

Abstract
Recent developments in single-molecule localization techniques using photoactivatable fluorescent proteins have allowed the probing of single-molecule motion in a living cell with high specificity, millisecond time resolution, and nanometer spatial resolution. Analyzing the dynamics of individual molecules at high densities in this manner promises to provide new insights into the mechanisms of many biological processes, including protein heterogeneity in the plasma membrane, the dynamics of cytoskeletal flow, and clustering of receptor complexes in response to signaling cues. Here we describe the method of single-molecule tracking photoactivated localization microscopy (sptPALM) and discuss how its use can contribute to a quantitative understanding of fundamental cellular processes.
1. Introduction
Dissecting the molecular mechanisms for biological processes lends great insight into how these systems function, both in health and disease states. Likewise, fluorescence imaging of specific proteins allows for the identification of dynamic processes and causal relationships. Single-molecule measurements aim to exploit and expand upon both of these powerful avenues by elucidating the dynamics of specific proteins at the molecular scale. By following the motions of single molecular motors such as myosin or kinesin with high spatial and temporal resolution, the mechanics of their work cycle have been revealed (Svoboda et al., 1993; Yildiz et al., 2003). For many biological processes, including receptor trafficking, cytoskeletal dynamics, and cellular signaling, the collective behaviors of molecules may be important, and it would be interesting to extend these measurements to many molecules. Moreover, obtaining information on statistically rare events requires the acquisition of data on large numbers of single molecules, ideally under multiple conditions. However, traditional single-molecule measurements using fluorescence imaging require an extremely low labeling density. This is due to the diffraction of light when passing through a lens, which spreads the fluorescence of a single molecule into an Airy disk nearly 100 times the size of the molecule itself. Thus, in the case of closely packed membrane proteins, only one protein in thousands can be labeled to distinguish individual molecules. Since many small structures such as clathrin-coated pits and viruses typically contain only 1000 copies or fewer of a single protein, the extension of single-molecule imaging to their study was not evident until the recent advent of photoswitchable fluorescent probes (Ando et al., 2002; Patterson and Lippincott-Schwartz, 2002). In this era of large data sets, new advances in particle tracking combined with photoactivation of fluorescence have enabled the study of the dynamics of these previously inaccessible systems. Here, we describe the method of single-particle tracking combined with single-molecule localization using photoactivated localization microscopy (PALM) (Betzig et al., 2006), called single-particle tracking PALM (sptPALM) (Manley et al., 2008). This approach permits the study of protein dynamics in the context of living cells where proteins exist in dense populations.
2. Description of the sptPALM Method
The technological basis underlying sptPALM is the localization of the positions of specific proteins at near-molecular spatial resolution. This method further exploits the switchable properties of photoactivatable fluorescent proteins (PA-FPs; Ando et al., 2002; Patterson and Lippincott-Schwartz, 2002; Wiedenmann et al., 2004) used in PALM, although the same principle (termed STORM) has been applied to imaging with antibody-targeted photoswitchable synthetic labels (Rust et al., 2006). In PALM (Fig. 5.1), PA-FPs are continuously activated, imaged, and bleached in order to temporally separate molecules that would otherwise be spatially indistinguishable.
Figure 5.1. Schematic of PALM analysis.
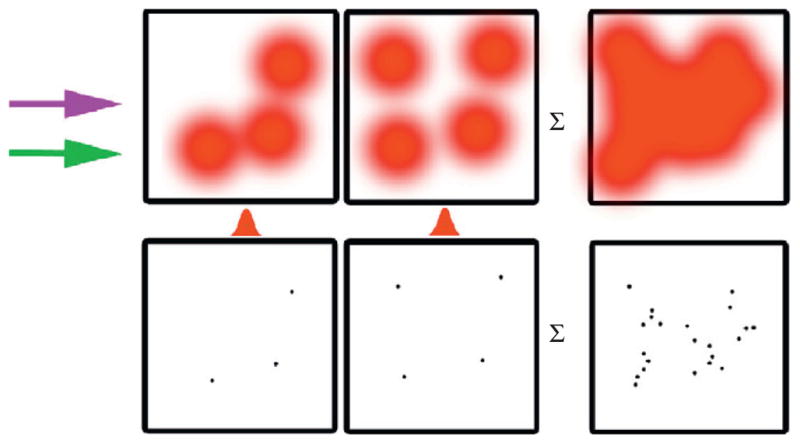
Raw data are acquired (top row) under continuous activation and excitation, then the image of each molecule is fit with a Gaussian to yield nanometric position information (bottom row). The summed information provides high-density, high-resolution molecular maps.
