

Abstract
The RB1 gene is the first tumor suppressor gene identified whose mutational inactivation is the cause of a human cancer, the pediatric cancer retinoblastoma. The twenty five years of research since its discovery has not only illuminated a general role for RB1 in human cancer, but also its critical importance in normal development. Understanding the molecular function of the RB1 encoded protein, pRb, is a long-standing goal that promises to inform our understanding of cancer, its relationship to normal development, and possible therapeutic strategies to combat this disease. Achieving this goal has been difficult, complicated by the complexity of pRb and related proteins. The goal of this review is to explore the hypothesis that, at its core, the molecular function of pRb is to dynamically regulate the location specific assembly or disassembly of protein complexes on the DNA in response to the output of various signaling pathways. These protein complexes participate in a variety of molecular processes relevant to DNA including gene transcription, DNA replication, DNA repair, and mitosis. Through regulation of these processes, RB1 plays a uniquely prominent role in normal development and cancer.
Keywords: retinoblastoma, stem cells, chromatin, differentiation, epigenetics
Introduction
If there is one lesson to be learned from the preceding three decades of cancer research, it is that there are a large variety of genetic, epigenetic, and chromosomal changes that accumulate in cancer cells. If only a small number of these changes actually drive malignant behavior, the possible combinations of contributing molecular alterations are dizzying. This molecular heterogeneity is perhaps why even the most successful therapeutic regimens fail in a significant fraction of patients, bedeviling efforts to understand and effectively treat cancer. How can this molecular complexity be distilled into important general principles that would advance understanding and predict successful therapeutic strategies?
Confronted by complexity, scientists have traditionally sought refuge in simple model systems. Simple systems are easier to study, and the general principles gleaned are often relevant to the more complicated cases. In the world of human cancer research, perhaps the simplest “model” system is the pediatric eye cancer retinoblastoma. A subset of retinoblastoma cases are clearly hereditary, with susceptibility transmitted as a simple autosomal dominant trait. The remaining cases arise sporadically. Hereditary cases are diagnosed earlier and exhibit tumors in both eyes while sporadic cases are diagnosed later and typically only occur in one eye. Mathematical modeling of the time to diagnosis suggests hereditary retinoblastoma as a one hit phenomenon while sporadic retinoblastoma is a two hit phenomenon (Knudson, 1971). This now famous “two-hit hypothesis” suggests that genetic mutation of both alleles of a single gene is the cause (Comings, 1973). The molecular cloning of the Rb1 gene has verified the essential features of this hypothesis (Friend et al., 1986; Fung et al., 1987; Lee et al., 1987). Mutational inactivation of both Rb1 alleles is both necessary and rate limiting, but likely not sufficient, for the genesis of retinoblastoma. The molecular etiology of retinoblastoma, therefore, is perhaps the simplest among all human cancers. In addition to its causative role in retinoblastoma, deregulation of the molecular pathway in which Rb1 functions occurs with substantial frequency in virtually every other type of cancer where it has been examined (Hanahan and Weinberg, 2000; Sherr and McCormick, 2002). Thus the possibility of learning important general principles relevant to cancer from a detailed molecular understanding of Rb1 has long been its siren song.
The relative genetic simplicity of retinoblastoma, however, belies the significant functional complexity of the Rb1 encoded protein (pRb). The first cellular function described for pRb, and the most thoroughly studied, is as a negative regulator of the cell cycle (Goodrich et al., 1991). Loss of pRb-mediated cell cycle control is frequently observed in cancer (Malumbres and Barbacid, 2001; Weinberg, 1995). As cancer is a disease of abnormal cell proliferation, it makes intuitive sense that the key function underlying pRb mediated tumor suppression is cell cycle regulation. However, pRb loss also has profound effects on many other cellular processes relevant to cancer including differentiation (Korenjak et al., 2005; McClellan et al., 2007b; Nguyen and McCance, 2005), survival (Chau et al., 2003; Delston et al., 2006; Hallstrom et al., 2009), senescence (Ben-Porath et al., 2005; Liu et al., 2004), and genome stability (Knudsen et al., 2006) to name a few. This functional complexity is mirrored by the variety of molecular interactions involving pRb. Rb1 protein interacts with a large and steadily growing list of cellular proteins, and an even greater number of genes. Deriving satisfying general cancer principles from the study of pRb thus remains elusive.
The goal of this review is to explore the emerging hypothesis that the core molecular function of pRb is to dynamically regulate the location specific assembly or disassembly of protein complexes on the DNA. In essence pRb serves as an adaptor, physically linking sequence specific DNA binding proteins with other proteins that influence chromatin and DNA in various ways. The pRb protein interactions central to this function are regulated by post translational modification, and these modifications represent the output of different signaling pathways. Rb1 protein thus integrates the output of signaling pathways and translates them into genome location specific changes in protein complexes that influence a variety of molecular processes relevant to DNA including gene transcription, DNA replication, DNA repair, and mitosis. Through its regulation of these important processes, Rb1 plays a uniquely prominent role in normal development and cancer.
The pocket proteins
Rb1 is the founding member of a small gene family that also includes the Rbl1 (p107) and Rbl2 (p130) genes. Of these three family members, Rb1 is clearly the focus of attention because of its well documented function as a tumor suppressor gene. Despite the fact that they share partially overlapping functions, Rbl1 and Rbl2 are rarely mutated in human cancer. Most of the experiments we will review here are focused on Rb1. For more coverage of the similarities and differences among the different pocket proteins, the reader is referred to comprehensive reviews on the gene family (Classon and Dyson, 2001; Claudio et al., 2002).
The proteins encoded by these three genes are structurally related with their defining feature being the “pocket,” a structural domain required for their established cellular and molecular functions. The pocket is composed of two highly conserved and well ordered subdomains separated by a less structured spacer and followed by a similarly unstructured tail (Figure 1). The structure of the sub domains has been solved by x-ray crystallography (Kim et al., 2001; Kim and Cho, 1997; Lee, 2002; Lee et al., 1998; Liu and Marmorstein, 2007; Xiao et al., 2003). Each sub domain bears similarity to the cyclin box fold (Kim and Cho, 1997). Thus the overall structure of the pocket includes tandem cyclin box folds separated by a spacer and followed by an unstructured tail. Cyclin folds are well characterized structures involved in mediating protein-protein interactions. The unstructured carboxy terminal tail of pRb also contributes to intermolecular protein interactions and can make intramolecular contacts with the tandem cyclin fold (Dick and Dyson, 2003; Rubin et al., 2005; Welch and Wang, 1993). These intramolecular contacts compete for access to a tandem cyclin fold protein interaction surface, thereby regulating intermolecular protein interactions. Interestingly, a similar tandem cyclin fold structure is present in the amino terminal half of pRb (Hassler et al., 2007). It too mediates intermolecular interactions with cellular proteins as well as intramolecular interactions with the pocket tandem cyclin fold. The amino terminal tandem cyclin fold thus adds additional protein interaction surfaces through which pRb can function and be regulated.
Figure 1. Structural organization of the Rb1 encoded protein.
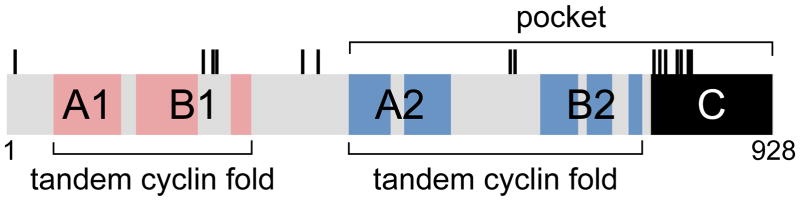
The schematic identifies major structural and functional elements including the “pocket,” the dual tandem cyclin folds (A1+B1, A2+B2), the unstructured tail (C), and threonine (T) and serine (S) residues whose phosphorylation regulates intermolecular and intramolecular protein interactions. The colored regions mark the positions of the five alpha helices that are characteristic of cyclin folds. Not shown are additional structural elements (i.e. alpha helices) that contribute to overall pRb structure. The schematic is approximately to scale.
