significant variation at P < 0.05 as measured by Student’s t-test.
DISCUSSION
SGPG has been characterized in SV-HCECs (Duvar et al., 2000), and our studies suggested that SGPG in ECs might act as a ligand for L-selectin and participate in T-cell adhesion (Kanda et al., 1995; Dasgupta et al., 2007). L-selectin, which is mostly distributed on blood leukocytes, is implicated in leukocyte homing under inflammation (Tedder et al., 1995; Winn et al., 1998) and mediates cell-cell adhesion of leukocytes-leukocytes, leukocytes-carcinoma cells, and leukocytes-ECs (Rosen, 2004). HEV in mouse and human systems expresses L-selectin. These ligands are sulfated oligosaccharides (Satomaa et al., 2002; van Zante and Rosen, 2003), and sulfation of sLex at C-6 of the GlcNAc moiety increases the binding of L-selectin to ECs, as observed in transfected cells that express 6-sulfo(GlcNAc)-sLex (Kanamori et al., 2002; van Zante and Rosen, 2003). In fact, sulfation of the carbohydrate epitope is apparently essential for L-selectin binding (Rosen, 1999; Bistrup et al., 1999). SGPG contains a sulfate group at the C-3 position of the glucuronic acid residue. It bears a structural resemblance to sulfated sLex, insofar as both contain a free–COOH and a sulfate group with a lactosamine backbone. The detailed enzymatic synthesis of SGPG is depicted in Figure 6. Studies have demonstrated that SGPG is a potential ligand for L-selectin (Needham and Schnaar, 1993; Kanda et al., 1995; Dasgupta et al., 2007), and further evaluation of the underlying mechanism by which SGPG mediates T-cell adhesion in inflammation is clearly warranted.
Fig. 6.

Enzymatic pathway for SGPG synthesis. Note that the primers for siRNA used in this study were designed to silence the gene for the ultimate enzyme, HNK-1ST, which regulates the final step for SGPG synthesis.
We have reported that the inflammatory cytokine IL-1β stimulates SGPG expression in bovine brain ECs and promotes T-cell adhesion (Kanda et al., 1995). Our recent study suggests that up-regulation of SGPG expression in SV-HCEC by IL-1β as well as by TNFα proceeds via stimulation of GlcAT-S and GlcAT-P genes (Dasgupta et al., 2007). In addition, we have proposed that SGPG acts as a ligand for L-selectin based on the following lines of evidence. First, SGPG and L-selectin were colocalized on T cells adhered to SV-HCEC. Second, we preincubated SV-HCECs with SGPG antibody (MAb NGR 50) after cytokine stimulation and/or preincubated T cells with anti-L-selectin (anti-CD62L) and observed an effective blocking of the cell adhesion. These results, however, did not show beyond doubt that SGPG was a specific ligand for T cells. The present investigations, on the other hand, have extended our previous observation by precisely regulating SGPG expression after silencing a gene that is involved in SGPG synthesis and then evaluating the role of SGPG in T-cell adhesion under inflammatory conditions. We have used the SV-HCECs for this study because 1) this cell line was prepared from brain microvascular cells of human origin and is suitable to study the blood–brain/nerve barrier in neurological disorders (Muruganandam et al., 1997); 2) it contains SGPG as one of the GSL components (Duvar et al., 2000; Dasgupta et al., 2007); 3) our previous study using this cell line indicates that inflammatory cytokines stimulate T-cell adhesion via SGPG up-regulation (Dasgupta et al., 2007), also reported in bovine brain primary ECs culture (Kanda et al., 1995); and 4) the cell line is routinely maintained and readily available in our laboratory.
