

Abstract
Hepatic cysts are increasingly found as a mere coincidence on abdominal imaging techniques, such as ultrasonography (USG), computed tomography (CT) and magnetic resonance imaging (MRI). These cysts often present a diagnostic challenge. Therefore, we performed a review of the recent literature and developed an evidence-based diagnostic algorithm to guide clinicians in characterising these lesions. Simple cysts are the most common cystic liver disease, and diagnosis is based on typical USG characteristics. Serodiagnostic tests and microbubble contrast-enhanced ultrasound (CEUS) are invaluable in differentiating complicated cysts, echinococcosis and cystadenoma/cystadenocarcinoma when USG, CT and MRI show ambiguous findings. Therefore, serodiagnostic tests and CEUS reduce the need for invasive procedures. Polycystic liver disease (PLD) is arbitrarily defined as the presence of > 20 liver cysts and can present as two distinct genetic disorders: autosomal dominant polycystic kidney disease (ADPKD) and autosomal dominant polycystic liver disease (PCLD). Although genetic testing for ADPKD and PCLD is possible, it is rarely performed because it does not affect the therapeutic management of PLD. USG screening of the liver and both kidneys combined with extensive family history taking are the cornerstone of diagnostic decision making in PLD. In conclusion, an amalgamation of these recent advances results in a diagnostic algorithm that facilitates evidence-based clinical decision making.
Keywords: Coincidental hepatic cystic lesions, Cystic liver disease, Complicated cyst, Polycystic liver disease, Diagnostic algorithm
Core tip: We performed a review of the recent literature, and through combining current consensus and recent advances, we developed an evidence-based diagnostic algorithm to guide clinicians in characterising hepatic cystic lesions. Serodiagnostic tests and microbubble contrast-enhanced ultrasound (CEUS) are invaluable in differentiating complicated cysts, echinococcosis and cystadenoma/cystadenocarcinoma when ultrasonography (USG), computed tomography and magnetic resonance imaging show ambiguous findings. As a result, serodiagnostic tests and CEUS reduce the need for invasive procedures. USG screening of the liver and both kidneys combined with extensive family history taking remains the cornerstone of diagnostic decision making in polycystic liver disease.
INTRODUCTION
Hepatic cystic lesions represent a comprehensive heterogeneous cluster with regard to pathogenesis, clinical presentation, diagnostic findings and therapeutic management (Table 1). Hepatic cystic lesions predominantly remain asymptomatic and are found as a mere coincidence on abdominal imaging techniques, such as ultrasonography (USG), computed tomography (CT) and magnetic resonance imaging (MRI)[1,2]. The use of these techniques has greatly increased over the last years, and as a corollary, there has been an increase in incidental findings of asymptomatic hepatic cystic lesions[3]. In most cases, hepatic cystic lesions will follow a benign course[4]. However, it is essential to differentiate benign cysts from potentially harmful cysts, such as echinococcosis, cystadenoma and cystadenocarcinoma, which require specific treatment[5,6]. Currently, clinicians must also be aware of changes in the epidemiology of certain hepatic cystic lesions. Echinococcosis has spread to previously non-endemic Western European countries[7,8]. For this reason, the early and accurate diagnosis of cysts is crucial. To facilitate the diagnostic process, we provide an overview of the wide spectrum of mono- and polycystic liver diseases based on literature published over the last five years.
Table 1.
Differential diagnosis of cystic lesions in the liver
| Monocytic disease | |
| Simple cyst | |
| Echinococcosis | |
| Cystic echinococcosis | |
| Alveolar echinococcosis | |
| Cystadenoma | |
| Cystadenocarcinoma | |
| Polycystic disease | |
| Autosomal dominant polycystic kidney disease | |
| Autosomal dominant polycystic liver disease | |
LITERATURE SEARCH
We searched the electronic database PubMed using the following search terms: “liver” and “cyst” and “diagnosis”. We limited our search to articles that were written in English, published between November 2007 and November 2012 and available in full text. A total of 992 articles were identified. For the purpose of this review, we included articles with a main focus on the evaluation of hepatic cystic lesions in humans. Screening the titles and abstracts identified 252 articles meeting these inclusion criteria (Figure 1). Additionally, we searched the reference lists from all eligible reviews for additional leads.
Figure 1.

Selection process of retrieved articles.
SIMPLE CYSTS
Pathogenesis
Simple cysts arise congenitally from aberrant bile duct cells and contain a clear, bile-like fluid[9]. Because bile duct epithelium covers the simple cyst inner lining, it is hypothesised that simple cysts arise during embryogenesis when intrahepatic ductules fail to connect with extrahepatic ducts[4,10].
Clinical features
The prevalence of simple cysts ranges from 2.5% to 18% and increases with age[11,12]. More than half of individuals older than 60 years are likely to have one or more simple cysts. Cysts are small in most patients but can grow to over 30 cm in selected cases. In a small fraction of patients, symptoms, such as abdominal pain, early satiety, nausea and vomiting, arise as a result of a mass effect[3]. Physical examination may reveal a palpable abdominal mass or hepatomegaly[1]. Complications such as haemorrhage, rupture and biliary obstruction are uncommon but are more likely in larger cysts[13]. Intracystic haemorrhage is a rare complication of simple cysts and usually presents with severe abdominal pain[14], although asymptomatic presentations are also observed[15,16].
Laboratory findings
Laboratory findings are predominantly normal, but a minority of patients have raised serum γ-glutamyl-transferase (γGT)[17]. Several studies have shown that serum and cyst fluid levels of carcinoembryonic antigen (CEA) and cancer antigen 19-9 (CA 19-9) may be elevated[18]. CA 19-9 is expressed in the simple cyst inner epithelial lining and leads to elevated cyst fluid and serum CA 19-9 levels[17]. CA 19-9 is not helpful in the differential diagnosis of intracystic haemorrhage[19].
Diagnostic features
Most simple cysts are diagnosed incidentally on USG (Figure 2A), CT (Figure 2B) or MRI. The diagnosis of a simple cyst is based on the following USG criteria: anechoic (i.e., fluid filled cavity), no septations, sharp smooth borders, strong posterior wall echoes (indicating a well-defined fluid/tissue interface), spherical or oval shaped and a relative accentuation of echoes beyond the cyst compared to echoes at a similar depth transmitted through normal adjacent hepatic tissue (Table 2)[20]. CT shows a sharply defined homogeneous hypodense lesion (Figure 2B)[21]. MRI T1-weighted sequence shows low signal intensity, whereas the T2-weighted sequence shows extremely high signal intensity, which does not enhance after contrast injection[22]. USG has a reported sensitivity and specificity of approximately 90% for diagnosing a simple cyst[23], and recent advances in CT and MRI technology might result in even higher sensitivity rates[12,22,24]. However, because CT is accompanied with a radiation load and both CT and MRI come at a significantly higher cost, USG remains the most accurate, non-invasive and cost-effective imaging modality for diagnosing simple cysts.
Figure 2.
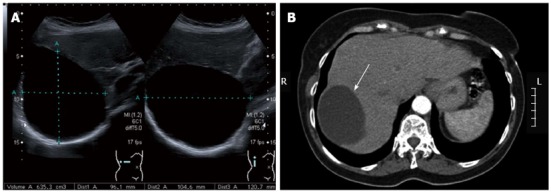
Simple cyst. A: On abdominal ultrasonography. Ultrasonography (USG) demonstrating a large simple cyst occupying the right hepatic lobe. Note the sharp and smooth border, oval shape, and anechoic echo pattern with the absence of septations and strong posterior wall echoes. The cyst size is indicated by the dotted lines; B: On abdominal computed tomography. Computed tomography demonstrating a sharply defined homogeneous hypodense cystic lesion (arrow) occupying the right hepatic lobe, which was diagnosed as a simple cyst.
Table 2.
Ultrasonography features for the diagnosis of monocytic diseases of the liver
| Simple cyst | Cystic echinococcosis | Alveolar echinococcosis | Cystadenoma and cystadenocarcinoma | |
| Border | Sharp and smooth | Laminated | Irregular | Irregular |
| Shape | Spherical or oval | Round or oval | Irregular | Round or oval |
| Echo pattern | Anechoic1 | Anechoic or atypical2 | Hyperechogenic outer ring and hypoechogenic centre | Hypoechogenic with hyperechogenic septations |
| Appearance | No septa | Multiseptated | Multivesicular | Septated and/or solid structures (papillary projections) |
| Wall | Strong posterior wall echoes | Wall enhancement | ||
| Posterior acoustic feature | Relative3 accentuation of echoes | Dorsal shadowing (calcified areas) | Dorsal shadowing (calcified areas) |
Fluid-filled cavity;
Snowflake-like inclusions or floating laminated membranes;
Compared to echoes at a similar depth transmitted through normal adjacent hepatic tissue.