Threading analysis suggests that all members of the Rb1 family have an analogous dual tandem cyclin fold structure (Hassler et al., 2007). However, it is clear that p107 and p130 (47% identity) are more closely related to each other than either one is to pRB (<21% identity). The greatest degree of similarity is within the pocket tandem cyclin fold (30–40% identity). The amino terminal tandem cyclin fold and the unstructured spacers and carboxy terminal tails are not as well conserved (10–20% identity). Differences in the dual tandem cyclin folds as well as the unstructured regions that make intramolecular contacts with the tandem cyclin folds conceivably could account for some of the functional differences between the pocket proteins. However, why pRb is often mutated in human cancer while p107 and p130 are not is an unresolved question.
It is clear from structure/function studies conducted over the last two decades that both the tandem cyclin folds and the unstructured tail that comprise the pocket are required for activity in cell culture based assays that have been used to define various aspects of pRb cellular function, including assays relevant to the cell cycle, differentiation, and apoptosis. The amino terminal tandem cyclin fold domain of pRb is much less studied, likely because it is generally not required for activity in such assays. However, there is evidence that the amino terminal tandem cyclin fold is required for normal function in vivo, although this conclusion is controversial (Goodrich, 2003). A small number of Rb1 missense point mutants have been identified from retinoblastoma patients that specifically affect the amino terminal tandem cyclin fold while sparing the carboxy terminal pocket. There are also a number of cellular proteins that interact specifically with the amino terminal tandem cyclin fold, some of which also play a role in functions regulated by pRb. It seems likely that additional amino terminal tandem cyclin fold interacting proteins will be discovered as more study is devoted to this domain. Finally, the amino terminal tandem cyclin fold is conserved among pocket protein orthologues in different species (Hassler et al., 2007).
The multiple protein interaction surfaces available within the overall pRb structure suggest it can interact with more than one protein at a time, making it ideally suited to serve as an adaptor. Function as an adaptor molecule is also in keeping with one of the defining characteristics of the pocket proteins, their ability to interact with a large variety of cellular and viral proteins (Goodrich, 2006; Morris and Dyson, 2001). Over 150 different pRb protein interactions have been documented in the literature to date, although the biological relevance of most of them has not been validated. The proteins that p107 and p130 interact with have not been well characterized. Nonetheless it is clear they can interact with some, but not all, of the proteins that interact with pRb. A significant majority of the proteins that interact with pRb are involved, in some way, in the regulation of RNA transcription and gene expression. Other protein interaction partners have roles in different processes involving DNA including DNA replication, chromosome condensation, and DNA repair. Thus it is likely that the pocket protein adaptor is a tool used in a number of biological contexts where location specific changes in protein complex assembly or activity are important.
The pRb/E2F cell cycle switch
The hypothesis that pRb functions as an adaptor is based on the paradigm of its well characterized interaction with the sequence specific DNA binding E2F transcription factors and the role of this complex in regulating cell proliferation. E2F1 is the founding member of the E2F gene family (Helin et al., 1992; Kaelin et al., 1992; Shan et al., 1992); the family is currently comprised of eight structurally related transcription factors, five of which (E2F1-5) interact with the pocket proteins (Chen et al., 2009a). E2Fs have traditionally been sub-classified as either transcriptional activators or repressors. Of the E2Fs that interact with pocket proteins, E2F1-3 are activators. They share a winged-helix DNA binding domain, a leucine zipper, a transcriptional activation domain, and marked box domains that specify binding to their obligate heterodimerization partners, the DP proteins (TFDP1-3). Within the transcriptional activation domain is a region required for pocket protein interaction. E2F1-3 proteins are most commonly found in complex with pRb, although p107 and p130 can bind E2F1-3 under certain conditions (Lee et al., 2002). E2F4-5 proteins are classified as transcriptional repressors. They have an overall structure similar to E2F1-3, but lack a nuclear localization signal and instead contain a nuclear export signal. Localization to the nucleus appears to depend on heterodimerization with DP proteins and interaction with pocket proteins. E2F4-5 are most frequently found in complex with p107 and p130, although pRb can also be found in complex with E2F4. The sub-division of E2F1-5 into activators and repressors is based largely on in vitro studies, reporter gene assays for example, and may not always reflect their actual role in vivo.
When bound to E2Fs, pocket proteins are able to negatively regulate transcription of E2F target genes by at least two different mechanisms. The transcriptional activation domain of E2F1 interacts with general transcription factors and histone acetylases that are necessary for transcriptional activation (Emili and Ingles, 1995; Hagemeier et al., 1993; Lang et al., 2001). Due to their overlapping interaction surfaces within the transcriptional activation domain, pRb competes with these factors for binding E2F thereby blocking transcriptional activation (Pearson and Greenblatt, 1997). Pocket proteins can also bind a host of factors that influence chromatin structure including histone deacetylases, histone methyltransferases, histone demethylases, DNA methyltransferases, and histone remodeling complexes (Table 1). In some cases such as the histone deacetylases, binding to pRb appears to be important for recruiting these factors to particular sites on the DNA (Brehm et al., 1998; Luo et al., 1998; Muzumdar et al., 2007). In other cases like the histone H4K20 methyltransferases SUV420H1 and SUV420H2, interaction with pRb appears to stabilize their localization to a specific location rather than to specify it (Gonzalo et al., 2005). pRb can also displace chromatin modifying factors from promoters (Benevolenskaya et al., 2005). For a comprehensive review of pRb interactions with such chromatin modifying factors, see (Frolov and Dyson, 2004). The typical net effect of pRb at these E2F bound promoters is alteration to a more closed chromatin structure that represses gene transcription. E2Fs regulate many genes important for the cell cycle (Ren et al. 2002; Ishida et al. 2001) and are required for a normal cell cycle in some cells (Humbert et al., 2000; Wu et al., 2001). Thus by both blocking E2F transcriptional activation and actively silencing expression of E2F target genes, pRb restrains cell proliferation.
Table 1.