We have used TNFα and IL-1β as inflammatory cytokines for this study for the following reasons. First, both TNFα and IL-1β are secreted in response to inflammation, which subsequently triggers nerve damage by activating NFκB signaling (Agarwal, 2003). Second, TNFα plays a critical role in pathogenic demyelination in PNS by mediating the systemic immune response (Jeffcote et al., 2005), which is further supported by genetic predisposition for high level of TNFα in demyelinating diseases of PNS and CNS such as Guillain-Barré syndrome and multiple sclerosis (Ma et al., 1998; McDonnell et al., 1999). Third, TNFα stimulates the activity of glycosyltransferases and sulfotransferases of GSLs and glycoprotein synthesis (Delmotte et al., 2002; Stricklett et al., 2002), and, finally, SGPG regulation in ECs is stimulated in these cells after exposure to TNFα and IL-1β (Kanda et al., 1995; Dasgupta et al., 2007). To inhibit the SGPG expression after cytokine stimulation, we introduced small interfering RNA (siRNA) for silencing the gene HNK-1 sulfotransferase, which catalyzes the final step for SGPG synthesis. By using an electroporation (Amaxa) method, we were successful in transfecting the ECs with the siHNK-1. A second set of cells was electroporated with the media composition (mock), whereas another set of cells was transfected with scrambled siRNA, and both sets were used as controls. We achieved approximately 80% (measured by real-time PCR, 2–ΔΔCt value between 2−3 and 2−2) inhibition of the gene expression in the siHNK-1-transfected cells, but no difference was observed between the two controls (data not shown). It is intriguing to note that siHNK-1 transfection also down-regulated the expression of two other genes, GlcAT-S and GlcAT-P (Fig. 1); their expression was no longer stimulated by cytokine exposure, as previously reported (Dasgupta et al., 2007). Both GlcAT-P and GlcAT-S genes regulate SGPG synthesis in an earlier step than HNK-1ST, and their expression is specifically elevated by cytokine stimulation (Dasgupta et al., 2007). It is likely that inhibition of HNK-1ST initiates the feedback inhibition to prevent the accumulation of glucuronic acid containing tetragloboside. Alternatively, this may stimulate other glycosyl-tranferase genes (for example, β-GlcNAc:galactosyltransferase) toward the synthesis of other lacto-series (repeating Gal-GlcNAc unit) GSLs. This type of positive (also the negative type) feedback loop in cell signaling is a common regulatory event in which down-regulation of expression of one gene interferes with the expression of other genes in the same loop (Brandman and Meyer, 2008).
Furthermore, the success of transfection is reflected by the quantitative estimation of SGPG in control and transfected cells; we observed a trend to resist the increase in SGPG concentration in siHNK-1-transfected cells after TNFα stimulation (Fig. 2, lane 6). This is consistent with the proposed pathway wherein HNK-1ST is a step-regulating enzyme for SGPG synthesis and is further supported by immunocytochemical localization of SGPG (Fig. 3). siHNK-1 tranfection prevented the SGPG elevation by inhibiting the gene expression responsible for SGPG synthesis (Figs. 1–3). Our data on cell adhesion further substantiated our hypothesis with the observation that a large percentage of Jurkat cells remained unattached after TNFα stimulation of the siHNK-1-transfected cells (Fig. 4A). Moreover, a significant reduction (approximately 40% or more) of CD4+ attachment was observed with siHNK-1-transfected ECs following cytokine stimulation (Fig. 4B). However, a partial inhibition (40%) of cell adhesion supports the concept that the adhesion mechanism is more complex than in our original hypothesis. In addition to SGPG, there are other molecules (for example, sLex, sulfated sLex) that could be stimulated by cytokine exposure and participate in T-cell adhesion. Taken together, our investigation unambiguously shows that SGPG participates in lymphocytes adhesion by serving as an EC ligand for Jurkat and CD4+ cells.