In case of an intracystic haemorrhage (i.e., complicated cyst), USG typically shows a hyperechogenic echo pattern combined with internal echoes that mimic septations or solid portions (Figure 3)[25]. In contrast, CT visualises intracystic haemorrhage as a high-density area[26], whereas MRI depicts it as a high signal intensity on T1- and T2-weighted sequences[27]. Neither CT nor MRI has additional diagnostic value compared to USG in the diagnosis of cystic bleeding[15]. The recent development of microbubble contrast-enhanced ultrasound (CEUS) enables us to visualise vascular flow within septa or solid components of cysts, which is absent in simple cysts with intracystic haemorrhage[28]. Therefore, CEUS can accurately characterise these cysts when USG, CT and MRI show ambiguous findings[29-31].
Figure 3.
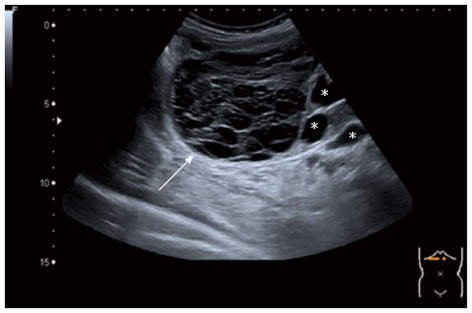
Complicated simple cyst on abdominal ultrasonography. Ultrasonography (USG) demonstrating a cystic lesion with a hyperechogenic echo pattern combined with internal echoes that mimic septations or solid portions (arrow) in a patient presenting with severe abdominal pain with a known history of multiple simple cysts (asterisks). Because of the known history of simple cysts, the lesion was diagnosed as a complicated simple cyst (i.e., intracystic haemorrhage).
Therapy
The management of most simple cysts relies on a “wait-and-see” policy, and no further treatment is required in these cases. If there are symptoms, aspiration-sclerotherapy is the preferred treatment[32,33]. Laparoscopic or open surgical fenestration techniques are similarly or even more effective in reducing symptoms[34,35] but have a significantly higher morbidity and mortality rate[36].
ECHINOCOCCOSIS
Echinococcosis is a zoonosis caused by larval stages of taeniid cestodes (tapeworms) belonging to the Echinococcus species. Two of the six known species cause solitary cystic lesions in humans: (1) Echinococcus granulosus (E. granulosus), responsible for cystic echinococcosis (CE); and (2) Echinococcus multilocularis (E. multilocularis), responsible for alveolar echinococcosis (AE)[6].
Echinococcosis-related deaths are uncommon in developed countries. For example, there were 41 echinococcosis-associated deaths in the United States over an 18-year study period[37]. However, echinococcosis is considered to be an emerging disease in Europe[38,39]. Thus, CE and AE are diseases with a considerable global disease impact, as indicated by a substantial loss in disability-adjusted life years[38,40].
Cystic echinococcosis
Pathogenesis: Humans become infected by acting as intermediate hosts of E. granulosus after ingestion of Echinococcus eggs, which are excreted by infected carnivores (dogs and other canids)[6]. Infection is typically observed in areas containing large numbers of the intermediate hosts of the parasite (sheep and goats) that are in close contact with the final host (herding dogs)[41-43].
Clinical features: Although CE has a worldwide geographic distribution, the highest prevalence of CE is found in the temperate zones, including the Mediterranean, Central Asia, Australia and some parts of America[44].
Because cyst growth in the liver is slow (ranging from 1-5 millimetres in diameter per year), CE can remain asymptomatic for a long time. In approximately 90% of cases, the primary presentation is a spherical, fluid-filled vesicle with an inner cellular layer and an outer laminated layer located in the liver, lungs or both[45]. Symptoms occur when cysts exert mass effects within the organ or surrounding tissues or rupture, often presenting as a sudden onset of abdominal pain. Secondary cholangitis (rupture into the biliary tree), biliary obstruction and intraperitoneal rupture followed by anaphylaxis are common complications of CE and require hospitalisation[6]. Worldwide mortality rate estimates vary between 2.2%-5.0%[45,46], although the exact mortality rate of CE in developed countries remains unknown.
Diagnostic features: The diagnosis of CE is based on the following criteria: endemic region history, clinical findings (e.g., abdominal pain, fever, chest pain, and dyspnea), pathognomonic USG features and positive immuno diagnostic tests[47]. USG shows a round or oval-shaped, anechoic or atypical (i.e., snowflake-like inclusions or floating laminated membranes) echo pattern with multiple septa confined by a laminated border (Table 2)[47]. USG has a reported specificity of 90% and is used in combination with CT when surgical treatment is considered. MRI has not been proven to be cost-effective and has no added value[48]. The currently used serodiagnostic tests to reveal E. granulosus antibodies have a sensitivity of 93.5% and specificity of 89.7%[49].
Therapy: The treatment of CE, including surgery (open or laparoscopic), percutaneous treatments [e.g., puncture aspiration injection re-aspiration (PAIR) method] and chemotherapy[50], is indicated to reduce symptoms and prevent complications[51]. PAIR is the treatment of choice for CE, as a recent review showed that PAIR resulted in parasitological clearance (i.e., negative serodiagnostic tests) in 95.8% of cases[52].
Alveolar echinococcosis
Pathogenesis: AE is endemic in the Northern hemisphere (e.g., North America, Asia, China, Japan and Europe). AE occurs when E. multilocularis eggs, found in the excrement of foxes, are ingested. The spread from endemic areas to previously non-endemic Western European countries is most likely due to an increasing fox population and spillover from these wild carnivores to domestic hosts[7,8].
Clinical features: The ingested eggs develop into an alveolar structure composed of numerous small vesicles that vary in diameter from smaller than 1 mm to 3 cm. Each vesicle has the same wall structure as CE. These vesicles grow slowly and are able to reach a maximum diameter of 15-20 cm, similar to simple cysts[53]. In approximately 99% of cases, the infection is initially confined to a solitary alveolar lesion in the liver[45]. After the primary infection, AE usually has an asymptomatic phase of 5-15 years prior to the development of symptoms. Symptoms are related to mass effect or are nonspecific, such as weight loss or fatigue[54]. In contrast to the encapsulated growth pattern of CE, AE eventually leads to liver failure due to an infiltrative neoplastic growth with potential metastasis to adjacent and distant (e.g., lungs, spleen, bone, and brain) organs[55,56].
Diagnostic features: Typical USG aspects are observed in 70% of cases and include irregular shape and border, hyperechogenic outer ring and hypoechogenic centre, multivesicular appearance and dorsal shadowing due to calcified areas (Table 2)[47]. Atypical USG aspects include small hyperechogenic nodules (amorphous AE), large lesions with massive necrosis (pseudocyst) and small calcified lesions (inert AE)[57]. In contrast to CE, MRI is superior to CT in detecting AE lesion margins[58,59]. Similar to CE, high diagnostic sensitivity (90%-100%) and specificity (95%-100%) are attained with serodiagnostic tests, and in 80%-95% of cases, AE can be differentiated from CE with the help of serologically obtained purified Echinococcus antigens[60].
Therapy: The approach to the management of AE resembles that of a hepatic malignancy. The cornerstone of treatment for AE includes radical surgery followed by a 2-year period of chemotherapy[6]. A recent study concluded that AE can be cured in 42% of cases by complete surgical removal of the parasitic mass. Early diagnosis could even improve this rate further[61].
CYSTADENOMA AND CYSTADENOCARCINOMA
Pathogenesis
Cystadenoma and cystadenocarcinoma are biliary cyst tumours that originate from the biliary epithelium[62]. Analogous to simple cysts, cystadenoma is considered to be a congenital disorder[63]. The exact mechanism of carcinogenesis in cystadenoma remains unknown. Several studies have suggested that cystadenocarcinoma develops from the ectopic remnants of primitive foregut sequestered within the liver[63]. In contrast, the malignant transformation of cystadenoma into cystadenocarcinoma is considered to be an alternative mechanism of carcinogenesis, as some cystadenocarcinomas may co-exist with cystadenoma[64]. This hypothesis is supported by the observation that the presence of cystadenoma increases the chance of developing cystadenocarcinoma[65].
