

Abstract
Benzobisthiazole derivatives were identified as novel helicase inhibitors through high throughput screening against purified S. aureus (Sa) and B. anthracis (Ba) replicative helicases. Chemical optimization has produced compound 59 with nanomolar potency against the DNA duplex strand unwinding activities of both B. anthracis and S. aureus helicases. Selectivity index (SI = CC50/IC50) values for 59 were greater than 500. Kinetic studies demonstrated that the benzobisthiazole-based bacterial helicase inhibitors act competitively with the DNA substrate. Therefore, benzobisthiazole helicase inhibitors represent a promising new scaffold for evaluation as antibacterial agents.
Keywords: Optimization, DNA helicase, DNA replication, Inhibitor
The increasing prevalence of antibiotic resistant strains of bacterial pathogens presents a major unmet medical need. For example, the incidence of skin and soft tissue infections with community acquired methicillin-resistant Staphylococcus aureus (MRSA) has increased over five-fold since 2003.1 In addition, a recent report revealed that 28% of enterococci cultured from 25 North American intensive care units (ICUs) were resistant to vancomycin (VRE).2 The development of new antibiotics against underexploited targets with novel mechanisms of action is a vital part of the solution to these problems because such antibiotics will not be affected by preexisting target based resistance mechanisms. The replicative DNA helicase is an essential component of the DNA replication pathway, acting early and catalyzing a rate limiting step in replication, but it is currently untargeted by antibacterial agents.
Replicative DNA helicase is a member of a drug-validated pathway and, along with gyrase, topoisomerase IV, and DNA polymerase III, is essential to bacteria.3–7 The primary structures of bacterial replicative helicases differ significantly from those of their eukaryotic and human counterparts,8,9 indicating that bacteria-specific inhibitors of helicase may be developed. These features make it particularly attractive as a target for the discovery of new antibacterial therapeutics.
The replicative DNA helicases from E. coli, S. aureus, and P. aeruginosa have been targeted previously in anti-infective screens,10–17 but few hits have been described, and none have progressed further in drug development due to poor potency and inadequate selectivity. Two distinct X-ray crystal structures have been reported: one shows a hexameric DnaB helicase in complex with a helicase binding fragment of primase,18 and another shows that the DnaB hexamer adopts a closed spiral staircase quaternary structure in complex with ATP mimic GDP-AlF4 and ssDNA.19 The two structures suggest that helicase may exist in both inactivated and activated forms during the bacterial DNA replication process. Structure-based approaches to target both the inactivated and activated forms of DnaB helicase may aid in the discovery of novel bacterial DNA helicase inhibitors.
We have previously discovered a coumarin-based DNA helicase inhibitor series through a high throughput screening campaign, and chemical optimization yielded compounds with antibacterial activities against several Gram-positive species including multiple clinically relevant ciprofloxacin-resistant MRSA strains.20,21 Herein we report chemical optimization and biological evaluation of a novel series of DNA bacterial helicase inhibitors based on a benzobisthiazole scaffold.
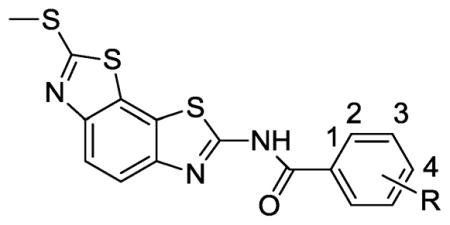
Benzobisthiazole derivatives were identified as novel inhibitors through high throughput screening against S. aureus (Sa) and B. anthracis (Ba) helicases. The screening hit compound 1 (Fig. 1) demonstrated antihelicase activities (IC50 (Ba) = 5 μM and IC50 (Sa) = 12 μM) and minimal cytotoxicity (CC50 > 100 μM). Initial investigation of structure-activity relationships (SARs) focused on the benzamide portion of the benzobisthiazole scaffold. Thirty-two analogs (compounds 4–35) with various substitutions on the phenyl ring of the benzamide portion and twelve analogs (compounds 36–47) with phenyl ring replacements were purchased from an outside vendor (Life Chemicals, Inc.). Their biological activities were evaluated in a fluorescence resonance transfer (FRET)-based helicase strand unwinding assay20 to measure concentration-dependent inhibition of Bacillus anthracis DNA replicative helicase, and the results are summarized in Tables 1 and 2.
Figure 1.
