

Abstract
Aminoacyl-tRNA synthetases (AARSs) are ligases (EC.6.1.1.-) that catalyze the acylation of amino acids to their cognate tRNAs in the process of translating genetic information from mRNA to protein. Their amino acid and tRNA specificity are crucial for correctly translating the genetic code. Glycine is the smallest amino acid and the glycyl-tRNA synthetase (GlyRS) belongs to Class II AARSs. The enzyme is unusual because it can assume different quaternary structures. In eukaryotes, archaebac-teria and some bacteria, it forms an α2 homodimer. In some bacteria, GlyRS is an α2β2 heterotetramer and shows a distant similarity to α2 GlyRSs. The human pathogen eubacterium Campylobacter jejuni GlyRS (CjGlyRS) is an α2β2 heterotetramer and is similar to Escherichia coli GlyRS; both are members of Class IIc AARSs. The two-step aminoacylation reaction of tetrameric GlyRSs requires the involvement of both α- and β-subunits. At present, the structure of the GlyRS α2β2 class and the details of the enzymatic mechanism of this enzyme remain unknown. Here we report the crystal structures of the catalytic α-subunit of CjGlyRS and its complexes with ATP, and ATP and glycine. These structures provide detailed information on substrate binding and show evidence for a proposed mechanism for amino acid activation and the formation of the glycyl-adenylate intermediate for Class II AARSs.
Keywords: Gly-tRNA synthetase, Catalytic subunit, ATP binding, Glycine binding
Introduction
The function of aminoacyl-tRNA synthetases (AARSs) is to pair cognate tRNAs with their specific amino acids and therefore translate the nucleic acid code into a protein sequence. This is crucial for the protein biosynthesis process and is accomplished in a two-step reaction. AARS first binds ATP and a specific amino acid to form an aminoacyl-adenylate intermediate with the release of pyrophosphate. In the second step, the AARS/aminoacyl-adenylate complex binds a cognate tRNA and the amino acid is transferred to the 3′ ribose of the cognate tRNA to form aminoacyl-tRNA. The amino acid specificity is accomplished by preferential binding of the specific substrate and selective removal of similar amino acids, often using dedicated domains. The tRNAs are similarly L-shaped and AARS specific recognition is achieved by binding unique regions of tRNA by dedicated domains. Subsequently the charged tRNA is transferred to a ribosome where the amino acid is added onto a growing peptide chain, therefore completing the translation of genetic information into a protein [1–4].
AARSs are grouped into two distinct classes, Class I and II, based on their primary sequences and structures [5, 6]. Class II, characterized by three sequence motifs, is further divided into three subtypes, IIa, IIb and IIc, according to their differences in quaternary structures [7]. Glycyl-tRNA synthetase (GlyRS), with two oligomeric types (α2 homodimer and α2β2 heterotetramer) found in different organisms, is regarded as an unusual synthetase and grouped into Class IIc [8–12]. The α2β2 heterotetrameric GlyRS is seemingly found in eubacteria only, while the α2 homodimeric form is more widely distributed and found in eukaryotes (including humans), archaebacteria and some eubacteria [13]. The only other α2β2 heterotetrameric AARS in this class is PheRS. These two enzymes show similar domain organization and tRNA recognition.
Campylobacter jejuni is a Gram-negative food-born pathogen and one of the leading causes of human diarrhea and enteritis. In recent years, significant progress has been made in understanding C. jejuni infections and virulence, and the complete sequence of C. jejuni NCTC11168 is now available [14]. The potential drug targets, including key enzymes in the protein biosynthesis of the pathogenic agent, remain to be fully investigated. AARSs are key enzymes in protein synthesis and are essential and therefore excellent targets for drug development. In the C. jejuni species, only LysRS and IleRS have been cloned and partially characterized [15, 16]. Both enzymes belong to Class IIb AARSs and show high sequence identities to their corresponding enzymes in E. coli. Similarly, GlyRS from C. jejuni (CjGlyRS) is expected to be an α2β2 heterotetr-amer (Class IIc) like its E. coli homologue (EcGlyRS). The CjGlyRS α- and β-subunits are 40 and 50 % identical to that of EcGlyRS, respectively.