Although we have demonstrated unequivocally that both TNFα and IL-1β up-regulate the SGPG expression in brain ECs by stimulating GlcAT-P/GlcAT-S gene expression (Dasgupta et al., 2007), the underlying mechanism of such overexpression has not been elucidated. Understanding the molecular signaling that regulates gene expression mediated by proinflammatory cytokines leading to EC activation would have clinical implications in developing an effective strategy for interfering T-cell adhesion (Meiler et al., 2002). The NFκB signaling pathway plays an important and evolutionarily conserved role in the immune system, and much of our understanding of NFκB is obtained by studying relevant immunological signaling pathways (Hayden and Ghosh, 2008). Numerous stimuli activate NFκB, which, in turn, regulates a large number of genes (Hayden and Ghosh, 2008). In addition, TNFR1 and Toll/IL1R signaling pathways have been implicated in NFκB activation (Hayden et al., 2006). Both TNFα and IL-1β initiate nerve damage by activating NFκB signaling (Aggarwal, 2003). Hence, we focused on the activation of NFκB signaling in SV-HCECs after TNFα and IL-1β stimulation. As expected, we observed a 2.5-fold increase in NFκB activity in SV-HCECs after cytokine stimulation compared with untreated cells, and the siHNK-1 transfection reduced the NFκB activity after exposing the cells to both TNFα and IL-1β (Fig. 5). Moreover, the NFκB activity was down-regulated to 0.5-fold after siHNK-1 transfection prior to cytokine stimulation, which indicated that SGPG might regulate the basal NFκB activity (Fig. 5). This is the first report indicating that the regulation of HNK-1 epitope bearing SGPG expression is mediated by NFκB signaling. Although the precise mechanism of SGPG-mediated NFκB regulation is unknown, we hypothesize that, being a negatively charged cell surface molecule, SGPG may promote the activation/orientation of cytokine receptors (TNFR/IL1R) and thus stimulate the NFκB activity, and inhibition of SGPG expression may interrupt the orientation process and, consequently, down-regulate the NFκB activity. Alternatively, SGPG may directly activate some regulatory kinases (for example, Akt or IKKB) of NFκB signaling, and a reduction of SGPG expression may down-regulate the NFκB activity.
In summary, we have established the regulation of SGPG under inflammatory conditions and its function as a ligand for T-cell adhesion. In addition, we have provided the first evidence that the up-regulation of GlcAT genes in SV-HCEC after cytokine stimulation is mediated by NFκB signaling, and conversely, inhibition of SGPG expression by HNK-1 silencing siRNA intercepts the NFκB activity. Because both TNFα and IL-1β up-regulate NFκB signaling and cause nerve damage (Aggarwal, 2003), interruption of this signaling pathway may prevent the detrimental action of proinflammatory cytokines and protect the ECs for maintaining the integrity of the blood–brain/nerve barrier. Moreover, NFκB signaling consists of a complex series of positive and negative regulatory elements, such as inducing stimuli for triggering IKK complex activation, degrading IκB proteins, and releasing NFκB dimers (Rel family; Hayden et al., 2006). A thorough understanding of NFκB signaling would be instrumental in designing pathway-specific inhibitors of NFκB for the treatment of specific human ailments (Tergaonkar, 2006). Hence, a detailed study of the NFκB signaling relevant to HNK-1ST or other gene expression responsible for SGPG synthesis could provide information useful in searching for a therapeutic strategy for inflammatory conditions that are triggered through T-cell-SGPG adhesion.
Acknowledgments
We acknowledge the helpful assistance of K. Krishnamurthy in performing some bench work and thoughtful criticisms and suggestions provided by Dr. E. Bieberich, Associate Professor, Medical College of Georgia. We also thank Dr. L. Ignatowicz, Associate Professor, Medical College of Georgia for helpful discussion with the project.
Contract grant sponsor: NIH; Contract grant number: NS026994 (to R.K.Y.); Contract grant number: NS11853 (to R.K.Y.).