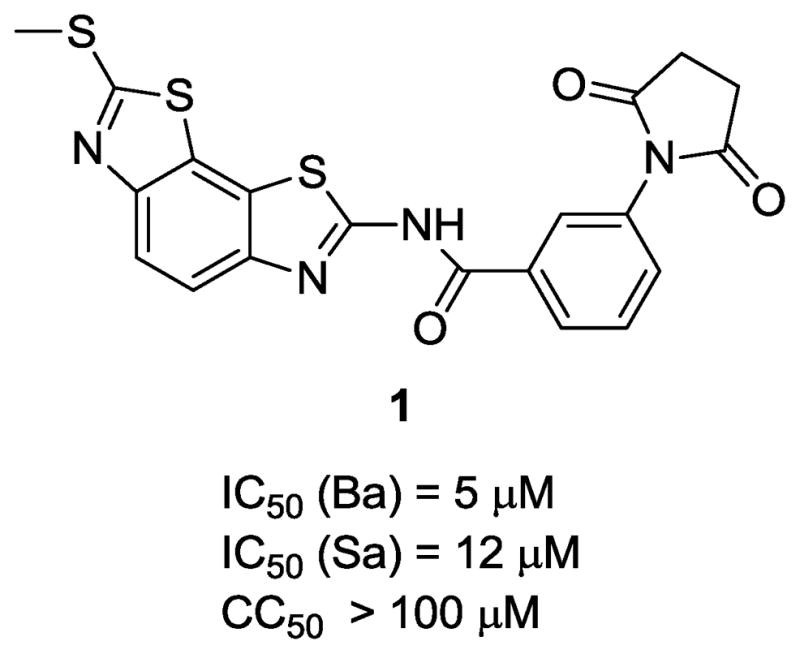
The structure of HTS hit 1.
Table 1.
Bacillus anthracis helicase inhibition by benzobisthiazole compounds 1–33.
| ||
|---|---|---|
| Compd. | R | IC50 Ba Helicase (μM) |
| 1 | 3-N[-(C=O)CH2CH2(C=O)-] | 5.0 |
| 2 | 4-N(CH2CH3)2 | >100 |
| 3 | 4-tBu | >100 |
| 4 | 4-OPh | >100 |
| 5 | 4-OiPr | >100 |
| 6 | 4-SO2N(CH2CH3)2 | >100 |
| 7 | 4-CH3 | 7.6 |
| 8 | 4-CN | 11.0 |
| 9 | 4-OCH2CH3 | 6.9 |
| 10 | 4-Cl | 5.0 |
| 11 | 4-COOCH3 | 3.7 |
| 12 | 4-N(CH3)2 | 2.4 |
| 13 | 4-OCH3 | 2.2 |
| 14 | 4-N[-(C=O)CH2CH2(C=O)-] | 1.3 |
| 15 | 3-N(CH3)2 | 36.0 |
| 16 | 3-OCH3 | 12.5 |
| 17 | 3-Cl | 5.3 |
| 18 | 3-F | 3.3 |
| 19 | 3-NO2 | 2.8 |
| 20 | 3-Br | 2.3 |
| 21 | 2-OPh | >100 |
| 22 | 2-Cl | >100 |
| 23 | 2-Br | >100 |
| 24 | 2-F | ~50 |
| 25 | 2-CH3 | 6.6 |
| 26 | 2,4-(OCH3)2 | >100 |
| 27 | 2,6-(OCH3)2 | >100 |
| 28 | 2,6-F2 | 64.5 |
| 29 | 3,4-(CH3)2 | 3.2 |
| 30 | 3,4-(OCH2CH2O) | 3.0 |
| 31 | 3,4-(OCH3)2 | 2.1 |
| 32 | 3,5-(OCH3)2 | 1.7 |
| 33 | 3,4,5-(OCH3)3 | 0.7 |
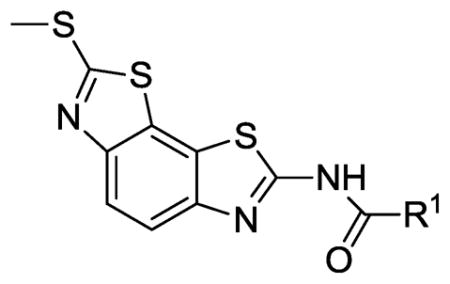
Table 2.