Both α- and β-subunits are required for the tRNA am-inoacylation activity of EcGlyRS [17, 18]. By comparison with α2β2 heterotetrameric PheRS the catalytic α-subunit is believed to be responsible for binding ATP and glycine while the β-subunit recognizes and binds tRNA. The interaction of the two subunits as well as the detailed mechanism of acylation is unknown. The crystal structures of the α2 dimeric GlyRSs from humans (HsGlyRS) and Thermus thermophilus (TtGlyRS) have been reported [19–22]. Mutations in HsGlyRS are associated with disease due to its functional expansion [21, 22]. The crystal structure of the apo-form α-subunit of α2β2 tetrameric GlyRS from Thermotoga maritima (TmGlyRS) has been deposited to the Protein Data Bank (PDB: 1J5W) without publication.
The structural information of the β-subunit of α2β2 GlyRSs remains unknown. Here we report the crystal structures of the α-subunit of CjGlyRS (α-CjGlyRS) in its apo-form and its complexes with ATP, and ATP and glycine. These structures provide detailed information on the α-subunit recognition of glycine and ATP, which is consistent with the currently accepted mechanism of aminoacylation by AARSs [23].
Experimental procedures
Protein cloning, expression, purification and characterization
The gene encoding the GlyRS α-subunit of C. jejuni subsp. jejuni NCTC 11168 was cloned into the pMCSG7 vector. The overexpression of α-CjGlyRS in E. coli BL21 (DE3) magic cells and its purification using Ni-affinity chromatography were the same as previously described [24]. However, cleavage of the His6-tag with recombinant TEV protease was unsuccessful [25]. Each recombinant α-CjGlyRS therefore included a vector-derived sequence, MHHHHHHSSGVDLGTENLYFQSNA, at its N-terminus. Size-exclusion chromatography was performed as described earlier to determine the oligomeric state of the protein in solution [25].
Protein crystallization
The α-CjGlyRS in crystallization buffer (200 mM NaCl, 20 mM HEPES pH 8.0, 2 mM DTT) was concentrated to 96 mg/mL using an Amicon Ultra centrifugal filter unit (Millipore). The protein was screened for crystallization conditions in the presence of 50 mM glycine with the help of a Mosquito liquid dispenser (TTP Labtech) using the sitting drop vapor diffusion technique in 96-well Crystal-Quick plates (Greiner). For each condition, 400 nL of protein (96 mg/mL) and 400 nL of crystallization formulation were mixed; the mixture was equilibrated against 135 μL of the reservoir solution. The MCSG- 1–4 (Microlytic, Inc.) crystallization screens were used and set up at 16 °C. Diffraction quality crystals of α-CjGlyRS–Gly appeared in the presence of 50 mM glycine under the condition of 0.1 M Bis–Tris propane:NaOH pH 7 and 0.7 M magnesium formate. Subsequently, the crystals of the α-CjGlyRS–ATP complex were grown in the presence of 20 mM ATP under the condition of 0.1 M Tris:HCl pH 8.5 and 0.3 M magnesium chloride, and the crystals of the α-CjGlyRS–ATP–Gly complex were grown in the presence of both ATP (20 mM) and glycine (50 mM) under the condition of 0.1 M Nα2HPO4:citrate acid pH 4.2, 0.2 M lithium sulfate and 20 % (w/v) PEG 1000. Prior to data collection, crystals were cryoprotected with glycerol (15–30 %) added to the crystallization buffer and were flash-cooled directly in liquid nitrogen.