pRb interacting genes products involved in chromatin regulation1
| Gene2 | Biochemical activity; role in chromatin biology | References |
|---|---|---|
| HDAC1, 2, 3 | Histone deacetylase; transcriptional repression | (Brehm et al., 1998; Lai et al., 2001; Lai et al., 1999; Luo et al., 1998; Magnaghi-Jaulin et al., 1998) |
| SIRT1 | NAD-dependent protein deacetylase; transcriptional repression | (Wong and Weber, 2007) |
| ARID4A (RBP1) | Component of Sin3-histone deacetylase complexes; imprinting, transcriptional repression | (Defeo-Jones et al., 1991; Fattaey et al., 1993) |
| PA2G4 (EBP1) | RNA binding protein, component of HDAC containing complexes; transcriptional repression | (Xia et al., 2001) |
| EP300 | Histone acetyltransferase; transcriptional activation | (Chan et al., 2001) |
| KAT5 (TIP60) | Histone acetyltransferase; transcriptional activation | (Leduc et al., 2006) |
| SUV420H1, 2 | Histone H4 methyltransferase; organization of heterochromatin | (Gonzalo et al., 2005; Isaac et al., 2006) |
| SUV39H1 | Histone H3 methyltransferase; organization of heterochromatin, transcriptional repression | (Nielsen et al., 2001; Vandel et al., 2001) |
| RBBP5 | Component of the MLL1/MLL Histone H3 methyltransferase complex; transcriptional activation | (Saijo et al., 1995) |
| KDM1A (LSD1) | Histone H3 demethylase; transcriptional repression or activation | (Chau et al., 2008) |
| KDM5A (RBP2) | Histone H3 demethylase; transcriptional repression or activation | (Benevolenskaya et al., 2005; Defeo-Jones et al., 1991; Fattaey et al., 1993) |
| DNMT1 | DNA methyltransferase; transcriptional silencing | (Robertson et al., 2000) |
| CBX1, 3, 5 (HP1) | Methyl-Lysine binding protein; organization of heterochromatin, transcriptional repression | (Nielsen et al., 2001) |
| MORF4L1 (MRG15) | Methyl-Lysine binding protein; component of histone acetylase and deacetylase complexes, transcriptional regulation | (Leung et al., 2001) |
| L3MBTL | Methyl-Lysine binding protein; nucleosome compaction | (Trojer et al., 2007) |
| RBBP7 (RpAp46) | Histone binding protein; component of multiple chromatin assembly, remodeling, and transcriptional repressor complexes | (Qian and Lee, 1995) |
| RBBP4 (RpAp48) | Histone binding protein; component of multiple chromatin assembly, remodeling, and transcriptional repressor complexes | (Qian et al., 1993) |
| SMARCA4 (BRG1) | ATP-dependent helicase, component of the SWI/SNF chromatin remodeling complex | (Dunaief et al., 1994) |
| SMARCA2 (BRM) | ATP-dependent helicase, component of the SWI/SNF chromatin remodeling complex | (Strober et al., 1996; Trouche et al., 1997) |
| NCAPD3 | Subunit of the condensin II complex, organization of chromosome condensation and cohesion during mitosis | (Longworth et al., 2008; Manning et al., 2010) |
| SMC4 | Part of the ATPase subunit of condensin complexes, chromosome condensation during mitosis | (Longworth et al., 2008) |
| RBBP8 (CtIP) | Interacts with CTBP/Polycomb transcriptional repressor complexes | (Dahiya et al., 2001; Meloni et al., 1999) |
This list is restricted to proteins where evidence supports direct binding to pRb. Additional proteins with roles in chromatin biology interact with pRb indirectly as part of multi-protein complexes. Other chromatin modifying complexes appear to interact primarily with other members of the pocket protein family (i.e. DREAM and LINC complexes).
Common alias in parenthesis.
Cyclin-dependent kinases (CDKs) can phosphorylate pRb in vitro. The phosphorylation status of pRb in cells correlates with the cyclic activity of CDKs during the cell cycle, and pRb is phosphorylated on a dozen or more sites that match the consensus target sites for proline directed kinases like CDKs. These observations suggest CDKs are physiological kinases for pRb. Phosphorylation sites are scattered throughout the length of pRb, but invariably cluster in the unstructured regions rather than the cyclin folds (Figure 1). The overall organization of the phosphorylation sites is similar among the different pocket proteins. While less is known about the phosphorylation of p107 and p130, regulation is likely to be analogous to that of pRb (Leng et al., 2002). Phosphorylation inhibits pRb/E2F complex formation (Dynlacht et al., 1994) and pRb-mediated cell cycle arrest activity (Connell-Crowley et al., 1997). How phosphorylation regulates other of the myriad pRb protein interactions is not well known, but most of the interactions studied to date are favored when pRb is relatively unphosphorylated.
High affinity pRb/E2F interaction involves two major contacts. One is between the transcriptional activation domain of E2F and the pocket tandem cyclin fold of pRb. This contact is modulated by two clusters of phosphorylation sites, S608/S612 and T356/T373. S608/S612 is located within the unstructured spacer between the pocket dual cyclin folds, and phosphorylation of these residues promotes an intramolecular interaction between the spacer and the dual cyclin fold that inhibits access to the E2F binding surface (Burke et al., 2010). T356/T373 is located within the linker between the amino terminal and pocket tandem cyclin folds. Phosphorylation of these residues induces an intramolecular interaction between the two tandem cyclin folds, also interfering with the interaction between pRb and the E2F transcriptional activation domain. The other major contact is between the flexible carboxy terminal tail of pRb and the marked box domains of the E2F/DP heterodimers. This contact is also modulated by two clusters of phosphorylation sites, S788/S795 and T821/T826. S788/S795 phosphorylation directly destabilizes one set of interactions while T821/T826 phosphorylation induces an intramolecular interaction between the tail and the dual cyclin fold that indirectly destabilizes the remaining interaction (Rubin et al., 2005).
All cell cycle CDKs that have been tested are able to phosphorylate pRb in vitro, and they do so with some site specificity (Connell-Crowley et al., 1997; Takaki et al., 2005; Zarkowska and Mittnacht, 1997). Other proline directed serine/threonine kinases can also phosphorylate pRb including the p38 stress activated kinase 1 (Hou et al., 2002; Nath et al., 2003), ERK1/2 (Guo et al., 2005), AMP-activated protein kinase (Dasgupta et al., 2009), protein kinase C beta (Suzuma et al., 2002), Aurora B (Nair et al., 2009), Chk1/2 (Inoue et al., 2007), and Raf-1 (Dasgupta et al., 2004). The particular subsets of sites phosphorylated by these kinases are overlapping, but typically different. While some phosphorylation sites appear to be critical for regulation (Connell-Crowley et al., 1997; Knudsen and Wang, 1996), how phosphorylation at individual sites or combinations of sites regulates pRb protein interactions and function has not been clearly defined (Barrientes et al., 2000; Brown et al., 1999; Knudsen and Wang, 1997). While dephosphorylation of pRb may be dynamic and occur throughout the cell cycle, pRb is converted to a largely unphosphorylated form as cells transit mitosis (Durfee et al., 1993; Ludlow et al., 1993).
These observations define the pRb mediated cell cycle switch that currently serves as the paradigm for Rb1 tumor suppressor function (Figure 2). In its unphosphorylated active state, pRb binds to E2F at the promoters of E2F regulated cell proliferation genes. Rb1 protein prevents assembly of protein complexes required for transcriptional transactivation and instead assembles alternative complexes that alter local chromatin structure to silence gene expression. Mitogenic signaling pathways impinge on pRb by altering its phosphorylation status. These phosphorylation events alter pRb protein interactions resulting in derepression and activation of E2F target cell cycle genes, including cyclins. CDK activity is enhanced increased cyclin levels, and increased CDK activity augments pRb phosphorylation forming a positive feedback loop. This positive feedback amplifies the mitogenic signals, inactivates the pRb mediated block to cell proliferation, and commits cells to a round of cell division. The switch is also subject to negative feedback control as the Rb1 gene itself is subject to transcriptional control by pRb/E2F (Burkhart et al., 2010). Increases in E2F transcriptional transactivation increase Rb1 expression which, in turn, dampens E2F activity. Positive and negative feedback control makes the pRb/E2F switch bistable (Yao et al., 2008). Negative feedback keeps the initial threshold of mitogenic stimulation necessary to derepress and activate transcription of E2F target genes high. Positive feedback through activation of CDK activity lowers the threshold of mitogenic stimulation required to maintain the switch in an “on” position. Thus the switch tends to exist in either an off or on state, spending less time in intermediate states. As cells transit mitosis, pRb is dephosphorylated. This resets the switch, making mitogenic stimulation necessary again for continued cell proliferation.
Figure 2. The pRb/E2F cell proliferation switch.

The pRb/E2F switch is bistable, existing primarily on one of two states, either permissive or non-permissive for cell proliferation. Bistability is established by both positive and negative feedback loops (see text). In its unphosphorylated state, pRb complexes with E2F transcription factors at gene promoters, blocks the ability of E2F to recruit transcriptional coactivators like histone acetylases (HAT), and recruits corepressors like histone deacetylases (HDAC). These coactivators and corepressors alter chromatin to a more open or condensed states, respectively. Mitogenic kinases phosphorylate pRb, altering its ability to interact with E2F and transcriptional corepressors. The resulting transcriptional derepression of genes like cyclins increases cyclin dependent kinase activity (CDK). Increased CDK activity, in turn, augments pRb phosphorylation thus creating a positive feedback loop that amplifies the mitogenic signal and triggers commitment to a round of cell division. Negative feedback is provided by the fact that the Rb1 promoter is itself regulated by E2F. As cells transit mitosis, pRb is dephosphorylated, resetting the switch to a non-proliferative state.