Because PA-FPs are activated stochastically, it is possible to ensure that highlighted molecules are sparse enough so that in each image only a single molecule (or less) is activated within a given diffraction-limited region. This is achieved by using a low intensity illumination at the activation wavelength (typically in the ultraviolet), while adjusting the excitation intensity to maintain a sparse pool of molecules by photobleaching. The position of each molecule is localized by fitting the measured photon distribution with the point spread function of the microscope (typically approximated with a Gaussian function). This has been demonstrated to provide superior accuracy in molecular position finding, relative to cross-correlation, sum-absolute difference, and centroid finding (Cheezum et al., 2001). The locations found during multiple imaging repetitions can then be summed to obtain a composite super-resolution PALM image that contains information about the location of many single molecules, even those sharing a single diffraction-limited region. In a typical PALM experiment, thousands of raw images are captured over a period of a few minutes, from which hundreds of thousands of molecules are localized. When performed in fixed cells, this approach provides a high-density map of a protein’s distribution at any particular time within the cell.
Proteins in living cells usually do not reside in a single place for very long, rather they are highly dynamic. Whether organized into multicomponent cytoskeletal structures, signaling ensembles or freely diffusing, proteins within cells are on the move. Understanding this motion is critical for providing a mechanistic basis for how proteins facilitate cell function. An approach for mapping the trajectories of individual molecules in living cells at very high densities is sptPALM (Manley et al., 2008). It combines the classic method of single-particle tracking with PALM. In sptPALM, fluorophores are stochastically activated (Fig. 5.2A) and imaged at video rate or slower, depending on the dynamics of the system. For slower processes, such as actin retrograde flow, slower imaging rates (commensurate with lower excitation power or incorporating a pause between images) are desirable to maintain fluorescence over significant motions. Molecular trajectories can be reconstructed by connecting the positions of photoactivated fluorophores in consecutive images (Fig. 5.2B). Thus, the information obtained can be used to create spatially resolved maps of mobility (Manley et al., 2008; Niu and Yu, 2008). In addition, maps of interacting proteins can be made by labeling different proteins with spectrally distinct fluorophores (Subach et al., 2009), although this has yet to be extended to sptPALM.
Figure 5.2. Schematic of sptPALM acquisition (A) and analysis (B).

Raw data are acquired (A) under continuous activation and excitation, then the image of each molecule is fit with a Gaussian to yield nanometric position information (B). Molecular positions in consecutive frames are then associated based on their proximity.
3. Labeling with Photoactivatable Fluorescent Probes
There are two primary means of labeling proteins for measurements with sptPALM, these are genetic encoding and expression of a PA-FP chimera, or targeting with an antibody or small molecule coupled to photocaged or photoswitchable dyes. In both cases, precautions must be taken to ensure minimal perturbations due to the labels. This generally means selecting cells for imaging with low expression levels, and demonstrating preservation of protein function in the chimeric form.