X-ray diffraction and structure determination
Single-wavelength diffraction data were collected at 100 K from α-CjGlyRS–Gly, α-CjGlyRS–ATP and α-CjGlyRS–ATP–Gly crystals at the 19ID beamline of the Structural Biology Center at the Advanced Photon Source at Argonne National Laboratory using the program SBCcollect. The reflection intensities were integrated and scaled with the HKL3000 program suite [26] (Table 1). The structure of α-CjGlyRS co-crystallized with glycine was first solved using the molecular replacement method with the program MolRep [27]. The structural template used was the A chain from TmGlyRS (PDB: 1J5W, 60 % sequence identity). Glycine added to the crystallization buffer was not observed in electron density maps and therefore we will refer to this structure as apo α-CjGlyRS in the manuscript and discuss ATP-dependent glycine-binding in α-CjGlyRS later. The structures of α-CjGlyRS–ATP and α-CjGlyRS–ATP–Gly were subsequently solved by performing the molecular replacement method with the apo α-CjGlyRS structure as the search model. Model re-building for each structure was performed manually using the program COOT [28]. The final refinement for all three structures was performed using the program Phenix.Refine [29] (Table 1). In each of these structures, there was an electron density near the opening of the active site of one subunit. The site is typically occupied by small molecules present in buffers during protein purification and crystallization procedures. This density was about 7–8 Å away from the ATP and glycine binding sites, may not be functionally relevant and will not be discussed in detail in this manuscript. The three structures have been deposited to the Protein Data Bank with access codes of 3RF1, 3UFG and 3RGL, respectively.
Table 1.
Crystallographic Statistics
| Data collection | apo-CjGlyRS | CjGlyRS + ATP | CjGlyRS + ATP + Glycine |
|---|---|---|---|
| Space group | H3 | H3 | H3 |
| Unit Cell (Å, °) | a = b = 112.7, c = 158.3 | a = b = 114.6, c = 157.7 | a = b = 112.7, c = 157.8 |
| MW Da (residue) | 33,058 (287)a | 33,058 (287)a | 33,058 (287)a |
| Mol (AU) | 2 | 2 | 2 |
| Wavelength(Å) | 0.9793 | 0.9792 | 0.9793 |
| Resolution(Å) | 39.0–2.2 | 42.0–2.55 | 42.0–2.45 |
| Number of unique reflections | 37970b | 24077b | 27496b |
| Redundancy | 3.7 (3.3)c | 3.0 (2.9)d | 3.3 (3.2)e |
| Completeness (%) | 99.7 (99.8)c | 95.9 (98.3)d | 99.8 (99.9)e |
| Rmerge (%) | 6.7 (67.5)c | 9.1 (58.1)d | 9.1 (62.7)e |
| I/σ(I) | 27.8 (1.8)c | 23.3 (1.4)d | 15.4 (1.2)e |
| Molecular replacement | |||
| wRfac (%) | 39.1 | 36.6 | 34.5 |
| Scor | 0.71 | 0.69 | 0.72 |
| Refinement | |||
| Resolution | 39.0–2.2 | 42.0–2.55 | 42.0–2.45 |
| Reflections (work/test) | 34,849/1,731 | 23,924/1,203 | 25,633/1,283 |
| Rcrystal/Rfree (%) | 16.9/21.4 | 17.4/25.5 | 17.3/23.3 |
| Rms deviation from ideal geometry | |||
| Bond length (Å)/angle (°) | 0.008/1.030 | 0.007/1.069 | 0.008/1.155 |
| No. of atoms (Protein/HETATM) | 4,562/221 | 4,579/190 | 4,575/212 |
| Mean B-value (Å2) (mainchain/sidechain) | 63.7/70.1 | 93.6/101.0 | 54.9/61.0 |
| Ramachandran plot statistic (%) | |||
| Residues in most favored regions, | 91.7 | 89.7 | 90.7 |
| In additional allowed regions | 8.3 | 10.1 | 9.3 |
| In generously allowed regions, | 0 | 0.2 | 0 |
| In disallowed region | 0 | 0 | 0 |
| PDB entry | 3RF1 | 3UFG | 3RGL |
Not including cloning artifact
Including Bijvoet pairs
Last resolution bin, 2.20–2.24 Å
Last resolution bin, 2.55–2.59 Å
Last resolution bin, 2.45–2.50 Å
Results
Overall structure of apo α-CjGlyRS
There are two α-CjGlyRSs in one asymmetrical unit, Fig. 1a, forming a pseudo two-fold dimer-like quaternary structure. Subunit interactions are quite extensive with an interface area of about 2,550 Å2 [30] and involve mostly hydrogen bonds (33 pairs) and salt bridges (12 pairs). No significant hydrophobic core is present at the interface. Size-exclusion chromatography indicates the α-CjGlyRS exists as a monomer in solution (data not shown). This suggests that the dimer-like quaternary structure in the crystal is likely to form at high protein concentrations and likely has no physiological significance.