Clinical features
Less than 5% of all cystic lesions of the liver are cystic neoplasms[2]. The clinical presentation of cystadenoma and cystadenocarcinoma is asymptomatic or tends to mimic symptoms of simple cysts or echinococcosis[66,67]. Studies have reported a predominance in women, with a mean age of onset varying from 40-60 years[64,65]. Cystadenomas appear to be slow growing, but exact growth rates are unknown. One case series evaluated 75 patients and recorded a variability in cyst size from 1.5-35 cm[68]. One study involving 63 cases diagnosed with cystadenocarcinoma demonstrated infiltrative growth in neighbouring organs in 33 cases (52%) and distant metastases in 15 cases (24%)[5]. For that reason, it is necessary to diagnose cystadenocarcinoma in an early stage.
Laboratory findings
In general, liver function tests are normal. A review of 13 cases found that serum concentrations of γGT and alkaline phosphatase (AP) were elevated in 3 cases[69]. One study reported a rise in serum levels of CEA in 3 of 22 cystadenocarcinoma cases (14%) and a rise in the serum concentration of CA 19-9 in 4 of 11 cases (36%)[5]. Similar results have been reported in cases with cystadenoma: one study showed elevated serum concentrations of CEA or CA 19-9 in 2 of 3 cases[63]. Consequently, laboratory studies are not helpful in differentiating cystadenoma and cystadenocarcinoma from complicated cysts or echinococcosis.
Diagnostic features
The USG characteristics of cystic neoplasms for both cystadenoma and cystadenocarcinoma are the following: a round or oval shape, irregular border, hypoechogenic echo pattern with hyperechogenic septations or solid structures (i.e., papillary projections), wall enhancement and dorsal shadowing due to calcified areas (Table 2)[2]. Because of these typical cystic neoplasmic features, which are absent in simple cysts, USG is a useful technique to easily discriminate between cystic neoplasms and simple cysts[2]. Like USG, CT and MRI show markedly similar characteristics for cystadenoma and cystadenocarcinoma: internal septations, thickened and irregular wall, papillary projections, calcifications and wall enhancements[62]. Cystadenomas predominantly have thinner septa and more regular walls[70], whereas solid structures, intracystic haemorrhage and vascularised septations on contrast-enhanced CT are more suspicious for cystadenocarcinoma[62]. However, in most cases, differentiation between cystadenoma and cystadenocarcinoma is not possible[1]. The same problem arises in differentiating echinococcosis and complicated cysts from cystadenoma and cystadenocarcinoma because in many cases, intracystic haemorrhage, calcifications and septations are present in these lesions[2,62].
Recent advances in technology have made diffusion-weighted magnetic resonance imaging (DWI) a promising MRI technique for liver lesion detection and characterisation[71]. DWI depicts the rate of diffusion of water molecules between tissues, given as the apparent diffusion coefficient (ADC)[72]. Generally, high ADC values are measured in cystic and necrotic tissue, which allow a relatively free diffusion of water, whereas low ADC values are an indication of cell-rich tissue (e.g., tumour tissue)[22,73,74]. However, because of an overlap of ADC values, differentiating cystic neoplasms, echinococcosis and complicated cysts is not possible with DWI[75]. Therefore, additional immunodiagnostic tests are needed to rule out echinococcosis. Fine needle aspiration (FNA) could be of additional help to exclude complicated cysts[76]; however, due to the risk of malignancy, FNA is generally not performed. In contrast, CEUS can be helpful in differentiating cystadenoma and cystadenocarcinoma from complicated cysts when USG, CT or MRI is inconclusive. CEUS characterises the vascular flow within septa in cystadenoma and cystadenocarcinoma, which is absent in complicated cysts[29-31]. Nonetheless, surgical resection remains the golden standard for diagnosing cystadenoma and cystadenocarcinoma when CEUS is not available.
Therapy
The primary treatment of cystadenoma and cystadenocarcinoma is hepatic resection. A study in which 66 cases of cystadenocarcinoma were subjected to hepatic resection described a 3-year survival rate of 74%[5].
PCLD AND ADPKD
Polycystic liver disease
Polycystic liver disease (PLD) is arbitrarily defined as the presence of > 20 liver cysts[77]. Autosomal dominant polycystic liver disease (PCLD) and autosomal dominant polycystic kidney disease (ADPKD) are two distinct genetic disorders associated with the development of polycystic livers[78]. Liver function, as judged by parameters of liver synthesis, is not affected in PLD, as functional hepatic tissue remains unaffected[77,79].
Pathogenesis
During embryogenesis, the intrahepatic bile ducts are formed from a cylindrical layer of cells (i.e., ductal plate) surrounding each portal vein. Incorrect involution of the ductal plate results in ductal plate malformation (DPM)[80,81]. DPM consists of excess embryonic bile duct structures in a ductal plate configuration that does not communicate with the normally developed intrahepatic bile ducts. The progressive dilatation of these excess intrahepatic structures during life results in multiple liver cysts[82]. Similar to simple cysts, these cysts contain a clear, bile-like fluid and an inner lining of cholangiocytes[83].
Genetics
PCLD was historically considered a phenotypic variant of ADPKD[84]. However, the presence of PLD in the absence of renal cysts led to the belief that PCLD should be regarded as a separate entity[85]. The discovery of a familial form of PLD[86], genetically distinct from the heterozygous mutation in genes PKD1 and PKD2 identified in ADPKD[87], ultimately led to the identification of heterozygous mutations in genes encoding SEC63 and PRKCSH[88-90]. Mutation analysis identified a heterozygous mutation in PRKCSH (15%) and SEC63 (5%) in approximately 20% of studied PCLD cases[91]. In contrast, a PKD1 mutation was found in 85% of cases of ADPKD, and a PKD2 mutation was found in the remaining cases[92].
PRKCSH and SEC63 encode hepatocystin and SEC63 proteins, respectively. Hepatocystin acts in the folding process of proteins, while SEC63 acts as part of the endoplasmic reticulum translocon[93]. Unfortunately, the exact mechanism of cystogenesis in PCLD remains unclear. Polycystin 1 and 2, encoded by PKD1 and PKD2, respectively, are important for adequate functioning of the primary cilium[94]. Its therefore suggested that primary cilia play a central pathogenic role in the mechanism of hepatic cystogenesis in ADPKD[78].
Clinical features
The extra polarisation of 137 identified PCLD cases in a specific adherence region (the Netherlands) led to an estimated PCLD prevalence of 1 per 158000[77]. This number is most likely an underestimation of the true prevalence because only symptomatic patients referred to tertiary centres were included in this study, and PCLD often remains asymptomatic[95]. ADPKD is the most common monogenetic disorder, with a world-wide estimated prevalence of 0.10%-0.25%[96], and it is responsible for approximately 8%-10% of cases with end-stage renal disease[97]. Although ADPKD is primarily characterised by the presence of renal cysts[98], liver cysts are considered the most prevalent extra-renal manifestation of ADPKD[99,100]. Indeed, one study involving 230 ADPKD cases found an overall prevalence of 83%[101]. However, the exact prevalence of PLD in ADPKD is still unknown. PCLD is predominantly confined to the liver, but a few renal cysts can also be present, which leads to difficulties in the accurate differentiation between PCLD and ADPKD[79,99]. Although renal cysts in ADPKD ultimately lead to renal failure, renal function remains unaffected in the presence of PCLD-associated renal cysts[102].
PLD is predominantly discovered during the fourth or fifth decade of life and is more severe in females[77,96,103,104]. PCLD tends to lead to a higher number and greater volume of liver cysts[79]. The number of pregnancies, increased age and severity of renal disease are considered additional risk factors for liver cyst growth in ADPKD[105]. PLD is mainly asymptomatic, but mechanical complaints can arise in a subset of patients[79,106]. Complications such as intracystic haemorrhage and infection are rare and typically occur in large cysts[106].
Laboratory findings
PLD causes increased γGT and AP levels in both PCLD and ADPKD patients[77]. Occasionally, increased serum aspartate aminotransferase (AST) is also found in ADPKD[79,107]. Renal function remains intact in PCLD, whereas ADPKD patients show a rise in serum creatinine due to impaired renal function[102].