3.1. Photoactivatable fluorescent proteins
The discovery and development of fluorescent proteins, which merited the 2008 Nobel Prize in chemistry, has proved to be a powerful tool for investigating the organization and dynamics of proteins in cells, tissue, and animals. The noninvasive nature of fluorescence imaging makes it ideal for studying living samples, and cells can readily be made to express DNA encoding proteins of interest labeled with PA-FP. Unlike affinity labeling with antibodies or small molecules, there is no background from nonspecific labeling. This can be of paramount importance when studying single molecules, whose fluorescence can easily be overcome by background fluorescence. Moreover, numerous PA-FPs have been developed in recent years (Shaner et al., 2007) beginning with PAGFP (Patterson and Lippincott-Schwartz, 2002), offering many possibilities for sptPALM probes. Thus far, EosFP (Manley et al., 2008), PAmCherry (Subach et al., 2009), and Dendra2 (Gurskaya et al., 2006) have been successfully used to map the dynamics of single molecules with sptPALM. Furthermore, single-molecule performance has now been used as a criterion for the selection of mutants to improve photon yields and signal-to-noise ratios (Subach et al., 2009), directly targeting the weaknesses of fluorescent proteins for sptPALM measurements.
3.2. Photocaged dyes
With any single-molecule measurement, the number of photons translates directly into how well a molecule can be localized (Thompson et al., 2002). Low photon yields can be an issue with molecular localization, and this problem is most severe for higher dimensional imaging, as in (x,y,z) three-dimensional imaging or (two-dimensional + time) live cell imaging (Shroff et al., 2008). Chemical dyes generally have increased photon yields compared to fluorescent proteins, but they have their own drawbacks. They cannot be genetically encoded and so must be targeted to the relevant protein, typically using antibodies. Antibodies have limited targeting efficiency and their large size adds uncertainty to the position of the target molecule. Also, because they cannot penetrate the plasma membrane, membranes must be permeabilized before imaging molecules inside the cell, creating potential artifacts and making this approach unsuitable for live cell imaging of intracellular structures.
An alternative approach is to use small molecule labeling with chemical tags, such as the human DNA repair protein O6-alkylguanine-DNA (hAGT) and its derivatives (CLIP) (Gautier et al., 2008; Keppler et al., 2004) or dihydrofolate reductase (DHFR) (Miller et al., 2005) conjugated with caged fluorophores. For a more complete list of small molecule targeting methods, see Fernandez-Suarez and Ting (2008). These labels have several advantages over FPs or antibody targeting. They are targeted to genetically expressed protein fusions with high affinity. There is the potential to make them membrane permeable and therefore compatible with live cell imaging. Finally, they can achieve the high photon yields of chemical fluorescent tags such as rhodamine.
4. Tracking Single Molecules
As indicated in Fig. 5.2B, there are two analyses involved in recreating multiple single-molecule trajectories. In the first, single molecules are identified and located. In the second, locations are connected to form trajectories.
4.1. Molecule identification
The image of a single molecule can be described by an Airy diffraction pattern, whose rings contain information about its vertical position. For two-dimensional imaging, as is frequently the case in membrane-bound molecular imaging and is enforced by total internal reflection fluorescence (TIRF) imaging, the molecular image can be fit with a Gaussian. This fit captures the central peak of the Airy diffraction pattern (Cheezum et al., 2001), and the associated errors with determining the molecule’s centroid position have been well explored (Thompson et al., 2002).
Further criteria for peak intensities and shapes are set to discard peaks corresponding to multiple molecules, which are too bright, broad, or lacking radial symmetry. An alternative criterion to identify single molecules is single-step photobleaching, which could conceivably be implemented into an automated identification algorithm.
As molecules move, the photons they emit are spread by their motion. Thus, it is best to acquire frames with the shortest possible exposure time to minimize blurring. At the same time, exposure times and excitation intensities must be high enough to allow for nanometer-scale localization, requiring hundreds of photons in each image. In addition to minimizing the collection time for single frames, the frame rate for data acquisition must be selected carefully to capture the dynamics of interest. That is, individual molecules should move distances between consecutive frames that are larger than the uncertainties in their positions to avoid unnecessary oversampling. Another critical parameter is the degree of photoactivation at each time frame. If too many molecules are photoactivated simultaneously, as occurs with high (fast) photoactivation, their diffraction-limited images will overlap, making the localization of single molecules impossible.