Fig. 1.
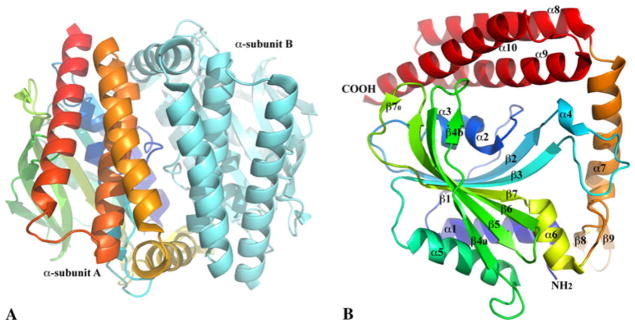
Ribbon diagrams of α-CjGlyRS structures. a α-CjGlyRS forms a pseudo twofold dimer-like structure in one asymmetric unit. To show the interface between two α-subunits and one of their main chain tracings, one subunit is colored in a rainbow spectrum from the N terminus (blue) to the C terminus (red) and the other in cyan. b Each α-CjGlyRS includes an N-terminal catalytic domain (from the α1 helix to α6 helix), a C-terminal three-helix bundle (α8–10 helices) and a linker between them (β8–β9 hairpin and α7 helix). One edge strand, β1, is short. The other edge strand, β4, is split into two segments (β4a and β4b) due to an interruption by a side chain to main chain hydrogen bond (OD1 of N114 on β4 to N of G126 on β5) in place of a regular main chain to main chain (O of D113 to N of G126) interaction. The middle strand is also interrupted, but by a bulge, dividing into a short strand (β70) antiparallel to the end of the β6 strand and a regular long strand (β7)
Each α-subunit is comprised of an N-terminal catalytic domain, a C-terminal three-helix bundle and a linker between them (Fig. 1b). Like the catalytic domain of other Class II AARSs, this domain includes a seven-strand β-sheet, in which only the short edge β1 strand is parallel to its neighboring β2 strand while all other strands are anti-parallel. The convex side of the twisted β-sheet is flanked by two anti-parallel helices, α1 and α5. The α1 helix covers most of the Class II AARS sequence motif-1 [31], Fig. 2a. On the concave side of the β-sheet, however, the linker between the β1- and β2-strands, including two short helices, α2 (310-helix) and α3, blocks the opening by t he edge β1 strand. Therefore it creates a large open pocket, forming the active site of the enzyme for substrate binding and aminoacylation reaction. The other two characteristic sequence motifs (motif-2 and -3) of Class II AARS [11, 12, 32], are located in this active site, Fig. 2a.
Fig. 2.