It is important to note that relatively normal cell cycles occur in the complete absence of the pocket protein family (Sage et al., 2000). Rather than execution of the cell cycle, their role is in regulating entry into and transit through different phases of the cell cycle. Thus pocket proteins translate the output of signaling pathways into binary go/no go decisions on whether to enter and traverse the cell cycle. This switch is important in many biological contexts that require coordination with the cell cycle including DNA damage responses, cellular differentiation, cell death signaling, contact inhibition, and tissue morphogenesis among others. Loss of pRb alters the dynamics of this switch, making the conditions necessary for cell proliferation in these different contexts more permissive. Permissive cell proliferation is presumably a major reason why pRb loss has profound effects on tumorigenesis.
While the pRb/E2F cell cycle switch is an intuitively satisfying explanation for Rb1 mediated tumor suppression, there is evidence that the switch is more versatile and can be used to regulate gene expression in biological contexts unrelated to cell cycle control. For example, compound loss of E2F3 rescues a cell cycle independent defect in neuronal migration observed in Rb1 null mice, presumably because the pRb/E2F switch regulates genes like neogenin with known roles in neuronal migration (McClellan et al., 2007a). Similarly, compound loss of E2F3a rescues a cell cycle independent defect in the differentiation of retinal starburst amacrine cells that occurs in Rb1 deficient mice (Chen et al., 2007). Rb1 loss also causes a defect in retinal rod cell differentiation that appears to be unrelated to the cell cycle (Zhang et al., 2004). Interestingly, the pRb/E2F switch is also used to regulate metabolic responses. pRb/E2F regulates insulin secretion by pancreatic β cells through modulation of KIR6.2 expression, a gene that encodes a K+ ATP channel that controls membrane polarization (Annicotte et al., 2009). CDK4 is activated in response to glucose in these cells, resulting in pRb phosphorylation, liberation of pE2F, and increased KIR6.2 transcription. This feedback loop involving the pRb/E2F switch thus contributes to blood glucose homeostasis. Such examples likely under represent the significance of cell cycle independent functions of the pRb/E2F switch as it is often difficult to disentangle direct effects from the indirect effects caused by deregulated cell proliferation. Thus it appears the pRb/E2F switch has been co-opted to regulate gene expression for a number of different purposes. How the pRb/E2F complexes are specifically directed to regulate unique subsets of genes in these different biological contexts remains unknown.
Evolutionary conservation of the switch
E2F and pocket proteins are conserved through evolution, with orthologues present in vertebrates, invertebrates, plants, and unicellular algae. The invertebrate orthologues most thoroughly studied are from Drosophila melanogaster and Caenorhabditis elegans where the reduced number of E2F and pocket protein family members has facilitated analysis. Studies of these model organisms have not only verified the evolutionary conservation of the pocket protein cell cycle switch, but have also yielded new potential roles for the switch in DNA replication and cell fate commitment.
Drosophila contains two pocket protein family members (RBF1-2), one activating E2F (dE2F1), one repressor E2F (dE2F2), and one DP partner for E2F (dDP) (van den Heuvel and Dyson, 2008). Activating dE2F1 is essential for normal cell proliferation in flies (Duronio et al., 1995), foreshadowing an analogous cell proliferation requirement for activating E2F1-3 observed in mouse fibroblasts (Wu et al., 2001). RBF1 is required for embryogenesis in Drosophila, and RBF1 deficient cells exhibit deregulated dE2F1 target gene expression, loss of normal cell cycle control, and increased apoptosis (Du and Dyson, 1999). These phenotypes are broadly similar to those observed in mice lacking Rb1 (Clarke et al., 1992; Jacks et al., 1992; Lee et al., 1992). Phenotypes observed in RBF1 deficient flies can be rescued by mutations in dE2F1, in particular mutations that specifically compromise its ability to transactivate transcription (Du, 2000). This observation suggests that the ability of pRb to block E2F transactivation, rather than its ability to actively silence gene expression, is paramount for control of cell proliferation in Drosophila. There is also evidence from studies in Drosophila that the pRb/E2F switch is responsive to signaling pathways. For example, Notch signaling promotes release of dE2F1 from RBF1, expression of dE2F1 cell cycle target genes, and cell proliferation (Baonza and Freeman, 2005).
Caenorhabditis elegans contains a single Rb1 like orthologue (lin-35), a single DP gene (DPL-1), and three possible E2F-like genes (EFL-1, EFL-2, F49E12.6). In worms, the pRb/E2F switch appears to have a less prominent role in the regulation of cell proliferation. Lin-35 mutant worms have subtle cell cycle defects (Grishok and Sharp, 2005; Ouellet and Roy, 2007), and the canonical pRb/E2F cell cycle switch is only revealed upon combining multiple mutations in switch components (Boxem and van den Heuvel, 2001; Boxem and van den Heuvel, 2002; Fay et al., 2002; Park and Krause, 1999). Rather, studies in C. elegans have emphasized a role for the pRb/E2F switch in cell fate commitment. Mutation of lin-35, dpl-1, or efl-1 all have similar phenotypes (Ceol and Horvitz, 2001; Page et al., 2001) suggesting they function as a unit to repress gene transcription (Kirienko and Fay, 2007). Thus in worms, it appears the ability of the pRb/E2F switch to actively silence gene expression may be most important for the cell fate commitment phenotypes observed. Of particular interest is that pRb/E2F may be involved in specifying the distinction between germline and somatic gene expression patterns. Germline specific expression patterns in worms are maintained by a number of different mechanisms including epigenetic chromatin modifications and RNA interference (Bender et al., 2006; Fong et al., 2002; Ketting et al., 1999; Sijen and Plasterk, 2003). Mutation of lin-35 causes a soma to germline transformation in expression pattern that is mediated, at least in part, by chromatin modifying factors like the MES-4 histone methyltransferase (Wang et al., 2005). In C. elegans, the pRb/E2F switch appears to facilitate a soma associated gene expression pattern by maintaining repressive chromatin at germline specific genes. Loss of this switch presumably leads to a relaxed chromatin environment permissive for transcription of these germline genes.
The essential features of the pRb/E2F cell cycle switch, including its role in regulating chromatin structure and gene expression, are also conserved in plants (Sanchez Mde et al., 2008; Shen, 2002). Arabidopsis thaliana contains a single Rb1 related gene (RBR). As in animals, studies in Arabidopsis have reinforced the notion that pocket proteins have context dependent roles in regulating both cell fate commitment and cell proliferation. RNA interference induced knockdown of RBR expression causes a defect in the ability of stem cells to commit to a differentiating lineage (Borghi et al., 2010; Wildwater et al., 2005). This initially results in expansion of the stem cell pool, but ultimately compromises stem cell maintenance causing an arrest of plant development. Increased expression of RBR has the opposite effect, decreasing the number of multipotent stem cells due to over commitment to differentiating lineages. While the phenotype of these plants is associated with disruption of the normal patterns of cell proliferation, the effects of RBR on stem cells appear to be primarily in cell fate commitment rather than deregulated cell proliferation. Loss of RBR also compromises gametogenesis (Chen et al., 2009b; Ebel et al., 2004; Ingouff et al., 2006). In this case, however, defects are attributed primarily to deregulated cell proliferation.
A comparative analysis of the pRb and pE2F function in evolutionarily divergent species reinforces the hypothesis that the pRb/E2F switch functions to control assembly and disassembly of protein complexes at specific regions of the genome. The switch has been adapted for various context dependent uses. A primary function is in the control of gene expression, and the genes it regulates can directly influence cell proliferation, differentiation, migration, and metabolic homeostasis to name a few.