4.2. Tracking algorithms
Tracking many single molecules requires identifying molecules, and determining their correspondence in consecutive frames. While this is straightforward for data with very sparse single molecules, and can be performed manually for few tracks, automation is necessary when tracking the hundreds or thousands of molecules that can be acquired in a single cell using sptPALM. For the purposes of sptPALM, data are typically acquired at high densities of molecules, such that a simple proximity-based algorithm that identifies each molecule with the nearest molecule in the subsequent frame is not sufficient. Numerous algorithms exist for particle tracking, with different probabilistic weightings or adjustable radial cutoffs designating molecular trajectories (Anderson et al., 1992; Crocker and Grier, 1996; Ghosh and Webb, 1994; Jaqaman et al., 2008). For a more complete review of the different multitarget tracking algorithms, see Kalaidzidis (2009). Automated tracking can be achieved by calculating the probability that identified molecules in one frame correspond to identified molecules in the next frame, on a frame-by-frame basis (Anderson et al., 1992; Crocker and Grier, 1996). Alternatively, a global probability can be calculated for all frames and optimized (Jaqaman et al., 2008). However, this method is computationally intensive and requires an underlying model to be introduced for the motions of molecules, to reduce the number of possible trajectories.
A useful criterion for diffusing molecules is that the probability for a molecule to diffuse a distance L or less in a time Δt is given by P(L, Δt) = 1 − exp(− L2/4DΔt), where D is the diffusion coefficient (Saxton, 1993). Thus, given the typical range of diffusion coefficients for membrane proteins of 0.1–0.5 μm2/s (Kenworthy et al., 2004), L can be chosen to yield an appropriate probability of detecting molecules.
5. Experimental Example: sptPALM on a Membrane Protein
In the example presented here, we begin by plating cells onto a glass coverslip. Coverglass thickness and index of refraction should be chosen for compatibility with your objective. For the purposes of studying the motion of membrane proteins, use of TIRF is implemented to reduce background light. Use of a camera with single-photon sensitivity is also necessary to collect single-molecule fluorescence emission.
Plate cells on a glass coverslip.
Transfect cells ~ 18 h before measuring. Grow to 80–90% confluency.
Fasten coverslip securely to microscope stage. Cells should be immersed in CO2 independent medium, unless a CO2 chamber is used for the microscope. We have built custom TIRF setups using both Olympus and Zeiss microscopes. In principle, any TIRF microscope that includes a 405 nm laser and the appropriate excitation wavelength for your chosen fluorophore should work.
Find a transfected cell, and center it in the field of view.
Take a DIC image prior to and after excitation to serve as a reference point for cell viability.
Begin streaming live cell images while illuminating with excitation wavelength.
Once molecules are sparse (initial molecules have bleached), gradually increase activation power to maintain a constant density of single molecules. This typically occurs within the first 100 images.
Localize molecules using fitting algorithm. Molecules should be subjected to strict criteria regarding their localization and the goodness of fit to a radially symmetric Gaussian. Several groups are now offering free software to do this (Hedde et al., 2009; Henriques et al., 2010).
Perform automated tracking. Several groups offer free software to do this, including Crocker, Weeks, and Grier (http://www.physics.emory.edu/~weeks/idl/index.html) and Jaqaman and Danuser (http://lccb.hms.harvard.edu/software.html).
At this stage of the analysis, many statistical measures may be applied to determine the kinds of dynamics present, whether they are diffusive, subdiffusive, or directed. Importantly, sufficiently long trajectories must be used to distinguish between modes of motion or to calculate diffusion coefficients. In the case of determining the diffusion coefficients of single vesicular stomatitis virus G (VSVG) proteins, we required trajectories to contain at least 15 positions to keep fractional errors in diffusion coefficients below 0.49 (Manley et al., 2008). The single-molecule trajectories and the associated diffusion coefficients are shown for a single cell in Fig. 5.3. The error associated with trajectories should be calculated to determine whether inferred differences are statistically significant (Qian et al., 1991). In addition, checks for a nonrandom distribution can be useful in determining whether molecules are clustered. Typical measures of clustering include the pair correlation function (Subach et al., 2009) or Ripley’s K-function (Appleyard et al., 1985), but caution must again be used to ensure that molecular blinking does not lead to artifactual identification of a single molecule as a cluster of distinct molecules. This is especially important if molecules are used that switch reversibly into dark states, such as Dronpa (Habuchi et al., 2005). An additional complication may arise in the case of TIRF microscopy, since undulations in the membrane may give the impression of fluctuations in molecular densities. This may be checked by using interference reflectance microscopy on the same cells, which will reveal where cell membranes are near or far from the coverslip (Verschuren, 1985). Likewise, in membrane sheet preparations, any patchiness in the lift-off procedure can give rise to an apparent clustering of remaining proteins.