Conserved structures of active site and substrate binding. a A ribbon diagram of the catalytic domain with conserved residues in the active site drawn in stick format. The three Class II AARS sequence motifs [11, 12, 31, 32] are colored differently, motif-1 in light green, motif-2 in cyan and motif-3 in pink. b ATP in the active site of α-CjGlyRS–ATP. All hydrogen bonds are drawn in magenta dashed lines with some bond distances labeled. In the α-CjGlyRS–ATP–Gly structure, glycine is not observed in one of the subunits (c). In the second subunit (d), both ATP and glycine bind. The ATP, glycine and active site residues are all drawn in stick format with different color schemes
The three-helix bundle (α8, α9 and α10) is unique compared to other known α2 dimer GlyRS structures [19, 20]. In this α-subunit structure, the helix bundle seemingly supports the formation of the dimer-like quaternary structure. In a functional α2β2 tetramer, it is likely to contribute to intersubunit interactions. The linker between the substrate binding domain and the helix bundle includes a β-hairpin and a long helix. In the TtGlyRS or HsGlyRS α2 GlyRS structures [19, 20], there is also a hairpin and an α-helix connecting the catalytic domain and the C-terminal domain. But, they are in very different conformations and the hairpin is more prominent while the helix has only a two-turn 310-helix [19, 20]. The α6 helix after the last strand (β7) in a very similar conformation is also found in the α2 sub-class of dimeric GlyRS structures and at least some residues from the helix, part of motif-3 [32] (Fig. 2a), are important to ATP binding as discussed below; we define the α6 helix as the last secondary element of the catalytic domain.
ATP and glycine binding
The active site of the catalytic domain is composed of highly conserved residues involved in the binding of two substrates, ATP and glycine (Fig. 2a). The adenosine ring of ATP assumes an identical position in all structures (Fig. 2b–d). The conserved Y74 in α2β2 GlyRSs forms π-π stacking with the adenine ring and it corresponds to an invariant phenylalanine in α2 GlyRSs (Fig. 3). The presence of a tyrosine in α2β2 GlyRSs can add one more hydrogen bond between its phenyl hydroxyl group to the O5 oxygen atom of the ribose (Fig. 2b–d). The amino group of the adenine and two hydroxyl groups of the ribose add at least four mostly conserved hydrogen bonds to their neighboring protein atoms. There is also a potential cation-π interaction between the adenine ring and the guanidine group of either R163 or R70. The two guanidine groups alternatively cover the adenine ring in the structures, seemingly providing a possible gating mechanism for ATP access and binding and product release. There is no conformational change of the α-CjGlyRS structure upon ATP or ATP and glycine binding. The apo-structure can be superimposed with two holo-structures with root-mean-square deviation (RMSD) values less than 0.28 Å without gaps.
Fig. 3.
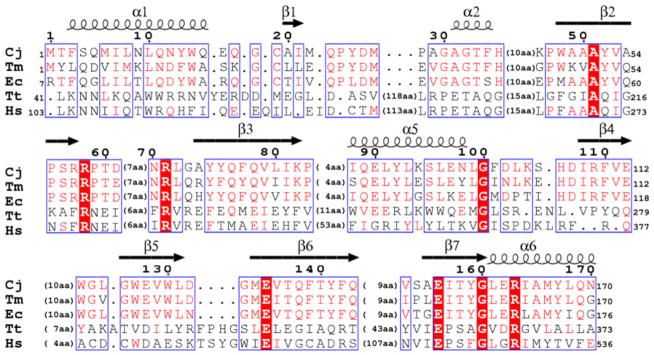
A structure-based sequence alignment of the catalytic domain of structurally known α2 and α2β2 GlyRSs. The catalytic domains of TmGlyRS (PDB: 1J5 W), TtGlyRS (PDB: 1ATI) and HsGlyRS (PDB: 2PME) were individually superimposed onto α-CjGlyRS with the program Secondary Structure Matching (SSM). The sequence of the most studied EcGlyRS is also included, which is sequentially aligned to α-CjGlyRS. Only the structurally aligned portions are displayed while the gaps are indicated with the number of residues in between brackets. The assignment of the secondary structures is based on the α-CjGlyRS structure with helices (coils) and strands (arrows) indicated above the appropriate sequences
In the α-CjGlyRS–ATP complex structure, the three phosphate groups of ATP are in an extended conformation with a γ-phosphate group reaching towards the glycine binding site (Fig. 2b, d). In the α-CjGlyRS–ATP–Gly structure, one subunit contains both ATP and glycine, the second subunit contains ATP only. The basis for this asymmetric binding is unknown even though the glycine concentration in the crystallization buffer was as high as 50 mM. In this structure the ATP is bended into a U-shape, a typical conformation observed in many Class II AARS structures. The U-shaped ATP is observed in both subunits of α-CjGlyRS in the presence (Fig. 2d) and absence of glycine (Fig. 2c), but with different conformations. The interactions between the phosphate groups and the highly conserved residues of R58 and E135 as well as residue Q138 (conserved in α2β2 GlyRSs, Fig. 3) is quite extensive (Fig. 2b–d). There is no electron density present near the phosphate groups of ATP, which could indicate metal binding (Mg2+ ion is observed in α2 TtGlyRS complex structures [20]).