Bacillus anthracis and Staphyloccocus aureus helicase inhibition by benzobisthiazole compounds 34–45.
| ||
|---|---|---|
| Compd. | R1 | IC50 Ba Helicase (μM) |
| 34 | cyclopropyl | >100 |
| 35 | CH2Ph | 47 |
| 36 | CH2CH2Ph | >100 |
| 37 |
|
>100 |
| 38 |
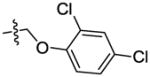
|
>100 |
| 39 |
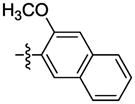
|
>50 |
| 40 |
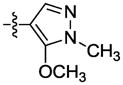
|
>100 |
| 41 | 2-furan | 102 |
| 42 | 2-thiophene | 90 |
| 43 |
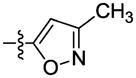
|
>50 |
| 44 |
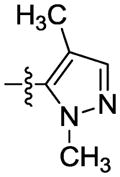
|
>50 |
| 45 | 2-pyrazine | 28 |
Substituents on the phenyl ring of the benzamide portion dramatically affected the antihelicase activity of the benzobisthiazoles (Table 1). In general, compounds with bulky substituents (compounds 2–6) were inactive vs B. anthracis helicase, while smaller substituents, such as F, Cl, Br, CN, CH3, CO2CH3, OCH3, and OCH2CH3 were tolerated at the 3- or 4-positions (compounds 7–20). Substituents at the 2-position of the phenyl ring were not tolerated except for the 2-CH3 group (compound 25). Disubstitution at the 3,4- or 3,5-positions with CH3 or OCH3 groups on the phenyl ring was tolerated. For example, compounds 29–32 with substituents 3,4-(CH3)2, 3,4-(OCH2CH2O), 3,4-(OCH3)2, and 3,5-(OCH3)2 displayed 1.7–3.2 μM IC50 values vs B. anthracis helicase, while compounds with disubstitution at the 2,4- or 2,6-positions (26–28) exhibited weak or no inhibitory activity. Compound 33, with 3,4,5-(OCH3)3 substitution on the phenyl ring, showed the best potency with an IC50 value of 0.7 μM in this initial investigation of probing the substitution effect on the antihelicase activity.
The effect of replacement of the phenyl ring with various groups was also investigated in the B. anthracis DNA helicase assay, and the results are shown in Table 2. Replacement of the phenyl ring with alkyl, arylalkyl, naphthyl or heteroaryl groups (compounds 34–44) significantly decreased potency, except for compound 45 with a pyrazine replacement, which exhibited modest activity (IC50 = 28 μM).
The most active B. anthracis helicase inhibitor, compound 33, also exhibited potent inhibitory activity vs S. aureus DNA helicase (IC50 = 0.4 μM) without detectable cytotoxicty (CC50 >100 μM), while compound 16, which bears a 3-OCH3 group on the phenyl ring, inhibited S. aureus DNA helicase with an IC50 value of 6.6 μM. To evaluate the SARs on the methylthio side of the benzobisthiazole core structure, we synthesized a series of analogs of two precursors 33 and 16, by further transforming the methylthio group to various amines, and the synthesis is shown in Scheme 1.
Scheme 1.

Reagents and conditions: a, benzoyl chlorides, CH2Cl2, yields 31–95%; b, aq. KMnO4, glacial HOAc, dioxane, 73% for 47, and 44% for 48; c, R2R3NH, DMF, 80–100 °C, yields 20–83%.
Commercially available aminobenzobisthiazole compound 46 was treated with corresponding benzoyl chlorides, to yield amides 16 and 33.22 Treatment of compounds 16 and 33 with KMnO4 under acidic conditions produced sulfones 47 and 48, respectively, which were treated with amines to produce benzobisthiazole analogs 49–63.23, 24 To evaluate whether the amide bond was required for antihelicase activity, amino compound 46 was converted to benzylamino analog 64 through reductive amination, and an imine analog 65 was also synthesized through a condensation reaction (Scheme 2).25 The biological activities of these analogs were evaluated vs both Bacillus anthracis and S. aureus DNA replicative helicases, and cytotoxicities were measured in a serum free MTT assay21. Results are summarized in Table 3.
Scheme 2.
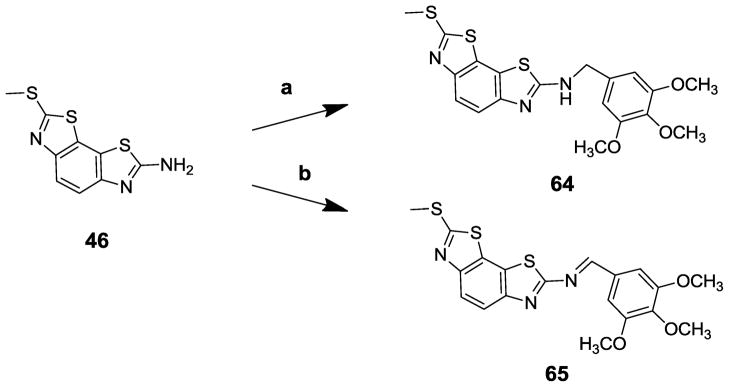
Reagents and conditions: a, 3,4,5-trimethoxybenzaldehyde, Na(OAc)3BH, glacial HOAc, DMSO, r.t., 48%; b, 3,4,5-trimethoxybenzaldehyde, toluene, reflux, 67%.