Figure 5.3.
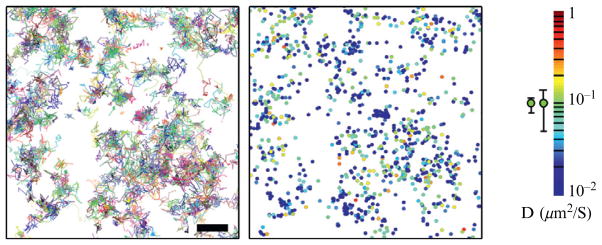
Example of sptPALM trajectories (left) and diffusion coefficient map (right) for the membrane protein VSVG. Color bar provides a scale for diffusion coefficients. Scale bar is 1 μm.
6. Conclusions
The method of sptPALM represents a step toward a statistical description of single proteins in cells, where information on thousands of molecules provides a more complete picture of the wide variety of molecular motions taking place in living cells. Thus far, the method of sptPALM has been used to study the diffusion of membrane proteins (VSVG), membrane-binding proteins (Gag), and cytoskeletal proteins (actin and FtsZ). These studies have revealed new information about these systems. In the case of VSVG and Gag, these proteins were found to have strikingly different diffusional behaviors on the plasma membrane: VSVG diffuses freely, while many Gag molecules are immobilized in domains the size of a budding viral particle (~100–200 nm diameter) (Manley et al., 2008). In the case of the actin-related protein, FtsZ, sptPALM revealed that the remodeling of FtsZ molecules in Z-rings within bacteria occurs by local binding and dissociation rather than by active transport or treadmilling behavior (Niu and Yu, 2008). These findings provide a glimpse of the types of questions and diverse biological systems that may be investigated using sptPALM.
In the near future, it will be possible to combine sptPALM with additional imaging modalities, which will greatly enhance the kinds of biological information obtained by these kinds of experiments. This includes multicolor (Subach et al., 2009) and three-dimensional (Shtengel et al., 2009; Vaziri et al., 2008) imaging capabilities, as well as functional imaging.
References
- Anderson CM, Georgiou GN, Morrisoni IEG, Stevenson GVW, Cherry RJ. Tracking of cell surface receptors by fluorescence digital imaging microscopy using a charge-coupled device camera. Low-density lipoprotein and influenza virus receptor mobility at 4 degrees C. J Cell Sci. 1992;101:415–425. doi: 10.1242/jcs.101.2.415. [DOI] [PubMed] [Google Scholar]
- Ando R, Hama H, Yamamoto-Hino M, Mizuno H, Miyawaki A. An optical marker based on the UV-induced green-to-red photoconversion of a fluorescent protein. Proc Natl Acad Sci USA. 2002;99:12651–12657. doi: 10.1073/pnas.202320599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appleyard ST, Witkowski JA, Ripley BD. A novel procedure for pattern analysis of features present on freeze-fractured plasma membranes. J Cell Sci. 1985;74:105–117. doi: 10.1242/jcs.74.1.105. [DOI] [PubMed] [Google Scholar]
- Betzig E, Patterson GH, Sougrat R, Lindwasser OW, Olenych S, Bonifacino JS, Davidson MW, Lippincott-Schwartz J, Hess HF. Imaging intracellular fluorescent proteins at nanometer resolution. Science. 2006;313:1642–1645. doi: 10.1126/science.1127344. [DOI] [PubMed] [Google Scholar]
- Cheezum MK, Walker WF, Guilford WH. Quantitative comparison of algorithms for tracking single fluorescent particles. Biophys J. 2001;81:2378–2388. doi: 10.1016/S0006-3495(01)75884-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crocker JC, Grier DG. Methods of digital video microscopy for colloidal studies. J Colloid Interface Sci. 1996;179:298–310. [Google Scholar]