The glycine substrate in the active site is hydrogen bonded to one oxygen atom of the α-phosphate group of ATP through its carboxyl group (Fig. 2d). The hydrogen bond distance is quite short (2.64 Å), indicating its potential involvement in the adenylate formation, the first reaction catalyzed by AARSs. The interaction of the glycine with its surrounding protein atoms is extensive and explains enzyme specificity. Its amino and carboxylate groups form a pair of hydrogen bonds to residue Q76; and its amino group is also hydrogen bonded to residue Q78. Both Q76 and Q78 are highly conserved residues in α2β2 GlyRSs (Fig. 3). Additionally, glycine is the smallest amino acid and has two α-hydrogens at the Ca carbon atom; the α-hydrogen is usually more acidic. The two α-hydrogens of glycine can potentially interact with two neighboring threonine residues, T33 and T158, which are also conserved in α2β2 GlyRSs. This interaction would not be possible with alanine, serine, cysteine or any other larger amino acid.
Conclusions
Both subunits α and β are necessary in the aminoacylation of tRNA by α2β2 GlyRSs [17, 18]. Only two AARSs, GlyRS and PheRS, have such a tetrameric organization. We have determined the structures of the CjGlyRS α-subunit and its complexes with ATP, and ATP and glycine. Each α-subunit is comprised of a catalytic domain that contains a typical Class II AARS active site, and a three-helix bundle domain that may contribute to the formation of an α2β2 heterotetramer. In the crystal, the α-CjGlyRS forms a homodimer with some contribution to the dimer interface from the three-helix bundle. However, α-CjGlyRS is a monomer in solution suggesting that a stable α2β2 CjGlyRS tetramer requires more extensive α- and β-subunit interactions perhaps similar to those observed in α2β2 PheRS structures [33, 34]. The requirement seems to be in agreement with the data that both α- and β-subunits are required for full enzymatic activities.
Glycine is the smallest amino acid and has no side chains. It seems that CjGlyRS shows a priority for ATP binding and ATP can assume two conformations: extended and U-shaped. The U-shaped conformation seems to represent a state that creates a space to bind glycine more efficiently. More importantly, the α- and γ-phosphate groups interact with glycine directly, with the α-phosphate group in position to form adenylate. However, in our structure the glycyl-adenylate is not present suggesting that other cofactors are also involved. The conformation of ATP and glycine in the active site before aminoacylation is in good agreement with the mechanism [20] proposed for TtGlyRS (an α2 GlyRS) and originated from the studies of other Class II AARSs. The only missing element of the mechanism to be employed by α-CjGlyRS is that the Mg2+ ion neighboring the phosphate groups of ATP is absent in these structures despite the fact that magnesium is present in the crystallization buffer. At present, it is unknown what the exact role the β-subunit plays in the enzyme’s activities (by analogy to PheRS, it may recognize and bind tRNA). However, for the α-subunit fully loaded with ATP and glycine, the β-subunit may be needed to attract a Mg2+ ion and/or provide essential residues to initiate the formation of the glycyl-adenylate as well as to contribute to the recognition of specific tRNA.
Acknowledgments
This project was funded by the National Institute of Health under Contract HHSN272200700058C and by the U. S. Department of Energy, Office of Biological and Environmental Research, under contract DE-AC02-06CH11357. The authors wish to thank Gekleng Chhor for proofreading of this manuscript.