References
- Aggarwal BB. Signaling pathways of the TNF superfamily: a double-edged sword. Nat Rev Immunol. 2003;3:745–756. doi: 10.1038/nri1184. [DOI] [PubMed] [Google Scholar]
- Arbones M, Ord DC, Li K, Ratech H, Maynard-Curry C, Otten G, Capon DJ, Tedder TF. Lymphocyte homing and leukocyte rolling and migration are impaired in L-selectin-deficient mice. Immunity. 1994;1:247–260. doi: 10.1016/1074-7613(94)90076-0. [DOI] [PubMed] [Google Scholar]
- Ariga T, Kohriyama T, Fredo L, Latov N, Saito M, Kon K, Ando S, Suzuki M, Hemling ME, Rinchart KL, Kusunoki S, Yu RK. Characterization of sulfated glucuronic acid containing glycolipids reacting with IgM M-proteins in patients with neuropathy. J Biol Chem. 1987;262:848–853. [PubMed] [Google Scholar]
- Bergelson LD. Serum gangliosides as endogenous immunomodulators. Immunol Today. 1995;16:483–486. doi: 10.1016/0167-5699(95)80032-8. [DOI] [PubMed] [Google Scholar]
- Bistrup A, Bhakta S, Lee JK, Belov YC, Gunn D. Sulfotransferases of two specificities function in the reconstitution of high-endothelial-cell ligands for L-selectin. J Cell Biol. 1999;145:899–910. doi: 10.1083/jcb.145.4.899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandman O, Meyer T. Feedback loops shape cellular signals in space and time. Science. 2008;322:390–395. doi: 10.1126/science.1160617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou DK, Jungalwala FB. Sulfoglucuronyl neolactoglycolipids in adult cerebellum: specific absence in murine mutants with Purkinje abnormality. J Neurochem. 1988;50:1655–1658. doi: 10.1111/j.1471-4159.1988.tb03056.x. [DOI] [PubMed] [Google Scholar]
- Chou DK, Ilyas AA, Evans JE, Costello C, Quarles RH, Jungalwala FB. Structure of sulfated glycolipids in nervous system reacting with HNK-1 antibody and some IgM paraproteins in neuropathy. J Biol Chem. 1986;261:11717–11725. [PubMed] [Google Scholar]
- Dasgupta S, Yanagisawa M, Krishnamurthy K, Liour S, Yu RK. Tumor necrosis factor up-regulates glucuronosyltransferase gene expression in human brain endothelial cells and promotes T cell adhesion. J Neurosci Res. 2007;85:1086–1094. doi: 10.1002/jnr.21214. [DOI] [PubMed] [Google Scholar]
- Delmotte P, Degroote S, Lafitte JJ, Lamblin G, Perini JM, Roussel P. Tumor necrosis factor alpha increases the expression of glycosyl-transferase responsible for biosynthesis of sialylated and/or sulfated Lewis × epitope in the human bronchial mucosa. J Biol Chem. 2002;277:424–431. doi: 10.1074/jbc.M109958200. [DOI] [PubMed] [Google Scholar]
- Duvar S, Suzuki M, Muruganandam A, Yu RK. Glycosphingolipid composition of a newly immortalized human cerebromicrovascular endothelial cell line. J Neurochem. 2000;75:1970–1976. doi: 10.1046/j.1471-4159.2000.0751970.x. [DOI] [PubMed] [Google Scholar]
- Hakomori S-I. Cell adhesion/recognition and signal transduction through glycosphingolipid microdomain. Glycoconj J. 2000;17:143–151. doi: 10.1023/a:1026524820177. [DOI] [PubMed] [Google Scholar]
- Hayden MS, Ghosh S. Shared principles in NFκB signaling. Cell. 2008;132:344–362. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- Hayden MS, West AP, Ghosh S. NF-kappa and the immune response. Oncogene. 2006;25:6758–6780. doi: 10.1038/sj.onc.1209943. [DOI] [PubMed] [Google Scholar]