Diagnostic features
PLD is detected with the use of USG, CT or MRI. USG, which is accurate, non-invasive and low cost, is the preferred imaging modality for both PCLD and ADPKD[108,109]. Currently, there are no generally accepted USG criteria for PCLD. One study suggested that the diagnosis can be made in case of a positive family history of PCLD and the presence of > 4 liver cysts[78]. However, diagnosing ADPKD is usually relatively straightforward when enlarged bilateral cystic kidneys are present in combination with a positive family history for ADPKD[102]. In case of a negative family history, screening direct family members with USG can be helpful to reveal asymptomatic ADPKD. Because mutation analysis for ADPKD has no clinical implications, its use is limited to family members of ADPKD patients involved in kidney donation programs. In 2009, the Pei USG criteria were developed because the original Ravine USG criteria for diagnosing ADPKD appeared to be insufficient[110,111]. Table 3 gives an overview of the USG criteria for diagnosing ADPKD when the causative gene is unknown. For example, in case of a positive ADPKD family history, diagnosis can be made when ≥ 3 renal cysts are unilaterally present in individuals aged 15 to 39 years[110]. ADPKD should be considered when there are > 10 bilateral renal cysts present in the absence of other renal or extra-renal disease that can cause renal cysts[108]. When PCLD or ADPKD criteria are not met, multiple simple cysts are most likely responsible for the hepatic cystic lesions.
Table 3.
Ultrasonography criteria for the diagnosis of autosomal dominant polycystic kidney disease
| Family history positive1 | ||
| Unknown genotype | ||
| Age (yr) | ||
| ≥ 15 and ≤ 39 | ≥ 3 unilateral renal cysts | |
| ≥ 40 and ≤ 59 | ≥ 2 bilateral renal cysts | |
| ≥ 60 | ≥ 4 bilateral renal cysts | |
| Family history negative | ||
| > 10 bilateral renal cysts, with the exclusion of renal or extra-renal disease causing renal cysts | ||
Exclude autosomal dominant polycystic kidney disease when < 2 unilateral renal cysts and ≥ 40 years of age.
ADPKD is characterised by an increased risk of developing vascular manifestations. Hypertension occurs in approximately 50%-70% of patients, and almost half of these hypertensive patients are reported to have left ventricular hypertrophy (LVH)[112]. Mitral valve prolapse is observed in 25% of patients and intracranial aneurysms in 4%-12% of patients[112]. As a result, magnetic resonance angiography (MRA) must be performed when ADPKD patients have a positive family history of intracranial aneurysms because the rupture of aneurysms is reported to be responsible for 4%-7% of deaths in affected ADPKD families[113]. In contrast to ADPKD, several studies have shown that PCLD patients do not appear to have an increased risk of vascular malformations. One study involving 19 PCLD cases reported hypertension in 10.5% of cases, mitral valve prolapse in 0% and aneurysms in 5.3%[79]. Another study involving 38 PCLD cases found mitral valve prolapse in 1 case (2.6%)[114]. Subsequently, targeted screening is not advised for PCLD.
Therapy
The main objective of therapy is to reduce liver cyst volume to diminish mass effect-related symptoms[115]. Hence, the only indication for reducing cyst volume is when a PLD patient reports symptoms that can be linked to the polycystic liver[116].
Surgical procedures, such as aspiration-sclerotherapy and fenestration, are indicated when PLD consists of large cysts confined to a limited part of the liver. In more extensive disease, segmental hepatic resection or even liver transplantation is imperative to relieve symptoms[117]. Future medical therapies include somatostatin analogues, as several clinical trials with lanreotide and octreotide achieved polycystic liver volume reduction in PCLD and ADPKD[118-123].
CONCLUSION
Cystic lesions of the liver encompass a wide spectrum of disorders. As a result of the frequent use of abdominal imaging techniques in recent years, the incidence of so-called coincidental cysts has increased. Simple cysts are the most prevalent and have a tendency to follow a benign course. However, complicated cysts, echinococcosis and cystic neoplasms (e.g., cystadenoma and cystadenocarcinoma), which cause a diagnostic enigma, demand accurate diagnosis in the early stage because specific treatment could be required. Furthermore, the presence of multiple hepatic cystic lesions must raise the suspicion of PCLD or ADPKD and requires further screening.
USG remains the most accurate, non-invasive and cost-effective imaging modality for diagnosing simple cysts. Despite recent advances (e.g., contrast-enhanced CT and DWI), distinguishing complicated cysts from echinococcosis and cystic neoplasms remains impossible with USG, CT or MRI alone. Because of an ever-increasing spread of Echinococcus to previously non-endemic regions and its initial quiescent phase after primary infection, it is necessary to exclude echinococcosis. Serodiagnostic tests have high sensitivity and specificity to reveal Echinococcus antibodies. Subsequently, CEUS can be used to accurately and reliably exclude cystic neoplasms by demonstrating the absence of any enhancement within the hepatic cystic lesion. Therefore, when CEUS is available, it reduces the need for surgical resection.
The detection of multiple liver cysts requires USG screening of both kidneys and extensive family history taking regarding the occurrence of ADPKD or PCLD. When PCLD or ADPKD criteria are not met, multiple simple cysts are most likely responsible for the hepatic cystic lesions. PCLD or ADPKD could eventually be diagnosed through USG follow-up.
To summarise, we developed a diagnostic algorithm by integrating recent advances with conventional diagnostic tools (Figure 4). Our diagnostic algorithm facilitates evidence-based clinical decision making when clinicians are confronted with coincidental hepatic cystic lesions on USG. Further development of USG- and MRI-based techniques, such as CEUS and DWI, will probably lead to further improvement of hepatic cystic lesion characterisation.
Figure 4.
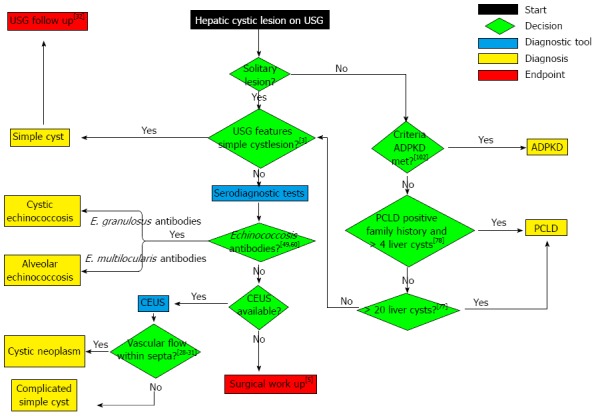
Diagnostic algorithm. Diagnosis of hepatic cystic lesions after detection on ultrasonography. E. granulosus: Echinococcus granulosus; E. multilocularis: Echinococcus multilocularis; CEUS: Contrast-enhanced ultrasound; PCLD: Polycystic liver disease; ADPKD: Autosomal dominant polycystic kidney disease.
ACKNOWLEDGMENTS
The authors wish to thank Melissa Chrispijn from the Department of Gastroenterology and Hepatology Radboud University Nijmegen Medical Center, the Netherlands, for her expert advice.