- Fernandez-Suarez M, Ting AY. Fluorescent probes for super-resolution imaging in living cells. Nat Rev Mol Cell Biol. 2008;9:929–943. doi: 10.1038/nrm2531. [DOI] [PubMed] [Google Scholar]
- Gautier A, Juillerat A, Heinis C, Correa IR, Jr, Kindermann M, Beaufils F, Johnsson K. An engineered protein tag for multiprotein labeling in living cells. Chem Biol. 2008;15:128–136. doi: 10.1016/j.chembiol.2008.01.007. [DOI] [PubMed] [Google Scholar]
- Ghosh RN, Webb WW. Automated detection and tracking of individual and clustered cell surface low density lipoprotein receptor molecules. Biophys J. 1994;66:1301–1318. doi: 10.1016/S0006-3495(94)80939-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurskaya NG, Verkhusha VV, Shcheglov AS, Staroverov DB, Chepurnykh TV, Fradkov AF, Lukyanov S, Lukyanov KA. Engineering of a monomeric green-to-red photoactivatable fluorescent protein induced by blue light. Nat Biotechnol. 2006;24:461–465. doi: 10.1038/nbt1191. [DOI] [PubMed] [Google Scholar]
- Habuchi S, Ando R, Dedecker P, Verheijen W, Mizuno H, Miyawaki A, Hofkens J. Reversible single-molecule photoswitching in the GFP-like fluorescent protein Dronpa. Proc Natl Acad Sci USA. 2005;102:9511–9516. doi: 10.1073/pnas.0500489102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedde PN, Fuchs J, Ozwald F, Wiedenmann J, Nienhaus GU. Online image analysis software for photoactivation localization microscopy. Nat Methods. 2009;6:689–690. doi: 10.1038/nmeth1009-689. [DOI] [PubMed] [Google Scholar]
- Henriques R, Lelek M, Fornasiero EF, Valtorta F, Zimmer C, Mhlanga MM. QuickPALM: 3D real-time photoactivation nanoscopy image processing in ImageJ. Nat Methods. 2010;7:339–340. doi: 10.1038/nmeth0510-339. [DOI] [PubMed] [Google Scholar]
- Jaqaman K, Loerke D, Mettlen M, Kuwata H, Grinstein S, Schmid SL, Danuser G. Robust single-particle tracking in live-cell time-lapse sequences. Nat Methods. 2008;5:695–702. doi: 10.1038/nmeth.1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalaidzidis Y. Multiple objects tracking in fluorescence microscopy. J Math Biol. 2009;58:57–80. doi: 10.1007/s00285-008-0180-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenworthy AK, Nichols BJ, Remmert CL, Hendrix GM, Kumar M, Zimmerberg J, Lippincott-Schwartz J. Dynamics of putative raft-associated proteins at the cell surface. J Cell Biol. 2004;165:735–746. doi: 10.1083/jcb.200312170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keppler A, Kindermann M, Gendreizig S, Pick H, Vogel H, Johnsson K. Labeling of fusion proteins of O6-alkylguanine-DNA alkyltransferase with small molecules in vivo and in vitro. Methods. 2004;32:437–444. doi: 10.1016/j.ymeth.2003.10.007. [DOI] [PubMed] [Google Scholar]
- Manley S, Gillette JM, Patterson GH, Shroff H, Hess HF, Betzig E, Lippincott-Schwartz J. High-density mapping of single-molecule trajectories with photoactivated localization microscopy. Nat Methods. 2008;5:155–157. doi: 10.1038/nmeth.1176. [DOI] [PubMed] [Google Scholar]
- Miller LW, Cai Y, Sheetz MP, Cornish VW. In vivo protein labeling with trimethoprim conjugates: A flexible chemical tag. Nat Methods. 2005;2:255–257. doi: 10.1038/nmeth749. [DOI] [PubMed] [Google Scholar]