- Hazra L, Evans AL, Chen M, Hyduk SJ, Collins T, Cybulsky MI. The NF-kappa B signal transduction pathway in aortic endothelial cells is primed for activation in regions predisposed to atherosclerotic lesion formation. Proc Natl Acad Sci U S A. 2000;97:9052–9057. doi: 10.1073/pnas.97.16.9052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffcote WJ, Game F, Cavanagh PR. The role of inflammatory cytokines in the cause of neuropathic osteoarthopathy (acute Charcoat foot) in diabetes. Lancet. 2005;366:2058–2061. doi: 10.1016/S0140-6736(05)67029-8. [DOI] [PubMed] [Google Scholar]
- Kanamori A, Kojima N, Uchimura K, Muramatsu T, Tamatani T, Berndt MC, Kansas GS, Kannagi R. Distinct sulfation requirements of selectin disclosed using cells that support rolling mediated by all three selectins under shear flow. L-selectin prefers carbohydrate 6-sulfation to tyrosine sulfation, whereas P-selectin does not. J Biol Chem. 2002;277:32578–32586. doi: 10.1074/jbc.M204400200. [DOI] [PubMed] [Google Scholar]
- Kanda T, Yoshino H, Ariga T, Imakawi M, Yu RK. Glycosphin-golipid antigens in cultured bovine brain microvascular endothelial cells: sulfoglucuronosyl paragloboside is a target of monoclonal IgM in demyelinating neuropathy. J Cell Biol. 1994;136:239–246. doi: 10.1083/jcb.126.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanda T, Imawaki M, Ariga T, Yu RK. Interleukin-1β up-regulates the expression of sulfoglucuronosyl paragloboside, a ligand for L-selectin, in brain microvascular endothelial cells. Proc Natl Acad Sci U S A. 1995;92:7897–7901. doi: 10.1073/pnas.92.17.7897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson K-A. Microbial recognition of target-cell glycoconjugates. Struct Biol. 1995;5:622–635. doi: 10.1016/0959-440x(95)80054-9. [DOI] [PubMed] [Google Scholar]
- Kawashima H, Petryniak B, Hiraoka N, Mitoma J, Huckaby V, Nakayama J, Uchimura K, Kadomatsu K, Muramatsu M, Lowe JB, Fukuda M. N-acetylglucosamine-6-sulfotransferases 1 and 2 cooperatively control lymphocyte homing through L-selectin ligand biosynthesis in high endothelial venules. Nat Immunol. 2005;6:1096–1104. doi: 10.1038/ni1259. [DOI] [PubMed] [Google Scholar]
- Kohriyama T, Kusunoki S, Ariga T, Yoshino JE, DeVries GH, Latov N, Yu RK. Subcellular localization of sulfated glucuronic acid-containing glycolipids reacting with anti-myelin-associated glycolipid antibody. J Neurochem. 1987;48:1516–1522. doi: 10.1111/j.1471-4159.1987.tb05694.x. [DOI] [PubMed] [Google Scholar]
- Kolter T, Sandhoff K. Sphingolipid metabolism in diseases. Biochim Biophys Acta. 2006;45:2057–2079. doi: 10.1016/j.bbamem.2006.05.027. [DOI] [PubMed] [Google Scholar]
- Liour SS, Dinkins M, Su C-Y, Yu RK. Spatiotemporal expression of GM1 in murine medial pallial neural progenitor cells. J Comp Neu-rol. 2005;491:330–338. doi: 10.1002/cne.20696. [DOI] [PubMed] [Google Scholar]
- Ma JJ, Nishimura M, Mine H, Kuroki S, Nukina M, Ohta M, Saji H, Obayashi H, Kawakami H, Saida T, Uchiyama T. Genetic contribution of tumor necrosis factor region in Guillain-Barré syndrome. Ann Neurol. 1998;44:815–818. doi: 10.1002/ana.410440517. [DOI] [PubMed] [Google Scholar]
- McDonnell GV, Kirk CW, Middleton D, Droogan AG, Hawkins SA, Patterson CC, Graham CA. Genetic association studies of tumor necrosis alpha and tumor necrosis factor receptor 1 and 2 polymorphism across the clinical spectrum of multiple sclerosis. J Neurol. 1999;246:1051–1058. doi: 10.1007/s004150050511. [DOI] [PubMed] [Google Scholar]
- Meiler SE, Hung RR, Gerszten RE, Gianetti J, Li L, Matsui T, Gim-brone MA, Jr, Rosenwig A. Endothelial IKKβ signaling is required for monocyte adhesion under laminar flow conditions. J Mol Cell Cardiol. 2002;34:349–359. doi: 10.1006/jmcc.2001.1519. [DOI] [PubMed] [Google Scholar]