Variations on the switch
The complexity of the pRb/E2F switch is considerable given that the above discussion has summarized a small subset of the literature published on the topic. This complexity brings to mind the principle that evolution often builds upon a good thing. Once a useful molecular tool is built to satisfy a particular need, it is often re-used to solve new problems in different biological contexts. By creating variations on the basic tool, its repertoire of possible functions can be enlarged. As an adaptor, pRb is ideally suited for functional diversity. Increasing the number of sequence specific DNA binding factors it interacts with will increase the variety of locations where it can be recruited. By increasing the variety of proteins it interacts with at those locations, the possible biological effects can be expanded and more finely tuned. The possible combinatorial permutations increase functional diversity further still.
Rb1 protein can interact with a number of sequence specific DNA binding transcription factors beyond E2F including MyoD (Gu et al. 1993), C/EBPs (Chen et al., 1996b), NF-IL6 (Chen et al., 1996a), ATF2 (Kim et al., 1992), Pax8 (Miccadei et al., 2005), CBFA1/RUNX2 (Thomas et al. 2001), Tbx2 (Vance et al., 2010), nuclear hormone receptors (Batsche et al., 2005; Lu and Danielsen, 1998; Singh et al., 1995), and GATA1 (Kadri et al., 2009). In a number of cases, loss of Rb1 compromises transcriptional activation and differentiation that is dependent on such transcription factors. Thus in contrast to the effects of the canonical pRb/E2F switch in blocking gene expression, pRb can also function as a transcriptional coactivator for lineage specific transcription factors. For example, pRb augments differentiation into muscle cells by binding muscle specific transcription factors like MyoD and augmenting their ability to drive expression of genes required for muscle differentiation and function (Gu et al., 1993; Novitch et al., 1996; Novitch et al., 1999; Schneider et al., 1994; Zacksenhaus et al., 1996). Likewise, Rb1 can function as a coactivator for the CBFA1/RUNX2 and the C/EBP transcription factors that participate in osteogenic and adipogenic differentiation, respectively (Chen et al., 1996b; Thomas et al., 2001). Importantly, the differentiation defects that result appear to be unrelated to cell proliferation. For example, adipogenesis defects are independent of cell proliferation since compound loss of E2F4 rescues cell proliferation defects, but does not rescue differentiation defects (Landsberg et al., 2003).
It seems likely that additional interactions between pRb and tissue specific transcription factors necessary for differentiation may exist. Rb1 deficiency in vivo affects the differentiation of a relatively large number of distinct tissues and cell types. Rb1 null mice die in midgestation (Clarke et al. 1992; Jacks et al. 1992; Lee et al. 1992) with defects in the eye lens, brain, peripheral nervous system, muscle, liver, placenta, and the hematopoietic system (Iavarone et al., 2004; Novitch et al., 1996; Spike et al., 2004; Tsai et al., 1998; Wu et al., 2003; Zacksenhaus et al., 1996). Conditional Rb1 loss in adult mice affects the differentiation of additional cell types and tissues including the epidermis (Balsitis et al., 2003; Ruiz et al., 2004), melanocytes (Yu et al., 2003), hair cells (Sage et al., 2005), prostate (Maddison et al., 2004), lung (Wikenheiser-Brokamp, 2004), cerebellum (Marino et al., 2003), pituitary (Vooijs et al., 1998), small intestine (Guo et al., 2009), and retina (Chen et al., 2004; MacPherson et al., 2004; Zhang et al., 2004) among others. The mechanisms responsible for these defects have not been completely defined. While the effects of deregulated E2F and cell proliferation probably contribute, it is possible that disruption of pRb interactions with currently unidentified tissue specific transcription factors also play a part in these phenotypes.
Rb1 function is thus extended by interaction with additional sequence specific transcription factors that recruit pRb to additional genome locations in different biological contexts. The observation that pRb can function as a transcriptional coactivator indicates that the net effect of pRb recruitment is not always to repress transcription. It is currently not clear how pRb functions as a transcriptional coactivator. One possible mechanism is that pRb can bind and block the activity of differentiation inhibitors like EID-1 (MacLellan et al., 2000; Miyake et al., 2000) and Id2 (Iavarone et al., 1994; Iavarone et al., 2004). EID-1 binds and inhibits p300 histone acetylase activity that normally functions as a coactivator for transcription factors like MyoD. When bound by pRb, EID-1 is targeted for proteolytic degradation. In this case, pRb functions as an inhibitor of an inhibitor of a transcriptional coactivator. In a similar way, pRb also inhibits the Id2 differentiation inhibitor. Id2 functions as a dominant negative helix-loop-helix protein that heterodimerizes with basic helix-loop-helix transcription factors. Heterodimerization with Id2 prevents these transcription factors from binding their E box response elements on DNA, thus preventing transcriptional activation. Locally recruited pRb can compete for binding Id2, thus shifting the equilibrium away from the inhibitory Id2/bHLH interaction. Thus pRb recruitment may disassemble inhibitory interactions that prevent transcriptional activation.
It is also conceivable that pRb may coactivate transcription by altering chromatin structure. While many repressive chromatin modifying factors are recruited or potentiated by pRb, it is also possible that pRb may oppose their activity. RBP2 is a histone demethylase specific for di- and tri-methylated H3K4. Tri-methylated H3K4 typically marks chromatin that is permissive for transcription (Christensen et al., 2007; Iwase et al., 2007; Klose et al., 2007). Removal of this methyl mark by RBP2 is expected to tip the balance in favor of repressive chromatin. Consistent with this, knockdown of RBP2 expression facilitates the transcription of genes necessary for differentiation in some contexts (Benevolenskaya et al. 2005). Interaction with pRb displaces RBP2 from promoters of genes such as osteocalcin, thus facilitating transcriptional activation. While pRb interaction opposes transcriptional repression by RBP2 at some promoters, they actually work together to activate transcription at other promoters. For example, binding of pRb and RBP2 to the BRD2 and BRD8 promoters coincides with increased expression of both native and reporter genes (Benevolenskaya et al., 2005). The mechanisms underlying this observed transcriptional activation are not currently known.
Such observations make clear that it is currently difficult to predict what the net effect of pRb will be on gene expression. While pRb recruitment typically results in transcriptional repression, there clearly are exceptions where pRb is required for robust transcriptional activation. Given that the preponderance of studies to date examine the effects of pRb/E2F interaction at cell cycle related genes, examples where pRb functions as a transcriptional activator may be under represented in the published literature. It is not well understood why pRb functions to repress transcription at some locations, but functions as a transcriptional activator at others. What is becoming clear is that the effects of pRb recruitment to a particular location are highly context dependent, probably reflecting the diversity of proteins present within the local environment. Current efforts to map the genome location of pocket proteins, transcription factors, chromatin modifying factors, and histone marks will likely inform these issues, especially when correlated with gene expression profiling.
Rb1 function beyond transcription
Studies in Drosophila have provided some of the earliest clues that the pRb/E2F switch, or variations of the switch, may be used for purposes beyond transcriptional regulation. While it is clear the switch can influence DNA replication indirectly through the regulation of gene expression, in a number of biological contexts the effects of pRb/E2F loss appear to be too rapid to be accounted for by changes in gene expression. A more direct role for pRb/E2F in DNA replication is suggested by the observation that Drosophila RBF and dE2f are found in complex with the origin recognition complex (DmORC) during chorion gene amplification (Bosco et al., 2001). Elimination of the RBF/dE2F complex by RBF or dE2F mutation increases DNA amplification and genomic replication without detectable effects on E2F target gene expression (Bosco et al., 2001; Royzman et al., 1999). Thus pRb/E2F appears to directly inhibit the activity DmORC.