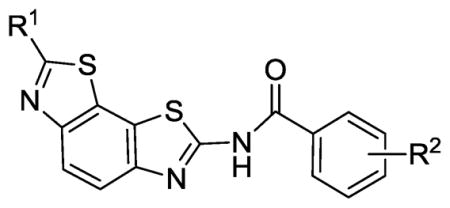
Table 3.
Bacillus anthracis and Staphyloccocus aureus helicase inhibition by benzobisthiazole compounds 16, 33 and 49–65.
| |||||
|---|---|---|---|---|---|
| Compd. | R1 | R2 | IC50 B. anthracis Helicase (μM) | IC50 S. aureus Helicase (μM) | HeLa CC50 in SFM (μM) |
| 16 | S-CH3 | 3-OCH3 | 12.5 | 6.6 | 12.4 |
| 49 | NH-CH3 | 3-OCH3 | 0.5 | 2.2 | >100 |
| 50 | NH-propyl | 3-OCH3 | 1.1 | 1.3 | <0.08; 15.6 |
| 51 | NH-isopropyl | 3-OCH3 | 65.0 | 80 | n.d. |
| 52 | NH-isobutyl | 3-OCH3 | 13.0 | 9.0 | 44 |
| 53 | NH-cyclohexyl | 3-OCH3 | 90 | >100 | n.d. |
| 54 | NH-Bn | 3-OCH3 | 75 | >100 | n.d. |
| 55 | NHCH2CH2N(CH3)2 | 3-OCH3 | 1.0 | 1.0 | |
| 56 |
|
3-OCH3 | 0.5 | 2.2 | 9.6 |
| 57 |
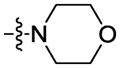
|
3-OCH3 | 2.3 | 3.0 | >100 |
| 58 | NHCH2CH2OH | 3-OCH3 | 2.2 | 4.2 | >100 |
| 33 | S-CH3 | 3,4,5-(OCH3)3 | 0.7 | 0.4 | >100 |
| 59 | NH-CH3 | 3,4,5-(OCH3)3 | 0.2 | 0.2 | >100 |
| 60 | NHCH2CH2N(CH3)2 | 3,4,5-(OCH3)3 | 0.2 | 0.2 | <0.78 |
| 61 |
|
3,4,5-(OCH3)3 | 0.2 | 0.2 | 5.7 |
| 62 |
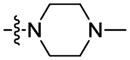
|
3,4,5-(OCH3)3 | 0.2 | 0.2 | 2.8 |
| 63 | NHCH2CH2OH | 3,4,5-(OCH3)3 | 0.2 | 0.3 | >100 |
| 64 |
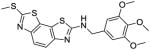
|
>100 | >100 | n.d. | |
| 65 |
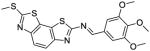
|
>100 | >100 | n.d. | |
Compared to the methylthio compound 16, methylamino analog 49 exhibited improved biological activity vs both Ba and Sa DNA helicases, and its cytotoxicity (CC50) was greater than 100 μM. Increasing the size of the alkylamino group revealed that propylamino was tolerated; however, isopropylamino, cyclohexylamino and benzylamino compounds (51, 53, 54) showed significantly reduced activity, while the isobutylamino compound 52 displayed modest inhibition vs both of the helicases. Interestingly, compounds 55–58, which bear additional polar groups, such as N(CH3)2, morpholino, or OH groups through a linker attaching to the aminobenzobisthiazole core, exhibited improved potency with IC50 values ranging between 1.0–2.3 μM and 1.0–4.2 μM vs Ba and Sa helicases, respectively. This finding suggests that the polar groups make additional, favorable interactions with the helicase enzymes.
Similarly, compared to the methylthio compound 33, methylamino analog 59 also displayed improved helicase inhibitory activity (Ba helicase IC50 = 0.2 μM; Sa helicase IC50 = 0.2 μM) without detectable cytotoxicity (CC50 >100 μM), and selectivity indices (CC50/IC50) were more than 500. Analogs 60–63 all inhibited Ba and Sa helicases with IC50 values between 0.2–0.3 μM. Furthermore, compound 63 did not show cytotoxicity (CC50 >100 μM). Compounds 64 and 65, in which the amide linker was replaced with -NHCH2- and -N=CH- linkers, lost activities vs both Ba and Sa helicases, suggesting that the amide bond is required for inhibitor binding, probably because of favorable hydrogen bonding interactions.