Footnotes
P- Reviewers de Oliveira C, Ramsay M, Silva ACS S- Editor Wen LL L- Editor A E- Editor Zhang DN
References
- 1.Cowles RA, Mulholland MW. Solitary hepatic cysts. J Am Coll Surg. 2000;191:311–321. doi: 10.1016/s1072-7515(00)00345-8. [DOI] [PubMed] [Google Scholar]
- 2.Del Poggio P, Buonocore M. Cystic tumors of the liver: a practical approach. World J Gastroenterol. 2008;14:3616–3620. doi: 10.3748/wjg.14.3616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bahirwani R, Reddy KR. Review article: the evaluation of solitary liver masses. Aliment Pharmacol Ther. 2008;28:953–965. doi: 10.1111/j.1365-2036.2008.03805.x. [DOI] [PubMed] [Google Scholar]
- 4.Choi BY, Nguyen MH. The diagnosis and management of benign hepatic tumors. J Clin Gastroenterol. 2005;39:401–412. doi: 10.1097/01.mcg.0000159226.63037.a2. [DOI] [PubMed] [Google Scholar]
- 5.Läuffer JM, Baer HU, Maurer CA, Stoupis C, Zimmerman A, Büchler MW. Biliary cystadenocarcinoma of the liver: the need for complete resection. Eur J Cancer. 1998;34:1845–1851. doi: 10.1016/s0959-8049(98)00166-x. [DOI] [PubMed] [Google Scholar]
- 6.Nunnari G, Pinzone MR, Gruttadauria S, Celesia BM, Madeddu G, Malaguarnera G, Pavone P, Cappellani A, Cacopardo B. Hepatic echinococcosis: clinical and therapeutic aspects. World J Gastroenterol. 2012;18:1448–1458. doi: 10.3748/wjg.v18.i13.1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eckert J, Deplazes P. Alveolar echinococcosis in humans: the current situation in Central Europe and the need for countermeasures. Parasitol Today. 1999;15:315–319. doi: 10.1016/s0169-4758(99)01476-3. [DOI] [PubMed] [Google Scholar]
- 8.Romig T, Dinkel A, Mackenstedt U. The present situation of echinococcosis in Europe. Parasitol Int. 2006;55 Suppl:S187–S191. doi: 10.1016/j.parint.2005.11.028. [DOI] [PubMed] [Google Scholar]
- 9.Sanfelippo PM, Beahrs OH, Weiland LH. Cystic disease of the liver. Ann Surg. 1974;179:922–925. doi: 10.1097/00000658-197406000-00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jones WL, Mountain JC, Warren KW. Symptomatic non-parasitic cysts of the liver. Br J Surg. 1974;61:118–123. doi: 10.1002/bjs.1800610211. [DOI] [PubMed] [Google Scholar]
- 11.Gaines PA, Sampson MA. The prevalence and characterization of simple hepatic cysts by ultrasound examination. Br J Radiol. 1989;62:335–337. doi: 10.1259/0007-1285-62-736-335. [DOI] [PubMed] [Google Scholar]
- 12.Carrim ZI, Murchison JT. The prevalence of simple renal and hepatic cysts detected by spiral computed tomography. Clin Radiol. 2003;58:626–629. doi: 10.1016/s0009-9260(03)00165-x. [DOI] [PubMed] [Google Scholar]
- 13.Hanazaki K, Wakabayashi M, Mori H, Sodeyama H, Yoshizawa K, Yokoyama S, Sode Y, Kawamura N, Miyazaki T. Hemorrhage into a simple liver cyst: diagnostic implications of a recent case. J Gastroenterol. 1997;32:848–851. doi: 10.1007/BF02936967. [DOI] [PubMed] [Google Scholar]
- 14.Salemis NS, Georgoulis E, Gourgiotis S, Tsohataridis E. Spontaneous rupture of a giant non parasitic hepatic cyst presenting as an acute surgical abdomen. Ann Hepatol. 2007;6:190–193. [PubMed] [Google Scholar]
- 15.Zhang YL, Yuan L, Shen F, Wang Y. Hemorrhagic hepatic cysts mimicking biliary cystadenoma. World J Gastroenterol. 2009;15:4601–4603. doi: 10.3748/wjg.15.4601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kitajima Y, Okayama Y, Hirai M, Hayashi K, Imai H, Okamoto T, Aoki S, Akita S, Gotoh K, Ohara H, et al. Intracystic hemorrhage of a simple liver cyst mimicking a biliary cystadenocarcinoma. J Gastroenterol. 2003;38:190–193. doi: 10.1007/s005350300032. [DOI] [PubMed] [Google Scholar]
- 17.Waanders E, van Keimpema L, Brouwer JT, van Oijen MG, Aerts R, Sweep FC, Nevens F, Drenth JP. Carbohydrate antigen 19-9 is extremely elevated in polycystic liver disease. Liver Int. 2009;29:1389–1395. doi: 10.1111/j.1478-3231.2009.02055.x. [DOI] [PubMed] [Google Scholar]
- 18.Choi HK, Lee JK, Lee KH, Lee KT, Rhee JC, Kim KH, Jang KT, Kim SH, Park Y. Differential diagnosis for intrahepatic biliary cystadenoma and hepatic simple cyst: significance of cystic fluid analysis and radiologic findings. J Clin Gastroenterol. 2010;44:289–293. doi: 10.1097/MCG.0b013e3181b5c789. [DOI] [PubMed] [Google Scholar]
- 19.Seo JK, Kim SH, Lee SH, Park JK, Woo SM, Jeong JB, Hwang JH, Ryu JK, Kim JW, Jeong SH, et al. Appropriate diagnosis of biliary cystic tumors: comparison with atypical hepatic simple cysts. Eur J Gastroenterol Hepatol. 2010;22:989–996. doi: 10.1097/MEG.0b013e328337c971. [DOI] [PubMed] [Google Scholar]
- 20.Spiegel RM, King DL, Green WM. Ultrasonography of primary cysts of the liver. AJR Am J Roentgenol. 1978;131:235–238. doi: 10.2214/ajr.131.2.235. [DOI] [PubMed] [Google Scholar]
- 21.Vachha B, Sun MR, Siewert B, Eisenberg RL. Cystic lesions of the liver. AJR Am J Roentgenol. 2011;196:W355–W366. doi: 10.2214/AJR.10.5292. [DOI] [PubMed] [Google Scholar]
- 22.Albiin N. MRI of Focal Liver Lesions. Curr Med Imaging Rev. 2012;8:107–116. doi: 10.2174/157340512800672216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Taylor KJ, Richman TS. Diseases of the liver. Semin Roentgenol. 1983;18:94–101. doi: 10.1016/0037-198x(83)90008-1. [DOI] [PubMed] [Google Scholar]
- 24.Hwang SH, Yu JS, Chung JJ, Kim JH, Kim KW. Diagnosing small hepatic cysts on multidetector CT: an additional merit of thinner coronal reformations. Korean J Radiol. 2011;12:341–350. doi: 10.3348/kjr.2011.12.3.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hagiwara A, Inoue Y, Shutoh T, Kinoshita H, Wakasa K. Haemorrhagic hepatic cyst: a differential diagnosis of cystic tumour. Br J Radiol. 2001;74:270–272. doi: 10.1259/bjr.74.879.740270. [DOI] [PubMed] [Google Scholar]
- 26.Yamaguchi M, Kuzume M, Matsumoto T, Matsumiya A, Nakano H, Kumada K. Spontaneous rupture of a nonparasitic liver cyst complicated by intracystic hemorrhage. J Gastroenterol. 1999;34:645–648. doi: 10.1007/s005350050388. [DOI] [PubMed] [Google Scholar]
- 27.Vilgrain V, Silbermann O, Benhamou JP, Nahum H. MR imaging in intracystic hemorrhage of simple hepatic cysts. Abdom Imaging. 1993;18:164–167. doi: 10.1007/BF00198056. [DOI] [PubMed] [Google Scholar]
- 28.Kim TK, Jang HJ, Wilson SR. Benign liver masses: imaging with microbubble contrast agents. Ultrasound Q. 2006;22:31–39. [PubMed] [Google Scholar]
- 29.Jang HJ, Yu H, Kim TK. Contrast-enhanced ultrasound in the detection and characterization of liver tumors. Cancer Imaging. 2009;9:96–103. doi: 10.1102/1470-7330.2009.0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sutherland T, Temple F, Lee WK, Hennessy O. Evaluation of focal hepatic lesions with ultrasound contrast agents. J Clin Ultrasound. 2011;39:399–407. doi: 10.1002/jcu.20847. [DOI] [PubMed] [Google Scholar]
- 31.Piscaglia F, Lencioni R, Sagrini E, Pina CD, Cioni D, Vidili G, Bolondi L. Characterization of focal liver lesions with contrast-enhanced ultrasound. Ultrasound Med Biol. 2010;36:531–550. doi: 10.1016/j.ultrasmedbio.2010.01.004. [DOI] [PubMed] [Google Scholar]
- 32.van Keimpema L, de Koning DB, Strijk SP, Drenth JP. Aspiration-sclerotherapy results in effective control of liver volume in patients with liver cysts. Dig Dis Sci. 2008;53:2251–2257. doi: 10.1007/s10620-007-0121-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moorthy K, Mihssin N, Houghton PW. The management of simple hepatic cysts: sclerotherapy or laparoscopic fenestration. Ann R Coll Surg Engl. 2001;83:409–414. [PMC free article] [PubMed] [Google Scholar]