Footnotes
The submitted manuscript has been created by UChicago Argonne, LLC, Operator of Argonne National Laboratory (“Argonne”). Argonne, a U.S. Department of Energy Office of Science laboratory, is operated under Contract No. DE-AC02-CH11357. The U.S. Government retains for itself, and others acting on its behalf, a paid-up nonexclusive, irrevocable worldwide license in said article to reproduce, prepare derivative works, distribute copies to the public, and perform publicly and display publicly, by or on behalf of the Government.
Contributor Information
Kemin Tan, Center for Structural Genomics of Infectious Diseases, University of Chicago, Chicago, IL 60637, USA. Computation Institute, University of Chicago, Chicago, IL 60637, USA. Structural Biology Center, Biosciences, Argonne National Laboratory, Argonne, IL 60439, USA.
Min Zhou, Center for Structural Genomics of Infectious Diseases, University of Chicago, Chicago, IL 60637, USA. Computation Institute, University of Chicago, Chicago, IL 60637, USA.
Rongguang Zhang, Center for Structural Genomics of Infectious Diseases, University of Chicago, Chicago, IL 60637, USA. Computation Institute, University of Chicago, Chicago, IL 60637, USA. Structural Biology Center, Biosciences, Argonne National Laboratory, Argonne, IL 60439, USA.
Wayne F. Anderson, Center for Structural Genomics of Infectious Diseases, University of Chicago, Chicago, IL 60637, USA. Molecular Pharmacology and Biological Chemistry, Northwestern University, 303 E. Chicago, Chicago, IL 60611-3008, USA
Andrzej Joachimiak, Center for Structural Genomics of Infectious Diseases, University of Chicago, Chicago, IL 60637, USA. Computation Institute, University of Chicago, Chicago, IL 60637, USA. Structural Biology Center, Biosciences, Argonne National Laboratory, Argonne, IL 60439, USA.
References
- 1.Carter CW., Jr Annu Rev Biochem. 1993;62:715–748. doi: 10.1146/annurev.bi.62.070193.003435. [DOI] [PubMed] [Google Scholar]
- 2.Szymanski M, Deniziak MA, Barciszewski J. Nucleic Acids Res. 2001;29:288–290. doi: 10.1093/nar/29.1.288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hausmann CD, Ibba M. FEMS Microbiol Rev. 2008;32:705–721. doi: 10.1111/j.1574-6976.2008.00119.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Park SG, Schimmel P, Kim S. Proc Natl Acad Sci USA. 2008;105:11043–11049. doi: 10.1073/pnas.0802862105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nagel GM, Doolittle RF. Proc Natl Acad Sci USA. 1991;88:8121–8125. doi: 10.1073/pnas.88.18.8121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eriani G, Delarue M, Poch O, Gangloff J, Moras D. Nature. 1990;347:203–206. doi: 10.1038/347203a0. [DOI] [PubMed] [Google Scholar]
- 7.Moras D. Trends Biochem Sci. 1992;17:159–164. doi: 10.1016/0968-0004(92)90326-5. [DOI] [PubMed] [Google Scholar]
- 8.Ostrem DL, Berg P. Biochemistry. 1974;13:1338–1348. doi: 10.1021/bi00704a006. [DOI] [PubMed] [Google Scholar]
- 9.Surguchov AP, Surguchova IG. Eur J Biochem. 1975;54:175–184. doi: 10.1111/j.1432-1033.1975.tb04127.x. [DOI] [PubMed] [Google Scholar]
- 10.Kern D, Giege R, Ebel JP. Biochemistry. 1981;20:122–131. doi: 10.1021/bi00504a021. [DOI] [PubMed] [Google Scholar]
- 11.Nada S, Chang PK, Dignam JD. J Biol Chem. 1993;268:7660–7667. [PubMed] [Google Scholar]