To determine the mode of inhibition for the benzobisthiazole scaffold, a dilution series of compound 49 was added to the helicase reaction in the presence of varying concentrations of the two substrates, ATP and oligonucleotide. The IC50 value of compound 49 varied less than 2-fold (2.0 to 3.8 μM) in response to changes in the ATP concentration (Fig. 2A), but varied considerably (>6.5-fold) when oligonucleotide concentrations were altered (Fig. 2B). These results suggest that compound 49 is noncompetitive with ATP but competitive with the DNA substrate.26 Indeed, a Dixon plot (Fig. 2C) of the DNA variation results is consistent with competitive inhibition with a Ki value of 2 μM, which is in good agreement with the IC50 value at the lowest oligonucleotide concentration tested (3 nM). Similar results were also obtained for compounds 59 and 63 in kinetic studies (data not shown). These results indicate that helicase inhibitors with the benzobisthiazole scaffold act with a different mechanism of action than the previously reported coumarin-based helicase inhibitors, which are noncompetitive with both ATP and DNA.21 However, neither the coumarin nor the benzobisthiazole inhibitors have any significant effect on the single-strand stimulated ATPase activity of S. aureus helicase at concentrations over 100-fold higher than their IC50 values (Fig. 2D).21 X-ray crystallography studies have revealed two distinct hexameric DnaB helicase structures: one showed a flat structure without DNA and NTP association, and the other formed a closed spiral staircase quaternary structure in complex with DNA and ATP mimic GDP-AIF4, suggesting that the former structure represents an inactivated form of DnaB helicase, and the latter one represents an activated form of DnaB helicase. The coumarin type helicase inhibitors are kinetically noncompetitive with both DNA and ATP substrates, suggesting that the coumarin-based inhibitors are likely bound to the inactivated form of the helicase, while the benzobisthiazole derivatives exhibit competitive kinetics with the DNA substrate, suggesting that the benzobisthiazole-based compounds may inhibit the activated form of the bacterial helicase. Structural information for the inhibitor-DnaB helicase complex is needed to further elucidate the mechanism of action of these small molecule helicase inhibitors and provide guidance for the discovery of novel bacterial helicase inhibitors.
Figure 2.
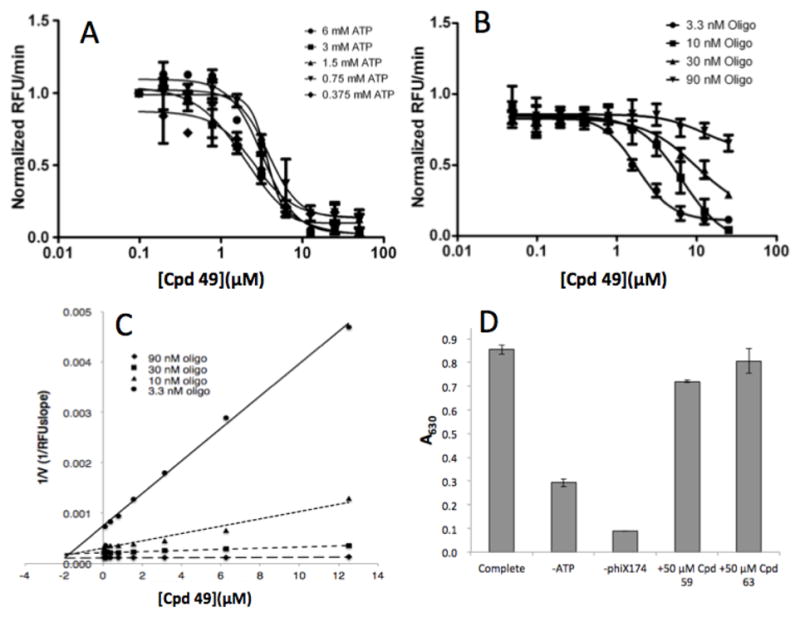
Mode of inhibition of benzobisthiazole 49. A dilution series of compound 49 was examined in the FRET-based S. aureus helicase assay20 with varying concentrations of ATP (A) or DNA (B) to determine IC50 values. Data were plotted in GraphPad Prism 5.0 using 4-parameter curve fitting. (C) The results from varying DNA are also shown in a Dixon plot (1/V vs [I]). (D) Compounds 49 and 59 failed to exhibit significant inhibition of the single-stranded DNA stimulated ATPase activity of S. aureus helicase as detected by malachite green.21
In addition to potently inhibiting replicative helicase, many benzobisthiazole analogs also inhibit the growth of bacterial cells (data not shown). However, the growth inhibition manifests primarily as a reproducible lengthened lag phase, delaying exponential growth, and is not sufficient to produce MIC values, possibly due to poor bacterial cellular permeability and/or short half-life of the parent compounds in cells. Nonetheless, incorporation of radiolabeled precursors into DNA in permeabilized preparations of B. anthracis cells 27 was reduced significantly by compounds 62 and 63 with IC50 values of 10 and 17 μM, respectively (Fig. S1). These results indicate that the benzobisthiazoles inhibit DNA synthesis in bacterial cells, and such inhibition could be responsible for the modest growth inhibition observed in live cells.