- 34.Fiamingo P, Tedeschi U, Veroux M, Cillo U, Brolese A, Da Rold A, Madia C, Zanus G, D’Amico DF. Laparoscopic treatment of simple hepatic cysts and polycystic liver disease. Surg Endosc. 2003;17:623–626. doi: 10.1007/s00464-002-9088-z. [DOI] [PubMed] [Google Scholar]
- 35.Hansman MF, Ryan JA, Holmes JH, Hogan S, Lee FT, Kramer D, Biehl T. Management and long-term follow-up of hepatic cysts. Am J Surg. 2001;181:404–410. doi: 10.1016/s0002-9610(01)00611-0. [DOI] [PubMed] [Google Scholar]
- 36.Gigot JF, Legrand M, Hubens G, de Canniere L, Wibin E, Deweer F, Druart ML, Bertrand C, Devriendt H, Droissart R, et al. Laparoscopic treatment of nonparasitic liver cysts: adequate selection of patients and surgical technique. World J Surg. 1996;20:556–561. doi: 10.1007/s002689900086. [DOI] [PubMed] [Google Scholar]
- 37.Bristow BN, Lee S, Shafir S, Sorvillo F. Human echinococcosis mortality in the United States, 1990-2007. PLoS Negl Trop Dis. 2012;6:e1524. doi: 10.1371/journal.pntd.0001524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Budke CM, Deplazes P, Torgerson PR. Global socioeconomic impact of cystic echinococcosis. Emerg Infect Dis. 2006;12:296–303. doi: 10.3201/eid1202.050499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dakkak A. Echinococcosis/hydatidosis: a severe threat in Mediterranean countries. Vet Parasitol. 2010;174:2–11. doi: 10.1016/j.vetpar.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 40.Torgerson PR, Keller K, Magnotta M, Ragland N. The global burden of alveolar echinococcosis. PLoS Negl Trop Dis. 2010;4:e722. doi: 10.1371/journal.pntd.0000722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Eckert J, Conraths FJ, Tackmann K. Echinococcosis: an emerging or re-emerging zoonosis? Int J Parasitol. 2000;30:1283–1294. doi: 10.1016/s0020-7519(00)00130-2. [DOI] [PubMed] [Google Scholar]
- 42.Grosso G, Gruttadauria S, Biondi A, Marventano S, Mistretta A. Worldwide epidemiology of liver hydatidosis including the Mediterranean area. World J Gastroenterol. 2012;18:1425–1437. doi: 10.3748/wjg.v18.i13.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Todorov T, Boeva V. Human echinococcosis in Bulgaria: a comparative epidemiological analysis. Bull World Health Organ. 1999;77:110–118. [PMC free article] [PubMed] [Google Scholar]
- 44.Mandal S, Mandal MD. Human cystic echinococcosis: epidemiologic, zoonotic, clinical, diagnostic and therapeutic aspects. Asian Pac J Trop Med. 2012;5:253–260. doi: 10.1016/S1995-7645(12)60035-2. [DOI] [PubMed] [Google Scholar]
- 45.McManus DP, Zhang W, Li J, Bartley PB. Echinococcosis. Lancet. 2003;362:1295–1304. doi: 10.1016/S0140-6736(03)14573-4. [DOI] [PubMed] [Google Scholar]
- 46.Craig PS, Larrieu E. Control of cystic echinococcosis/hydatidosis: 1863-2002. Adv Parasitol. 2006;61:443–508. doi: 10.1016/S0065-308X(05)61011-1. [DOI] [PubMed] [Google Scholar]
- 47.Eckert J. WHO/OIE manual on echinococcosis in humans and animals: a public health problem of global concern. Paris: World Organisation for Animal Health; 2001. pp. 20–72. [Google Scholar]
- 48.Sayek I, Onat D. Diagnosis and treatment of uncomplicated hydatid cyst of the liver. World J Surg. 2001;25:21–27. doi: 10.1007/s002680020004. [DOI] [PubMed] [Google Scholar]
- 49.Sbihi Y, Rmiqui A, Rodriguez-Cabezas MN, Orduña A, Rodriguez-Torres A, Osuna A. Comparative sensitivity of six serological tests and diagnostic value of ELISA using purified antigen in hydatidosis. J Clin Lab Anal. 2001;15:14–18. doi: 10.1002/1098-2825(2001)15:1<14::AID-JCLA3>3.0.CO;2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brunetti E, Junghanss T. Update on cystic hydatid disease. Curr Opin Infect Dis. 2009;22:497–502. doi: 10.1097/QCO.0b013e328330331c. [DOI] [PubMed] [Google Scholar]
- 51.Buttenschoen K, Carli Buttenschoen D. Echinococcus granulosus infection: the challenge of surgical treatment. Langenbecks Arch Surg. 2003;388:218–230. doi: 10.1007/s00423-003-0397-z. [DOI] [PubMed] [Google Scholar]
- 52.Smego RA, Sebanego P. Treatment options for hepatic cystic echinococcosis. Int J Infect Dis. 2005;9:69–76. doi: 10.1016/j.ijid.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 53.Eckert J, Deplazes P. Biological, epidemiological, and clinical aspects of echinococcosis, a zoonosis of increasing concern. Clin Microbiol Rev. 2004;17:107–135. doi: 10.1128/CMR.17.1.107-135.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Moro P, Schantz PM. Echinococcosis: a review. Int J Infect Dis. 2009;13:125–133. doi: 10.1016/j.ijid.2008.03.037. [DOI] [PubMed] [Google Scholar]
- 55.Kern P. Clinical features and treatment of alveolar echinococcosis. Curr Opin Infect Dis. 2010;23:505–512. doi: 10.1097/QCO.0b013e32833d7516. [DOI] [PubMed] [Google Scholar]
- 56.Takci E, Sengul G, Akar A, Uslu H, Alper F, Erdogan F, Aydin IH. Alveolar echinococcosis of the brain in five patients. J Clin Neurosci. 2008;15:1105–1109. doi: 10.1016/j.jocn.2007.09.020. [DOI] [PubMed] [Google Scholar]
- 57.Bresson-Hadni S, Delabrousse E, Blagosklonov O, Bartholomot B, Koch S, Miguet JP, André Mantion G, Angèle Vuitton D. Imaging aspects and non-surgical interventional treatment in human alveolar echinococcosis. Parasitol Int. 2006;55 Suppl:S267–S272. doi: 10.1016/j.parint.2005.11.053. [DOI] [PubMed] [Google Scholar]
- 58.Kodama Y, Fujita N, Shimizu T, Endo H, Nambu T, Sato N, Todo S, Miyasaka K. Alveolar echinococcosis: MR findings in the liver. Radiology. 2003;228:172–177. doi: 10.1148/radiol.2281020323. [DOI] [PubMed] [Google Scholar]
- 59.Harman M, Arslan H, Kotan C, Etlik O, Kayan M, Deveci A. MRI findings of hepatic alveolar echinococcosis. Clin Imaging. 2003;27:411–416. doi: 10.1016/s0899-7071(03)00006-8. [DOI] [PubMed] [Google Scholar]
- 60.Brunetti E, Kern P, Vuitton DA. Expert consensus for the diagnosis and treatment of cystic and alveolar echinococcosis in humans. Acta Trop. 2010;114:1–16. doi: 10.1016/j.actatropica.2009.11.001. [DOI] [PubMed] [Google Scholar]
- 61.Buttenschoen K, Carli Buttenschoen D, Gruener B, Kern P, Beger HG, Henne-Bruns D, Reuter S. Long-term experience on surgical treatment of alveolar echinococcosis. Langenbecks Arch Surg. 2009;394:689–698. doi: 10.1007/s00423-008-0392-5. [DOI] [PubMed] [Google Scholar]
- 62.Delis SG, Touloumis Z, Bakoyiannis A, Tassopoulos N, Paraskeva K, Athanassiou K, Safioleas M, Dervenis C. Intrahepatic biliary cystadenoma: a need for radical resection. Eur J Gastroenterol Hepatol. 2008;20:10–14. doi: 10.1097/MEG.0b013e3282f16a76. [DOI] [PubMed] [Google Scholar]
- 63.Hai S, Hirohashi K, Uenishi T, Yamamoto T, Shuto T, Tanaka H, Kubo S, Tanaka S, Kinoshita H. Surgical management of cystic hepatic neoplasms. J Gastroenterol. 2003;38:759–764. doi: 10.1007/s00535-003-1142-7. [DOI] [PubMed] [Google Scholar]
- 64.Wheeler DA, Edmondson HA. Cystadenoma with mesenchymal stroma (CMS) in the liver and bile ducts. A clinicopathologic study of 17 cases, 4 with malignant change. Cancer. 1985;56:1434–1445. doi: 10.1002/1097-0142(19850915)56:6<1434::aid-cncr2820560635>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 65.Devaney K, Goodman ZD, Ishak KG. Hepatobiliary cystadenoma and cystadenocarcinoma. A light microscopic and immunohistochemical study of 70 patients. Am J Surg Pathol. 1994;18:1078–1091. [PubMed] [Google Scholar]