While it is not well understood how pRb/E2F inhibits DmORC, some possibilities have been suggested. DNA replication initiation during Drosophila chorion gene amplification is associated with acetylation of histones at the origin (Hartl et al., 2007). Chromatin structure is known to regulate replication origin activity in Drosophila (Aggarwal and Calvi, 2004). Loss of RBF results in the persistence of acetylated histones and increased DNA replication, perhaps due to lack of histone deacetylase activity normally recruited by pRb. This suggests that the pRb/E2F adaptor is used in this case to alter chromatin structure for regulation of DNA replication rather than gene transcription. Other possible mechanisms have been suggested by mammalian systems. For example, pRb interacts with replication factors such as MCM7 and replication factor C, blocking activation of the pre-replication complex (Mukherjee et al., 2010; Pennaneach et al., 2001; Sterner et al., 1998). Rb1 protein has also been suggested to impede the loading of the DNA replication processivity factor PCNA (Angus et al., 2004). Consistent with these findings, pRb can inhibit DNA replication in vitro, and this pRb mediated activity is regulated by CDK phosphorylation (Gladden and Diehl, 2003; Sterner et al., 1998). Thus targeting pRb to replication origins may impede assembly or activation of protein complexes necessary for initiation and elongation of DNA replication.
Convincing evidence has recently been accumulating that pRb may also play a direct role in mitosis. Loss of pRb has long been known to compromise genome stability, and one component of this instability is reflected in the appearance of aneuploidy. Chromosome losses occur with a frequency three orders of magnitude higher in embryonic stem cells lacking pRb compared to wild type cells (Zheng et al., 2002). Acute loss of pRb in human or mouse primary fibroblasts causes supernumerary centrosomes, aneuploidy, and micronuclei (Amato et al., 2009; Iovino et al., 2006). Both loss of normal spindle assembly checkpoint control and defects in centrosome homeostasis can contribute to such effects. The pRb/E2F switch does regulate a number of genes involved in these processes (Ishida et al., 2001; Ren et al., 2002), and deregulated expression of these genes does contribute to chromosome instability observed upon pRb loss. For example, the mitotic checkpoint gene MAD2 arrests cells in metaphase by blocking activation of the anaphase promoting complex that is required for destruction of the chromosome cohesion regulator Securin. MAD2 expression is controlled by the pRb/E2F switch, and MAD2 over expression resulting from pRb loss leads to aneuploidy and tumorigenesis (Hernando et al., 2004; Sotillo et al., 2007).
While transcriptional effects of the pRb/E2F switch are clearly important for chromosome stability, pRb also plays a direct, non-transcriptional role in chromosome structure and mitosis. Loss of Drosophila RBF causes a defect in normal chromosome condensation in mitotic cells; chromosomes in these animals contain regions of condensed chromatin interspersed with regions of uncondensed chromatin. Both genetic and biochemical data indicate this effect is mediated by direct interaction of pRb with the condensin II complex subunit CAP-D3 (Longworth et al., 2008). RBF appears to be required for optimal loading of CAP-D3 onto chromosomes. This function appears to be specific for CAP-D3 as other condensin subunits localize normally to chromosomes in the absence of RBF. As the two condensin complexes load to distinct regions of chromatin, the specificity of the pRb/CAP-D3 interaction may explain the region specific chromatin condensation defect.
This pRb/CAP-D3 interaction is also observed in human and rodent cells (Longworth et al., 2008; Manning et al., 2010; van Harn et al., 2010). RNAi mediated silencing of Rb1 in immortalized rat pigment epithelial cells causes a number of subtle mitotic defects including delayed progression through mitosis, an increased intercentromeric distance, premature loss of sister chromatid cohesion, a defect in chromosome congression, and lagging chromosomes in anaphase. These defects are associated with a deficiency in the loading of cohesin and CAP-D3 to centromeric locations. The net effect of these defects is to cause chromosome missegregation at a frequency comparable to that observed in human tumor cell lines, suggesting that chromosome instability upon pRb loss may contribute to tumorigenesis.
Many chromatin regulators contain the so called “LXCXE” amino acid motif that binds pRb through an interaction surface in the pocket tandem cyclin fold. Alanine substitutions at three critical amino acids in this pRb interaction surface disrupts interactions with LXCXE containing proteins, and a knock-in Rb1 allele containing these mutations has been created in the mouse (Isaac et al., 2006). Mice homozygous for the mutant allele are viable without pronounced cell cycle or differentiation defects. However cells from these mice have subtle defects in chromatin structure, particularly a reduction in trimethylated H4K20 at pericentric heterochromatin. The mutant pRb is unable to bind CAP-D3, and mutant cells show defects in chromosome loading of CAP-D3 and other condensin II components. Because of these defects, mutant cells take longer to complete mitosis and exhibit aneuploidy. Importantly, the mutant Rb1 allele exacerbates tumorigenesis in TP53 deficient mice (Coschi et al., 2010). The median time to death is reduced in these double mutant mice, compared to mice deficient only for TP53, and tumors from these mice exhibit increased numbers of chromosomal changes.
These observations reinforce the hypothesis that pRb has direct functions in organizing chromosome structure during mitosis, and these functions are independent of the canonical pRb/E2F switch. Importantly, loss of these functions in mitosis can contribute directly to tumorigenesis. While these studies demonstrate that pRb plays a role in recruiting condensin II complexes, other mechanisms of regulating chromosome condensation during mitosis exist (Takemoto et al., 2009). Further, it is clear that pocket proteins are not strictly required for mitosis, but rather contribute to the fidelity of the process. It is currently unknown how pRb is located to particular regions of chromosomes during mitosis to assist in recruitment of condensin complexes, or whether pRb has additional effects on the function of these complexes once recruited to the chromosome. A possibly relevant observation is that pRb is also known to directly interact with the targeting subunits of E3 ubiquitin ligases like SCFskp2 and APCcdh1 (Binne et al., 2007; Ji et al., 2004) and such interactions are important for tumorigenesis (Wang et al., 2010). Regulated protein degradation is required for proper execution of mitosis. Thus pRb can recruit biochemical activities that may be relevant to mitosis.
Most of the examples reviewed here have emphasized the effects of pRb on local chromatin structure and gene expression. The effects of pRb deficiency on chromosome structure during mitosis suggest it may also contribute to the organization of chromatin domains over larger regions (Longworth and Dyson, 2009). The emerging roles of pRb in cellular senescence are consistent with this possibility. Cellular senescence is a physiological response to a variety of stresses that results in long term or permanent cessation of cell proliferation. It provides an important barrier to tumorigenesis (Prieur and Peeper, 2008). Rb1 and TP53 represent two key tumor suppressor gene pathways that are required for enforcing this senescence response (Courtois-Cox et al., 2008). It is not obvious whether pRb is essential for the initiation or maintenance of senescence, or both. Loss of all three pocket protein family members is sufficient to trigger cell cycle reentry in senescent mouse embryonic fibroblasts (Sage et al., 2003), suggesting they are at least required for the maintenance. Ectopic expression of pRb in some cell lines can induce a senescence response, suggesting pRb may also be involved in initiation (Hinds et al., 1992; Huang et al., 1988; Templeton et al., 1991). The molecular mechanisms underlying pRb’s role in cellular senescence are not completely defined, but the pRb/E2F switch appears to be involved. The pRb/E2F switch regulates a subset E2F target genes, particularly those involved in DNA replication, specifically in the context of cellular senescence (Chicas et al., 2010). The regulation of these genes is unique to pRb among the pocket proteins.
Of note is that in order to enforce the stability of the non-proliferative state, cellular senescence is accompanied by global reorganization of chromatin domains. In particular, senescence is associated with the appearance of microscopically visible foci of heterochromatin called senescence associated heterochromatin foci (SAHF) (Narita et al., 2003). These foci are highly enriched for trimethylated H3K9, the methyl-lysine binding proteins HP1, the histone variant macroH2A, chromatin regulators HIRA and ASF1a, and high mobility group A proteins (Funayama et al., 2006; Narita et al., 2006; Zhang et al., 2005). These factors participate in the formation of the transcriptionally repressive facultative heterochromatin at SAHF. A number of the genes within SAHF are E2F target genes, and both pRb and marks of heterochromatin can be found at these genes. As discussed above, pRb can interact with a number of chromatin modifying factors involved in SAHF formation, including the H3K9 methylase SUV39H1, HP1, and histone deacetylases (Nielsen et al., 2001; Vaute et al., 2002).