In conclusion, we have identified a novel class of potent and selective benzobisthiazole-based bacterial DnaB helicase inhibitors through high throughput screening. Subsequent structure-activity relationship analysis and chemical optimization led to substantial improvement of biological activity and identification of nanomolar compounds with selectivity indices of more than 500. To the best of our knowledge, these compounds are the most potent bacterial replicative helicase inhibitors reported to date. Further optimization of the benzobisthiazole-based helicase inhibitors may provide a novel small molecule drug for antibacterial therapy.
Supplementary Material
Acknowledgments
We thank Ms. Shawna M. Rotoli and Dr. Subhasis B. Biswas (Department of Molecular Biology, University of Medicine and Dentistry of New Jersey (UMDNJ)) and Dr. Esther E. Biswas-Fliss (UMDNJ and Department of Bioscience Technologies, Jefferson School of Health Professions, Thomas Jefferson University) for providing B. anthracis helicase, for radiometric assays of the B. anthracis helicase inhibition potency, and for helpful discussions. We thank Mr. Steven C. Cardinale at Microbiotix for assistance with the malachite green-based ATPase assay. This work was supported by the National Institutes of Health/National Institute of Allergy and Infectious Diseases (AI064974). The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Moran GJ, Krishnadasan A, Gorwitz RJ, Fosheim GE, McDougal LK, Carey RB, Talan DA. N Engl J Med. 2006;355:666. doi: 10.1056/NEJMoa055356. [DOI] [PubMed] [Google Scholar]
- 2.Streit JM, Jones RN, Sader HS, Fritsche TR. Int J Antimicrob Agents. 2004;24:111. doi: 10.1016/j.ijantimicag.2003.12.019. [DOI] [PubMed] [Google Scholar]
- 3.Collin F, Karkare S, Maxwell A. Appl Microbiol Biotechnol. 2011;92:479. doi: 10.1007/s00253-011-3557-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Domagala JM, Hanna LD, Heifetz CL, Hutt MP, Mich TF, Sanchez JP, Solomon M. J Med Chem. 1986;29:394. doi: 10.1021/jm00153a015. [DOI] [PubMed] [Google Scholar]
- 5.Butler MM, Dudycz LW, Khan NN, Wright GE, Brown NC. Nucleic Acids Res. 1990;18:7381. doi: 10.1093/nar/18.24.7381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Butler MM, Skow DJ, Stephenson RO, Lyden PT, LaMarr WA, Foster KA. Antimicrob Agents Chemother. 2002;46:3770. doi: 10.1128/AAC.46.12.3770-3775.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tarantino PM, Jr, Zhi C, Wright GE, Brown NC. Antimicrob Agents Chemother. 1999;43:1982. doi: 10.1128/aac.43.8.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Patel SS, Picha KM. Annu Rev Biochem. 2000;69:651. doi: 10.1146/annurev.biochem.69.1.651. [DOI] [PubMed] [Google Scholar]
- 9.Moyer SE, Lewis PW, Botchan MR. Proc Natl Acad Sci U S A. 2006;103:10236. doi: 10.1073/pnas.0602400103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dubaele S, Jahnke W, Schoepfer J, Fuchs J, Chene P. Bioorg Med Chem Lett. 2006;16:923. doi: 10.1016/j.bmcl.2005.10.110. [DOI] [PubMed] [Google Scholar]