- 66.Hernandez Bartolome MA, Fuerte Ruiz S, Manzanedo Romero I, Ramos Lojo B, Rodriguez Prieto I, Gimenez Alvira L, Granados Carreño R, Limones Esteban M. Biliary cystadenoma. World J Gastroenterol. 2009;15:3573–3575. doi: 10.3748/wjg.15.3573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yi B, Cheng QB, Jiang XQ, Liu C, Luo XJ, Dong H, Zhang BH, Wu MC. A special growth manner of intrahepatic biliary cystadenoma. World J Gastroenterol. 2009;15:6134–6136. doi: 10.3748/wjg.15.6134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kim HG. [Biliary cystic neoplasm: biliary cystadenoma and biliary cystadenocarcinoma] Korean J Gastroenterol. 2006;47:5–14. [PubMed] [Google Scholar]
- 69.Ahanatha Pillai S, Velayutham V, Perumal S, Ulagendra Perumal S, Lakshmanan A, Ramaswami S, Ramasamy R, Sathyanesan J, Palaniappan R, Rajagopal S. Biliary cystadenomas: a case for complete resection. HPB Surg. 2012;2012:501705. doi: 10.1155/2012/501705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Teoh AY, Ng SS, Lee KF, Lai PB. Biliary cystadenoma and other complicated cystic lesions of the liver: diagnostic and therapeutic challenges. World J Surg. 2006;30:1560–1566. doi: 10.1007/s00268-005-0461-7. [DOI] [PubMed] [Google Scholar]
- 71.Taouli B, Koh DM. Diffusion-weighted MR imaging of the liver. Radiology. 2010;254:47–66. doi: 10.1148/radiol.09090021. [DOI] [PubMed] [Google Scholar]
- 72.Yamada I, Aung W, Himeno Y, Nakagawa T, Shibuya H. Diffusion coefficients in abdominal organs and hepatic lesions: evaluation with intravoxel incoherent motion echo-planar MR imaging. Radiology. 1999;210:617–623. doi: 10.1148/radiology.210.3.r99fe17617. [DOI] [PubMed] [Google Scholar]
- 73.Inan N, Arslan A, Akansel G, Anik Y, Sarisoy HT, Ciftci E, Demirci A. Diffusion-weighted imaging in the differential diagnosis of simple and hydatid cysts of the liver. AJR Am J Roentgenol. 2007;189:1031–1036. doi: 10.2214/AJR.07.2251. [DOI] [PubMed] [Google Scholar]
- 74.Fowler KJ, Brown JJ, Narra VR. Magnetic resonance imaging of focal liver lesions: approach to imaging diagnosis. Hepatology. 2011;54:2227–2237. doi: 10.1002/hep.24679. [DOI] [PubMed] [Google Scholar]
- 75.Kele PG, van der Jagt EJ. Diffusion weighted imaging in the liver. World J Gastroenterol. 2010;16:1567–1576. doi: 10.3748/wjg.v16.i13.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pinto MM, Kaye AD. Fine needle aspiration of cystic liver lesions. Cytologic examination and carcinoembryonic antigen assay of cyst contents. Acta Cytol. 1989;33:852–856. [PubMed] [Google Scholar]
- 77.Van Keimpema L, De Koning DB, Van Hoek B, Van Den Berg AP, Van Oijen MG, De Man RA, Nevens F, Drenth JP. Patients with isolated polycystic liver disease referred to liver centres: clinical characterization of 137 cases. Liver Int. 2011;31:92–98. doi: 10.1111/j.1478-3231.2010.02247.x. [DOI] [PubMed] [Google Scholar]
- 78.Drenth JP, Chrispijn M, Bergmann C. Congenital fibrocystic liver diseases. Best Pract Res Clin Gastroenterol. 2010;24:573–584. doi: 10.1016/j.bpg.2010.08.007. [DOI] [PubMed] [Google Scholar]
- 79.Hoevenaren IA, Wester R, Schrier RW, McFann K, Doctor RB, Drenth JP, Everson GT. Polycystic liver: clinical characteristics of patients with isolated polycystic liver disease compared with patients with polycystic liver and autosomal dominant polycystic kidney disease. Liver Int. 2008;28:264–270. doi: 10.1111/j.1478-3231.2007.01595.x. [DOI] [PubMed] [Google Scholar]
- 80.Brancatelli G, Federle MP, Vilgrain V, Vullierme MP, Marin D, Lagalla R. Fibropolycystic liver disease: CT and MR imaging findings. Radiographics. 2005;25:659–670. doi: 10.1148/rg.253045114. [DOI] [PubMed] [Google Scholar]
- 81.Roskams T, Desmet V. Embryology of extra- and intrahepatic bile ducts, the ductal plate. Anat Rec (Hoboken) 2008;291:628–635. doi: 10.1002/ar.20710. [DOI] [PubMed] [Google Scholar]
- 82.Desmet VJ. Ludwig symposium on biliary disorders--part I. Pathogenesis of ductal plate abnormalities. Mayo Clin Proc. 1998;73:80–89. doi: 10.4065/73.1.80. [DOI] [PubMed] [Google Scholar]
- 83.Patterson M, Gonzalez-Vitale JC, Fagan CJ. Polycystic liver disease: a study of cyst fluid constituents. Hepatology. 1982;2:475–478. doi: 10.1002/hep.1840020414. [DOI] [PubMed] [Google Scholar]
- 84.Torres VE, Watson ML. Polycystic kidney disease: antiquity to the 20th century. Nephrol Dial Transplant. 1998;13:2690–2696. doi: 10.1093/ndt/13.10.2690. [DOI] [PubMed] [Google Scholar]
- 85.Karhunen PJ, Tenhu M. Adult polycystic liver and kidney diseases are separate entities. Clin Genet. 1986;30:29–37. doi: 10.1111/j.1399-0004.1986.tb00565.x. [DOI] [PubMed] [Google Scholar]
- 86.Pirson Y, Lannoy N, Peters D, Geubel A, Gigot JF, Breuning M, Verellen-Dumoulin C. Isolated polycystic liver disease as a distinct genetic disease, unlinked to polycystic kidney disease 1 and polycystic kidney disease 2. Hepatology. 1996;23:249–252. doi: 10.1002/hep.510230208. [DOI] [PubMed] [Google Scholar]
- 87.Harris PC, Torres VE. Polycystic kidney disease. Annu Rev Med. 2009;60:321–337. doi: 10.1146/annurev.med.60.101707.125712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Davila S, Furu L, Gharavi AG, Tian X, Onoe T, Qian Q, Li A, Cai Y, Kamath PS, King BF, et al. Mutations in SEC63 cause autosomal dominant polycystic liver disease. Nat Genet. 2004;36:575–577. doi: 10.1038/ng1357. [DOI] [PubMed] [Google Scholar]
- 89.Drenth JP, te Morsche RH, Smink R, Bonifacino JS, Jansen JB. Germline mutations in PRKCSH are associated with autosomal dominant polycystic liver disease. Nat Genet. 2003;33:345–347. doi: 10.1038/ng1104. [DOI] [PubMed] [Google Scholar]
- 90.Li A, Davila S, Furu L, Qian Q, Tian X, Kamath PS, King BF, Torres VE, Somlo S. Mutations in PRKCSH cause isolated autosomal dominant polycystic liver disease. Am J Hum Genet. 2003;72:691–703. doi: 10.1086/368295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Waanders E, Venselaar H, te Morsche RH, de Koning DB, Kamath PS, Torres VE, Somlo S, Drenth JP. Secondary and tertiary structure modeling reveals effects of novel mutations in polycystic liver disease genes PRKCSH and SEC63. Clin Genet. 2010;78:47–56. doi: 10.1111/j.1399-0004.2009.01353.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Rossetti S, Consugar MB, Chapman AB, Torres VE, Guay-Woodford LM, Grantham JJ, Bennett WM, Meyers CM, Walker DL, Bae K, et al. Comprehensive molecular diagnostics in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2007;18:2143–2160. doi: 10.1681/ASN.2006121387. [DOI] [PubMed] [Google Scholar]
- 93.Janssen MJ, Waanders E, Woudenberg J, Lefeber DJ, Drenth JP. Congenital disorders of glycosylation in hepatology: the example of polycystic liver disease. J Hepatol. 2010;52:432–440. doi: 10.1016/j.jhep.2009.12.011. [DOI] [PubMed] [Google Scholar]
- 94.Yoder BK, Mulroy S, Eustace H, Boucher C, Sandford R. Molecular pathogenesis of autosomal dominant polycystic kidney disease. Expert Rev Mol Med. 2006;8:1–22. doi: 10.1017/S1462399406010362. [DOI] [PubMed] [Google Scholar]