- Niu L, Yu J. Investigating intracellular dynamics of FtsZ cytoskeleton with photoactivation single-molecule tracking. Biophys J. 2008;95:2009–2016. doi: 10.1529/biophysj.108.128751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson GH, Lippincott-Schwartz J. A photoactivatable GFP for selective photolabeling of proteins and cells. Science. 2002;297:1873–1877. doi: 10.1126/science.1074952. [DOI] [PubMed] [Google Scholar]
- Qian H, Sheetz MP, Elson EL. Single particle tracking: Analysis of diffusion and flow in two-dimensional systems. Biophys J. 1991;60:910–921. doi: 10.1016/S0006-3495(91)82125-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rust MJ, Bates M, Zhuang X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM) Nat Methods. 2006;3:793–795. doi: 10.1038/nmeth929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxton MJ. Lateral diffusion in an archipelago: Single-particle diffusion. Biophys J. 1993;64:1766–1780. doi: 10.1016/S0006-3495(93)81548-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaner NC, Patterson GH, Davidson MW. Advances in fluorescent protein technology. J Cell Sci. 2007;120:4247–4260. doi: 10.1242/jcs.005801. [DOI] [PubMed] [Google Scholar]
- Shroff H, Galbraith CG, Galbraith JA, Betzig E. Live-cell photoactivated localization microscopy of nanoscale adhesion dynamics. Nat Methods. 2008;5:417–423. doi: 10.1038/nmeth.1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shtengel G, Galbraith JA, Galbraith CG, Lippincott-Schwartz J, Gillette JM, Manley S, Sougrat R, Waterman CM, Kanchanawong P, Davidson M, Fetter R, Hess HF. Interferometric flourescent super-resolution microscopy resolves 3D cellular nano-architecture. Proc Natl Acad Sci USA. 2008;106(9):3125–3130. doi: 10.1073/pnas.0813131106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subach FV, Patterson GH, Manley S, Gillette JM, Lippincott-Schwartz J, Verkhusha VV. Photoactivatable mCherry for high-resolution two-color fluorescence microscopy. Nat Methods. 2009;6:153–159. doi: 10.1038/nmeth.1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svoboda K, Schmidt CF, Schnapp BJ, Block SM. Direct observation of kinesin stepping by optical trapping interferometry. Nature. 1993;365:721–727. doi: 10.1038/365721a0. [DOI] [PubMed] [Google Scholar]
- Thompson RE, Larson DR, Webb WW. Precise nanometer localization analysis for individual fluorescent probes. Biophys J. 2002;82:2775–2783. doi: 10.1016/S0006-3495(02)75618-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaziri A, Tang J, Shroff H, Shank CV. Multilayer three-dimensional super resolution imaging of thick biological samples. Proc Natl Acad Sci USA. 2008;105:20221–20226. doi: 10.1073/pnas.0810636105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verschuren H. Interference reflection microscopy in cell biology: Methodology and applications. J Cell Sci. 1985;75:279–301. doi: 10.1242/jcs.75.1.279. [DOI] [PubMed] [Google Scholar]
- Wiedenmann J, Ivanchenko S, Oswald F, Schmitt F, Rocker C, Salih A, Spindler KD, Nienhaus GU. EosFP, a fluorescent marker protein with UV-inducible green-to-red fluorescence conversion. Proc Natl Acad Sci USA. 2004;101:15905–15910. doi: 10.1073/pnas.0403668101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yildiz A, Forkey JN, McKinney SA, Ha T, Goldman YE, Selvin PR. Myosin V walks hand-over-hand: Single fluorophore imaging with 1.5-nm localization. Science. 2003;300:2061–2065. doi: 10.1126/science.1084398. [DOI] [PubMed] [Google Scholar]