- 11.Earnshaw DL, Moore KJ, Greenwood CJ, Djaballah H, Jurewicz AJ, Murray KJ, Pope AJ. J Biomol Screen. 1999;4:239. doi: 10.1177/108705719900400505. [DOI] [PubMed] [Google Scholar]
- 12.Earnshaw DL, Pope AJ. J Biomol Screen. 2001;6:39. doi: 10.1177/108705710100600106. [DOI] [PubMed] [Google Scholar]
- 13.Griep MA, Blood S, Larson MA, Koepsell SA, Hinrichs SH. Bioorg Med Chem. 2007;15:7203. doi: 10.1016/j.bmc.2007.07.057. [DOI] [PubMed] [Google Scholar]
- 14.McKay GA, Reddy R, Arhin F, Belley A, Lehoux D, Moeck G, Sarmiento I, Parr TR, Gros P, Pelletier J, Far AR. Bioorg Med Chem Lett. 2006;16:1286. doi: 10.1016/j.bmcl.2005.11.076. [DOI] [PubMed] [Google Scholar]
- 15.Zhang B, Zhang AH, Chen L, Xi XG. J Biochem. 2008;143:773. doi: 10.1093/jb/mvn026. [DOI] [PubMed] [Google Scholar]
- 16.Zhang L, Schwartz G, O’Donnell M, Harrison RK. Anal Biochem. 2001;293:31. doi: 10.1006/abio.2001.5093. [DOI] [PubMed] [Google Scholar]
- 17.Zhang Y, Yang F, Kao YC, Kurilla MG, Pompliano DL, Dicker IB. Anal Biochem. 2002;304:174. doi: 10.1006/abio.2002.5627. [DOI] [PubMed] [Google Scholar]
- 18.Bailey S, Eliason WK, Steitz TA. Science. 2007;318:459. doi: 10.1126/science.1147353. [DOI] [PubMed] [Google Scholar]
- 19.Itsathitphaisarn O, Wing RA, Eliason WK, Wang J, Steitz TA. Cell. 2012;151:267. doi: 10.1016/j.cell.2012.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aiello D, Barnes MH, Biswas EE, Biswas SB, Gu S, Williams JD, Bowlin TL, Moir DT. Bioorg Med Chem. 2009;17:4466. doi: 10.1016/j.bmc.2009.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li B, Pai R, Di M, Aiello D, Butler MM, Tashjian T, Peet NP, Bowlin TL, Moir D. J Med Chem. 2012;55:10896. doi: 10.1021/jm300922h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Synthetic procedures for preparation of compounds 16 and 33. Compound 46 (10 g, 39.5 mmol) was suspended in pyridine (200 mL) and cooled to 0 °C in an ice bath. Diisopropylethylamine (11.1 g, 86 mmol) was added, followed by dropwise addition of 3-methoxybenzoyl chloride (6.8 g, 39.9 mmol). After stirring at room temperature for 3 days the solvent was evaporated. The crude product was sonicated with ethyl acetate (200 mL) and water (100 mL) for 10 minutes and filtered. The precipitate was stirred in chloroform (400 mL) at room temperature and filtered. The filtrate was washed thrice with water, brine, dried over sodium sulphate and evaporated to give a white solid 16 (9.4 g, yield 62%), mp = 237–238 °C, Rf = 0.71 (2:98::MeOH:CH2Cl2); 1H NMR (300 MHz, DMSO-d6) δ 13.08 (s, 1H), 7.93 (d, J = 8.7 Hz, 1H), 7.86 (d, J = 8.7 Hz, 1H), 7.73 (d, J = 2.7 Hz, 2H), 7.48 (t, J = 8.1 Hz, 1H), 7.23 (d, J = 8.1 Hz, 1H), 3.87 (s, 3H), 2.84 (s, 3H). LC/MS m/z 388.3 (M+1)+. Compound 33 was synthesized using the same procedure. Yield 96%, mp = 231–232 °C, Rf = 0.76 (5:95::MeOH:CH2Cl2); 1H NMR (300 MHz, DMSO-d6) δ 13.05 (s, 1H), 7.94 (d, J = 8.7 Hz, 1H), 7.85 (d, J = 8.4 Hz, 1H), 7.53 (s, 2H), 3.90 (s, 6H), 3.77 (s, 3H), 2.84 (s, 3H).