These observations have led to the hypothesis that pRb can specify the regional organization of chromatin into SAHF during senescence. Recruitment of pRb to specific genomic location potentiates H3K9 methylation that attracts HP1. HP1 recruitment is augmented by local histone deacetylases. HP1 facilitates the formation of local heterochromatin as well as heterochromatin spreading (Verschure et al., 2005). This hypothesis is supported by evidence indicating that loss of pRb changes the dynamics of HP1 localization, the sensitivity of chromatin to Micrococcal nuclease digestion, and changes in histone H1 phosphorylation (Herrera et al., 1996; Siddiqui et al., 2007). These changes are consistent with a more open chromatin structure in the absence of pRb. Thus pRb may participate in the organization of chromatin structure in contexts as diverse as cellular senescence and mitosis.
Rb1, stem cells, and the cell of tumor origin
How does pRb loss initiate retinoblastoma? Given the diversity of pRb function reviewed, the answer is certainly not obvious. As much of this functional diversity is context dependent, it is difficult to learn how pRb loss initiates tumorigenesis without knowing the type of cell it is lost in. Thus it is useful to know the cell of tumor origin. Cancer is thought to be a clonal disease in an evolutionary sense. It arises from a single ancestor cell that undergoes rounds of genetic and epigenetic alteration followed by selection, eventually evolving into a malignant cancer. Thus identifying the ancestor cell of origin for a particular type of cancer is important as it allows identification of the initiating genetic mutations and how those mutations affect the cells most relevant to tumor initiation.
In the case of retinoblastoma, the initiating genetic mutation is known. Despite the simple molecular etiology of retinoblastoma, however, the cell of retinoblastoma origin has remained controversial for more than a hundred years since the initial attempts at assignment based on similarity to photoreceptors (Wintersteiner, 1897). The crux of the problem is that the cell of origin is inferred from the morphological, molecular, and functional properties of the presenting tumors, and retinoblastoma exhibits features of multiple cell types. Recent work has provided support for the hypothesis that a photoreceptor is the cell of origin (Xu et al., 2009), but other work has suggested that an interneuron (Johnson et al., 2007; Laurie et al., 2006) or a progenitor cell is the tumor originating cell (Kyritsis et al., 1984). This is not just due to the mixture of multiple cell types within a heterogeneous tumor, as a recent unpublished examination of human and mouse retinoblastoma suggests individual retinoblastoma cells simultaneously exhibit molecular, cellular, and neurochemical features of multiple cell types (M. Dyer, personal communication). The cells express markers that are specific for progenitor, interneuron, and photoreceptor cells. The expression of such markers is normally mutually exclusive. This suggests it may not be possible to infer the retinoblastoma cell of origin from examination of retinoblastoma cells as they take on the identity of multiple cell lineages. What can be inferred, however, is that retinoblastoma cells have lost the ability to establish or maintain differentiated gene expression patterns associated with these distinct cell lineages. Thus we are left to guess the cell of retinoblastoma origin, but this guess is informed by the knowledge that it will most likely be a cell where pRb loss has the greatest potential to disrupt cell fate commitment and elaboration of lineage specifying gene expression programs.
We have reviewed evidence that supports the hypothesis that the core function of pRb is to serve as a signaling pathway responsive adaptor that regulates the assembly or activity of protein complexes at specific locations in the genome. Most typically, this results in alteration of local and regional chromatin structure to a more closed confirmation that is repressive for gene transcription. Based on this, we suggest that a relatively uncommitted stem or progenitor cell is the most likely candidate for the retinoblastoma cell of origin. Loss of pRb is expected to cause a loosening of local and regional chromatin making it more permissive for transcription. We surmise that stem or progenitor cells are more sensitive to such effects as they exhibit a neutral chromatin structure at important lineage specifying genes. This neutral chromatin exhibits histone modifications associated with both permissive and repressive chromatin suggesting it is poised for either transcriptional activation or repression in accordance with cell fate commitment (Hemberger et al., 2009; Mohn and Schubeler, 2009; Sang et al., 2009). Loss of pRb in such cells is expected to tip the balance toward transcriptional activation by preventing stable transcriptional repression. This would potentially allow simultaneous expression of genes that are normally restricted to distinct lineages. Loss of pRb in more committed, differentiated cells would be expected to have less effect on gene expression patterns as permissive or repressive chromatin states are reinforced by multiple redundant mechanisms, some of which may not be impacted by pRb loss. Thus the relative plasticity of stem or progenitor cells, as reflected in the programming of their chromatin structure, may make them particularly sensitive to pRb loss.
This is not to say that loss of pRb cannot initiate retinoblastoma in lineage committed cells. Indeed, mouse models of retinoblastoma have been created that are comprised of interneurons with a striking degree of morphological, molecular, and functional differentiation (Ajioka et al., 2007). Rather, we suggest the probability of transformation decreases with increasing lineage commitment and differentiation. Of course the lower probability of transformation may be offset by larger numbers of lineage committed cells. One potentially salient observation is that retinoblastoma is never observed in adults, even if they have inherited an Rb1 mutation (the penetrance of hereditary retinoblastoma is very high, but not 100%). This suggests the cell of retinoblastoma origin exists only during a narrow window of retinal development, past which the cells change or differentiate in such a way as to make them resistant to the effects of Rb1 loss. This argues against the candidacy of cell types that persist into adulthood for the cell of retinoblastoma origin. While we favor this interpretation, there may be something unique about the pediatric retinal microenvironment that facilitates the genesis of retinoblastoma. In this case, the cell tumor origin persists into adulthood, but the microenvironment cannot support neoplastic transformation and tumorigenesis.
Beyond retinoblastoma, small cell lung cancer (SCLC) exhibits the highest rate of Rb1 mutation in human cancers, up to 90% (Wistuba et al., 2001). Hereditary retinoblastoma patients do exhibit an increased risk of lung cancer (Marees et al., 2008). Thus Rb1 mutation may be an initiating event in small cell lung carcinoma. Consistent with this notion, compound loss of pRb and p53 in the mouse lung generates tumors with strong resemblance to human SCLC (Meuwissen et al., 2003). SCLC express genes characteristic of the neuroendocrine lineage like ASCL1, synaptophysin, and NCAM1. They also express markers of stem/progenitor cells (Meuwissen et al., 2003; Moreira et al., 2010). Interestingly, Rb1 loss is required for this unique SCLC phenotype as mouse lung tumors retaining pRb are phenotypically adenocarcinomas. Thus in more than one type of human tissue, stem or progenitor cells may be particularly sensitive to loss of pRb in the context of tumorigenesis.
There is a fair amount of current evidence that supports the hypothesis that stem or progenitor cells are uniquely susceptible to pRb loss. As reviewed above, studies in plants and invertebrates have demonstrated that pRb deficiency causes defects in stem cell pool size and cell fate commitment. There are hints that similar effects occur upon Rb1 loss in vertebrate stem and progenitor cells. For example, Rb1 null mouse embryonic stem cells show defects in differentiation in vitro (Papadimou et al., 2005). Loss of pRb in mouse trophoblast stem cells causes an abnormal expansion of this stem cell pool (Wenzel et al., 2007). Such defects are not observed when pRb is ablated in more differentiated trophoblast derivatives. Loss of pRb causes stem or progenitor cell related defects in a number of additional mouse tissues including the skin (Ruiz et al., 2004), bone (Gutierrez et al., 2008), small intestine (Guo et al., 2009), and blood (Macleod, 2008). However, the effects of pRb loss on stem/progenitor cells are likely context dependent. Rb1 status does not apparently affect the rescue of differentiating prostate tissue by transplantation of stem cell enriched embryonic urogenital sinus (Day et al., 2002). Other pocket protein family members, like p107, may also serve the predominant role in regulating stem cells in some biological contexts (Vanderluit et al., 2004).