- 12.Ge Q, Trieu EP, Targoff IN. J Biol Chem. 1994;269:28790–28797. [PubMed] [Google Scholar]
- 13.Tang SN, Huang JF. FEBS Lett. 2005;579:1441–1445. doi: 10.1016/j.febslet.2005.01.045. [DOI] [PubMed] [Google Scholar]
- 14.Parkhill J, Wren BW, Mungall K, Ketley JM, Churcher C, Basham D, Chillingworth T, Davies RM, Feltwell T, Holroyd S, Jagels K, Karlyshev AV, Moule S, Pallen MJ, Penn CW, Quail MA, Rajandream MA, Rutherford KM, van Vliet AH, Whitehead S, Barrell BG. Nature. 2000;403:665–668. doi: 10.1038/35001088. [DOI] [PubMed] [Google Scholar]
- 15.Chan VL, Bingham HL. J Bacteriol. 1992;174:695–701. doi: 10.1128/jb.174.3.695-701.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hong Y, Wong T, Bourke B, Chan VL. Microbiology. 1995;141(Pt 10):2561–2567. doi: 10.1099/13500872-141-10-2561. [DOI] [PubMed] [Google Scholar]
- 17.Toth MJ, Schimmel P. J Biol Chem. 1990;265:1000–1004. [PubMed] [Google Scholar]
- 18.Toth MJ, Schimmel P. J Biol Chem. 1990;265:1005–1009. [PubMed] [Google Scholar]
- 19.Logan DT, Mazauric MH, Kern D, Moras D. EMBO J. 1995;14:4156–4167. doi: 10.1002/j.1460-2075.1995.tb00089.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arnez JG, Dock-Bregeon AC, Moras D. J Mol Biol. 1999;286:1449–1459. doi: 10.1006/jmbi.1999.2562. [DOI] [PubMed] [Google Scholar]
- 21.Cader MZ, Ren J, James PA, Bird LE, Talbot K, Stammers DK. FEBS Lett. 2007;581:2959–2964. doi: 10.1016/j.febslet.2007.05.046. [DOI] [PubMed] [Google Scholar]
- 22.Xie W, Nangle LA, Zhang W, Schimmel P, Yang XL. Proc Natl Acad Sci USA. 2007;104:9976–9981. doi: 10.1073/pnas.0703908104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guo RT, Chong YE, Guo M, Yang XL. J Biol Chem. 2009;284:28968–28976. doi: 10.1074/jbc.M109.030692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim Y, Dementieva I, Zhou M, Wu R, Lezondra L, Quartey P, Joachimiak G, Korolev O, Li H, Joachimiak A. J Struct Funct Genomics. 2004;5:111–118. doi: 10.1023/B:JSFG.0000029206.07778.fc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tan K, Li H, Zhang R, Gu M, Clancy ST, Joachimiak A. J Struct Biol. 2008;162:94–107. doi: 10.1016/j.jsb.2007.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Minor W, Cymborowski M, Otwinowski Z, Chruszcz M. Acta Crystallogr A. 2006;62:859–866. doi: 10.1107/S0907444906019949. [DOI] [PubMed] [Google Scholar]
- 27.Vagin AA, Teplyakov A. J Appl Crystallogr. 1997;30:1022–1025. [Google Scholar]
- 28.Emsley P, Cowtan K. Acta Crystallogr A. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 29.Murshudov GN, Vagin AA, Dodson EJ. Acta Crystallogr A. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 30.Krissinel E, Henrick K. J Mol Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 31.Cusack S. Biochimie. 1993;75:1077–1081. doi: 10.1016/0300-9084(93)90006-e. [DOI] [PubMed] [Google Scholar]
- 32.Shiba K, Schimmel P, Motegi H, Noda T. J Biol Chem. 1994;269:30049–30055. [PubMed] [Google Scholar]
- 33.Mosyak L, Reshetnikova L, Goldgur Y, Delarue M, Safro MG. Nat Struct Biol. 1995;2:537–547. doi: 10.1038/nsb0795-537. [DOI] [PubMed] [Google Scholar]
- 34.Reshetnikova L, Moor N, Lavrik O, Vassylyev DG. J Mol Biol. 1999;287:555–568. doi: 10.1006/jmbi.1999.2617. [DOI] [PubMed] [Google Scholar]