- 95.Qian Q. Isolated polycystic liver disease. Adv Chronic Kidney Dis. 2010;17:181–189. doi: 10.1053/j.ackd.2009.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Torres VE, Harris PC. Autosomal dominant polycystic kidney disease: the last 3 years. Kidney Int. 2009;76:149–168. doi: 10.1038/ki.2009.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Perrone RD. Extrarenal manifestations of ADPKD. Kidney Int. 1997;51:2022–2036. doi: 10.1038/ki.1997.276. [DOI] [PubMed] [Google Scholar]
- 98.Harris PC, Bae KT, Rossetti S, Torres VE, Grantham JJ, Chapman AB, Guay-Woodford LM, King BF, Wetzel LH, Baumgarten DA, et al. Cyst number but not the rate of cystic growth is associated with the mutated gene in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2006;17:3013–3019. doi: 10.1681/ASN.2006080835. [DOI] [PubMed] [Google Scholar]
- 99.Qian Q, Li A, King BF, Kamath PS, Lager DJ, Huston J, Shub C, Davila S, Somlo S, Torres VE. Clinical profile of autosomal dominant polycystic liver disease. Hepatology. 2003;37:164–171. doi: 10.1053/jhep.2003.50006. [DOI] [PubMed] [Google Scholar]
- 100.Pirson Y. Extrarenal manifestations of autosomal dominant polycystic kidney disease. Adv Chronic Kidney Dis. 2010;17:173–180. doi: 10.1053/j.ackd.2010.01.003. [DOI] [PubMed] [Google Scholar]
- 101.Bae KT, Zhu F, Chapman AB, Torres VE, Grantham JJ, Guay-Woodford LM, Baumgarten DA, King BF, Wetzel LH, Kenney PJ, et al. Magnetic resonance imaging evaluation of hepatic cysts in early autosomal-dominant polycystic kidney disease: the Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease cohort. Clin J Am Soc Nephrol. 2006;1:64–69. doi: 10.2215/CJN.00080605. [DOI] [PubMed] [Google Scholar]
- 102.Pei Y, Watnick T. Diagnosis and screening of autosomal dominant polycystic kidney disease. Adv Chronic Kidney Dis. 2010;17:140–152. doi: 10.1053/j.ackd.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Alvaro D, Mancino MG, Onori P, Franchitto A, Alpini G, Francis H, Glaser S, Gaudio E. Estrogens and the pathophysiology of the biliary tree. World J Gastroenterol. 2006;12:3537–3545. doi: 10.3748/wjg.v12.i22.3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hateboer N, v Dijk MA, Bogdanova N, Coto E, Saggar-Malik AK, San Millan JL, Torra R, Breuning M, Ravine D. Comparison of phenotypes of polycystic kidney disease types 1 and 2. European PKD1-PKD2 Study Group. Lancet. 1999;353:103–107. doi: 10.1016/s0140-6736(98)03495-3. [DOI] [PubMed] [Google Scholar]
- 105.Gabow PA, Johnson AM, Kaehny WD, Manco-Johnson ML, Duley IT, Everson GT. Risk factors for the development of hepatic cysts in autosomal dominant polycystic kidney disease. Hepatology. 1990;11:1033–1037. doi: 10.1002/hep.1840110619. [DOI] [PubMed] [Google Scholar]
- 106.Torres VE, Harris PC, Pirson Y. Autosomal dominant polycystic kidney disease. Lancet. 2007;369:1287–1301. doi: 10.1016/S0140-6736(07)60601-1. [DOI] [PubMed] [Google Scholar]
- 107.Que F, Nagorney DM, Gross JB, Torres VE. Liver resection and cyst fenestration in the treatment of severe polycystic liver disease. Gastroenterology. 1995;108:487–494. doi: 10.1016/0016-5085(95)90078-0. [DOI] [PubMed] [Google Scholar]
- 108.Belibi FA, Edelstein CL. Unified ultrasonographic diagnostic criteria for polycystic kidney disease. J Am Soc Nephrol. 2009;20:6–8. doi: 10.1681/ASN.2008111164. [DOI] [PubMed] [Google Scholar]
- 109.Nicolau C, Torra R, Badenas C, Vilana R, Bianchi L, Gilabert R, Darnell A, Brú C. Autosomal dominant polycystic kidney disease types 1 and 2: assessment of US sensitivity for diagnosis. Radiology. 1999;213:273–276. doi: 10.1148/radiology.213.1.r99oc05273. [DOI] [PubMed] [Google Scholar]
- 110.Pei Y, Obaji J, Dupuis A, Paterson AD, Magistroni R, Dicks E, Parfrey P, Cramer B, Coto E, Torra R, et al. Unified criteria for ultrasonographic diagnosis of ADPKD. J Am Soc Nephrol. 2009;20:205–212. doi: 10.1681/ASN.2008050507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ravine D, Gibson RN, Walker RG, Sheffield LJ, Kincaid-Smith P, Danks DM. Evaluation of ultrasonographic diagnostic criteria for autosomal dominant polycystic kidney disease 1. Lancet. 1994;343:824–827. doi: 10.1016/s0140-6736(94)92026-5. [DOI] [PubMed] [Google Scholar]
- 112.Ecder T, Schrier RW. Cardiovascular abnormalities in autosomal-dominant polycystic kidney disease. Nat Rev Nephrol. 2009;5:221–228. doi: 10.1038/nrneph.2009.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Schrier RW. Optimal care of autosomal dominant polycystic kidney disease patients. Nephrology (Carlton) 2006;11:124–130. doi: 10.1111/j.1440-1797.2006.00535.x. [DOI] [PubMed] [Google Scholar]
- 114.Gevers TJ, de Koning DB, van Dijk AP, Drenth JP. Low prevalence of cardiac valve abnormalities in patients with autosomal dominant polycystic liver disease. Liver Int. 2012;32:690–692. doi: 10.1111/j.1478-3231.2011.02683.x. [DOI] [PubMed] [Google Scholar]
- 115.Drenth JP, Chrispijn M, Nagorney DM, Kamath PS, Torres VE. Medical and surgical treatment options for polycystic liver disease. Hepatology. 2010;52:2223–2230. doi: 10.1002/hep.24036. [DOI] [PubMed] [Google Scholar]
- 116.Temmerman F, Missiaen L, Bammens B, Laleman W, Cassiman D, Verslype C, van Pelt J, Nevens F. Systematic review: the pathophysiology and management of polycystic liver disease. Aliment Pharmacol Ther. 2011;34:702–713. doi: 10.1111/j.1365-2036.2011.04783.x. [DOI] [PubMed] [Google Scholar]
- 117.Russell RT, Pinson CW. Surgical management of polycystic liver disease. World J Gastroenterol. 2007;13:5052–5059. doi: 10.3748/wjg.v13.i38.5052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Gevers TJ, Drenth JP. Somatostatin analogues for treatment of polycystic liver disease. Curr Opin Gastroenterol. 2011;27:294–300. doi: 10.1097/MOG.0b013e328343433f. [DOI] [PubMed] [Google Scholar]
- 119.van Keimpema L, Nevens F, Vanslembrouck R, van Oijen MG, Hoffmann AL, Dekker HM, de Man RA, Drenth JP. Lanreotide reduces the volume of polycystic liver: a randomized, double-blind, placebo-controlled trial. Gastroenterology. 2009;137:1661–8.e1-2. doi: 10.1053/j.gastro.2009.07.052. [DOI] [PubMed] [Google Scholar]
- 120.Caroli A, Antiga L, Cafaro M, Fasolini G, Remuzzi A, Remuzzi G, Ruggenenti P. Reducing polycystic liver volume in ADPKD: effects of somatostatin analogue octreotide. Clin J Am Soc Nephrol. 2010;5:783–789. doi: 10.2215/CJN.05380709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hogan MC, Masyuk TV, Page LJ, Kubly VJ, Bergstralh EJ, Li X, Kim B, King BF, Glockner J, Holmes DR, et al. Randomized clinical trial of long-acting somatostatin for autosomal dominant polycystic kidney and liver disease. J Am Soc Nephrol. 2010;21:1052–1061. doi: 10.1681/ASN.2009121291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hogan MC, Masyuk TV, Page L, Holmes DR, Li X, Bergstralh EJ, Irazabal MV, Kim B, King BF, Glockner JF, et al. Somatostatin analog therapy for severe polycystic liver disease: results after 2 years. Nephrol Dial Transplant. 2012;27:3532–3539. doi: 10.1093/ndt/gfs152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Chrispijn M, Drenth JP. Everolimus and long acting octreotide as a volume reducing treatment of polycystic livers (ELATE): study protocol for a randomized controlled trial. Trials. 2011;12:246. doi: 10.1186/1745-6215-12-246. [DOI] [PMC free article] [PubMed] [Google Scholar]