- 23.Synthetic procedures for preparation of compounds 47 and 48. Compound 16 (3.0 g, 10.0 mmol) was suspended in a mixture of glacial acetic acid (100 mL) and dioxane (40 mL). A solution of KMnO4 (6.0 g, 38.0 mmol) in water (15 mL) was added dropwise while stirring. After stirring at 55 °C for 24 hours the mixture was poured into a solution of NaHSO3 (15 g in 250 mL) and stirred for 15 minutes. The resulting precipitate was collected by filtration, washed with water and dried at 55 °C under vacuum overnight to give the desired product. The crude product was refluxed with MeOH (250 mL) for 1 hour and filtered while hot. The precipitate was washed with MeOH and dried at 55 °C overnight under vacuum to give compound 47 (3.15 g, yield 73%), mp = 284–285 °C, Rf = 0.35 (1:1::EtOAc:Hexane); 1H NMR (300 MHz, DMSO-d6) δ 13.26 (s, 1H), 8.27 (d, J = 8.7 Hz, 1H), 8.06 (d, J = 9.0 Hz, 1H), 7.73-7.72 (m, 2H), 7.48 (t, J = 8.4 Hz, 1H), 7.23 (dd, J = 2.1, 8.1 Hz, 1H), 3.87 (s, 3H), 3.65 (s, 3H). LC/MS m/z 420 (M+1)+. Compound 48 was synthesized using the same procedure. Yield 44%, mp = 160–161 °C, Rf = 0.12 (1:1::EtOAc:Hexane); 1H NMR (300 MHz, DMSO-d6) δ 13.27 (s, 1H), 8.33 (d, J = 8.7 Hz, 1H), 8.11 (d, J = 8.7 Hz, 1H), 7.56 (s, 2H), 3.91 (s, 6H), 3.77 (s, 3H), 3.63 (s, 3H). LC/MS m/z 479.8 (M+1)+.
- 24.General synthetic procedures for preparation of compounds 49–63. Compounds 47 and 48 (6.3 mmol) was treated with 20 eq. of corresponding amine in a sealed tube at 70 °C for 24 hour. Then the reaction mixture was evaporated to dryness and dried at 50 °C under vacuum overnight. The resulting crude mixture was purified by high-pressure liquid chromatography to obtain compounds 49–63. Characterization data for compound 49: Yield 62%, mp = 301–302 °C, Rf = 0.1 (1:1::EtOAc:Hexane); 1H NMR (300 MHz, DMSO-d6) δ 13.17 (s, 1H), 8.73 (s, 1H), 7.74 (m, 3H), 7.56 (d, J = 9.0 Hz, 1H), 7.48 (t, J = 9.0 Hz, 1H), 7.23 (dd, J = 8.1 Hz, 1H), 3.88 (s, 3H), 2.52 (s, 3H). LC/MS m/z 371.2 (M+1)+. Characterization data for compound 59: Yield 38%, mp = 238–239 °C, Rf = 0.87 (80:18:2::CHCl3:CH3OH:CH3NH2); 1H NMR (300 MHz, DMSO-d6) δ 8.24 (br, 1H), 7.67 (d, J = 9.0 Hz, 1H), 7.54 (s, 1H), 7.51 (d, J = 0.9 Hz, 2H), 3.88 (s, 6H), 3.74 (s, 3H), 2.97 (s, 3H). LC/MS m/z 431.1 (M+1)+.
- 25.Characterization data for compounds 64 and 65. For compound 64: Yield 48%, mp = 168–169 °C, Rf = 0.38 (50:50::EtOAc:n-Hexanes), 1H NMR (300 MHz, DMSO-d6) δ 8.67 (s, 1H), 7.72 (d, J = 8.7 Hz, 1H), 7.51 (d, J = 8.7 Hz, 1H), 6.73 (s, 2H), 4.54 (d, J = 4.8 Hz, 2H), 3.75 (s, 6H), 3.62 (s, 3H), 2.78 (s, 3H). LC/MS m/z 434.1 (M+1)+. For compound 65: Yield 67%, mp = 207–208 °C, Rf = 0.68 (5:95::MeOH:CH2Cl2); 1H NMR (300 MHz, DMSO-d6) δ 9.13 (s, 1H), 7.99 (s, 2H), 7.46 (s, 2H), 3.89 (s, 6H), 3.79 (s, 3H), 2.85 (s, 3H), LC/MS m/z 431.6 (M+1)+.
- 26.Wei M, Wynn R, Hollis G, Liao B, Margulis A, Reid BG, Klabe R, Liu PC, Becker-Pasha M, Rupar M, Burn TC, McCall DE, Li Y. J Biomol Screen. 2007;12:220. doi: 10.1177/1087057106296679. [DOI] [PubMed] [Google Scholar]
- 27.Brown NC, Weisseman CL, III, Matsushita T. Nat New Biol. 1972;237:72. doi: 10.1038/newbio237072a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.