- Mitoma J, Bao X, Petryanik B, Schareli P, Gauget J-M, Yu S-Y, Kawa-shima H, Saito H, Ohtsubo K, Marth JD, Khoo K-H, von Andrian UH, Lowe JB, Fukuda M. Critical functions of N-glycans in L-selectin-mediated lymphocyte homing and recruitment. Nat Immunol. 2007;8:409–418. doi: 10.1038/ni1442. [DOI] [PubMed] [Google Scholar]
- Muruganandam A, Herx LM, Monette R, Durkin J, Stanimirovic DV. Development of immortalized human cerebromicrovascular endothelial cell line as in vitro model of human blood–brain barrier. FASEB J. 1997;11:1187–1197. doi: 10.1096/fasebj.11.13.9367354. [DOI] [PubMed] [Google Scholar]
- Needham L, Schnaar R. The HNK-1 reactive sulfoglucuronosyl glycolipids are ligands for L- and P-selectins but not for E-selectins. Proc Natl Acad Sci U S A. 1993;90:1359–1363. doi: 10.1073/pnas.90.4.1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Neucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riboni L, Viani P, Bassi R, Prinetti A, Tettamanti G. The role of sphingolipid in the process of signal transduction. Prog Lipid Res. 1997;36:153–195. doi: 10.1016/s0163-7827(97)00008-8. [DOI] [PubMed] [Google Scholar]
- Rosen SD. Endothelial ligands for L-selectin: from lymphocyte recirculation to allograft rejection. Am J Pathol. 1999;155:1013–1020. doi: 10.1016/S0002-9440(10)65201-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen SD. Ligands for L-selectin: homing, inflammation and beyond. Annu Rev Immunol. 2004;22:129–156. doi: 10.1146/annurev.immunol.21.090501.080131. [DOI] [PubMed] [Google Scholar]
- Satomaa T, Renkonen O, Helin J, Kirveskari J, Maketie A, Renkonen R. O-glycans on human endothelial CD34 putatively participating in L-selectin recognition. Blood. 2002;99:2609–2611. doi: 10.1182/blood.v99.7.2609. [DOI] [PubMed] [Google Scholar]
- Schwarting GA, Jungalwala FB, Chou DKH, Boyer AM, Yamamoto M. Glucuronic acid and sulfate containing glycoconjugates are developmentally and spatially regulated antigens in developing mammalian central nervous system. Dev Biol. 1987;120:65–76. doi: 10.1016/0012-1606(87)90104-7. [DOI] [PubMed] [Google Scholar]
- Stricklett PK, Hughes AK, Ergonul Z, Kohan DF. Molecular basis for up-regulation by inflammatory cytokines of Shiga toxin 1 cytotoxic-ity and globotriosylceramide expression. J Infect Dis. 2002;186:976–982. doi: 10.1086/344053. [DOI] [PubMed] [Google Scholar]
- Tedder TF, Steeber DA, Chen A, Engel P. The selectins: vascular adhesion molecules. FASEB J. 1995;9:866–873. [PubMed] [Google Scholar]
- Tergaonkar V. NFkappaB pathway: a good signaling paradigm and therapeutic target. Int J Biochem Cell Biol. 2006;38:1647–1653. doi: 10.1016/j.biocel.2006.03.023. [DOI] [PubMed] [Google Scholar]
- Uchimura K, Gauguet JM, Singer MS, Tsay D, Kannagi R, Muramatsu T, von Andrian UH, Rosen SD. A major class of L-selectin ligands is eliminated in mice deficient in two sulfotransferases expressed in high endothelial venules. Nat Immunol. 2005;6:1105–1113. doi: 10.1038/ni1258. [DOI] [PubMed] [Google Scholar]
- van Zante A, Rosen SD. Sulphated endothelial cell ligands for L-selectin in lymphocyte homing and inflammation. Biochem Soc Trans. 2003;31:313–317. doi: 10.1042/bst0310313. [DOI] [PubMed] [Google Scholar]
- Winn R, Vedder N, Ramamurthy C, Sharar S, Harlan J. Endothe-lial and leukocyte adhesion molecules in inflammation and diseases. Blood Coagul Fibrinolysis. 1998;9(Suppl 2):S17–S23. [PubMed] [Google Scholar]