There is also evidence that stem or progenitor cells are particularly sensitive to pRb loss in the context of neoplastic transformation. In the mouse, the effects of conditional Rb1 deletion have been examined in a number of different tissues. Typically this is not sufficient for tumorigenesis, but tumors often do occur when Rb1 deletion is combined with other mutations, commonly deletion of TP53. For example, conditional deletion of floxed Rb1 and TP53 alleles using a probasin promoter based Cre transgene causes metastatic prostate cancer (Zhou et al., 2006). Analogous to retinoblastoma, individual prostate tumor cells express markers of multiple lineages, in this case both luminal epithelial and neuroendocrine lineages. The probasin promoter is androgen responsive and maximally expressed in differentiated prostate epithelial cells in post-pubescent males. Despite frequent deletion of Rb1 and TP53 and neoplastic changes in these differentiated epithelial cells, lethal tumors first arise in an anatomical region enriched for prostatic stem cells (Zhou et al., 2007). Early neoplastic lesions in this region co-express stem cell, epithelial, and neuroendocrine markers, as do a small number of normal cells that exist there. These observations suggest that prostatic stem or progenitor cells are the cell of origin for the first lethal tumors, and thus that they are more sensitive to neoplastic transformation upon pRb loss than differentiated epithelial cells. In a similar situation, deletion of Rb1 and TP53 in the skin causes squamous cell carcinoma that first arises in the hair follicle, an anatomical location enriched with epidermal stem cells (Martinez-Cruz et al., 2008). Early neoplastic lesions in the hair follicle express stem cell markers. It has also been observed that deletion of Rb1 and TP53 in brain stem cells, but not mature astrocytes, causes brain tumors (Jacques et al., 2010). The resulting tumors have a relatively undifferentiated neuroectodermal phenotype. Mouse models of osteosarcoma (Berman et al., 2008) and gastrointestinal tract cancer (Kucherlapati et al., 2008) have provided additional evidence consistent with the involvement of stem, progenitor, or immature precursor cells. These observations reinforce the notion that there may be an inverse relationship between the degree of cellular differentiation and susceptibility to loss of pRb.
Rb1 and tumor progression
Rb1 is clearly important for the initiation of retinoblastoma and likely for other tumors like SCLC. Mouse models have demonstrated that Rb1 deletion can contribute to the initiation of tumorigenesis in a wide variety of tissues. For many common human malignancies, however, it is clear that Rb1 mutation is a rather late event in tumor progression (Burkhart and Sage, 2008). This indicates Rb1 mutation does not typically contribute to tumor initiation in some cancers. Assuming late Rb1 mutation is a driver of malignant behavior, why is it selected for late, rather than early, in tumor progression?
One possibility is that a cell type susceptible to tumor initiation upon pRb loss does not exist in a particular tissue, or may be too rare to support a significant probability of tumorigenesis. In such tissues, tumorigenesis is initiated in an Rb1 resistant cell by other genetic alterations. Once the cell is transformed, however, it may become sensitive to the effects of pRb loss, thus providing selection for acquisition of Rb1 mutations that further drive tumor progression. A common feature of tumorigenesis is progressive loss of differentiated features. If this is accompanied by increased plasticity in the programming of chromatin structure, then loss of pRb may be expected to have greater effect on gene expression patterns in such cells than in the original cells from which transformation is initiated. This possibility has not been tested as the cell types susceptible to transformation upon pRb loss have not been well characterized in most tissues, and because mouse models that test the effects of pRb loss in pre-existing neoplastic lesions currently do not exist. However, pRb has been implicated in a number of processes that are particularly relevant to the progression of pre-existing tumors. For example, autophagy is induced in hypoxic tumors. The pRb/E2F switch regulates a number of genes important for hypoxia induced autophagy (Polager et al., 2008; Tracy et al., 2007). The pRb/E2F switch also regulates genes involved in angiogenesis (Gabellini et al., 2006). Loss of pRb could thus alleviate hypoxic stress as tumors grow and also influence the responses to that stress. We have noted above that pRb functions to enforce cellular senescence and the fidelity of mitosis. Increased chromosome instability and loss of senescence responses in the absence of pRb will also facilitate tumor progression.
Another possibility is that there may be selection against Rb1 mutation early in tumorigenesis that is ultimately relaxed later in tumorigenesis. This would suggest a paradoxical oncogenic role for pRb in early stages of tumorigenesis in some tissues. Surprisingly, there is experimental precedent for such an oncogenic role for pRb. E2F1 activity is a well known mediator of apoptotic cell death in response to pRb loss (Tsai et al., 1998), and the regulation of E2F1 mediated apoptosis may be a normal physiological function of pRb during the DNA damage response (Inoue et al., 2007; Markham et al., 2006). E2F1 mediated apoptosis appears to be regulated by a unique interaction surface in the pRb carboxy terminal tail (Dick and Dyson, 2003). Thus in some contexts, pRb mediated suppression of apoptosis may facilitate the survival of neoplastic cells, particularly since they are prone to DNA damage (Halazonetis et al., 2008). Consistent with this, constitutive activation of pRb in the mouse mammary gland initiates tumorigenesis (Jiang and Zacksenhaus, 2002). Further, pRb appears to be required for transformation by some oncogenes, in some cases (Williams et al., 2006).
Summary
Here we have reviewed the hypothesis that the core function of pRb is as a signaling pathway responsive adaptor that facilitates the assembly or disassembly of location specific protein complexes on the DNA. Rb1 protein interacts with a number of different sequence specific DNA binding factors that determine its location. The presence of pRb affects the composition or activity of protein complexes at those locations in a variety of ways. It can recruit new proteins to the location or stabilize their occupancy. The primary example of this is the recruitment of chromatin modifying factors like histone deacetylases. Recruitment of such factors results in the alteration of chromatin structure, typically to a closed state that is repressive for transcription. Rb1 protein can also displace proteins from a location, or inhibit the activity of a protein at that location. In this way pRb can function as a transcriptional coactivator by blocking the effects of transcriptional inhibitors. In sum, pRb is a major regulator of chromatin structure that establishes gene expression patterns necessary for cell cycle control, cell fate commitment, cellular differentiation, and development. This function is presumed to be a key reason why Rb1 mutation has such profound effects on normal development and cancer.
While the function of pRb as a programmer of chromatin structure is a major theme, another theme is the diversity of pRb function. The pRb adaptor is a tool that has been co-opted for use in many disparate biological contexts relevant to DNA metabolism. For example, the pRb adaptor is not only used to control local chromatin structure at individual genes, but it has also been adapted for use in the regional organization of chromatin structure that is important for mitosis and cellular senescence. The pRb adaptor has also been used to influence DNA replication and DNA repair, either indirectly through modulation of chromatin or directly by modulating the protein complexes that carry out these biochemical processes. As an adaptor, the range of its potential functions is quite large given the number of DNA binding factors it interacts with, the number of DNA modifying proteins it interacts with, the number of signaling pathways that impinge upon these interactions, and the potential combinatorial permutations.
An important implication that follows from this functional diversity is that the consequences of pRb loss will be highly context dependent. As an adaptor, pRb function will be largely defined by the subset of proteins available for interaction in a particular cell or sub-cellular location, and the signals impinging on that cell which influence pRb post-translational modification. The state of chromatin in a cell at the time of pRb loss will also heavily influence the resulting effects. Chromatin structure is regulated by multiple and redundant mechanisms. As cells become committed to a particular differentiated cell type, their chromatin tends to become progressively fixed to stably maintain the necessary gene expression pattern. Thus the impact of pRb loss on gene expression is likely to be greater in a cell with a more plastic chromatin state. The diverse, context dependent functions of pRb suggest there may be no universal mechanism of pRb mediated tumor suppression. How pRb loss impacts tumorigenesis is likely to differ in different tissues.
An important motivation for studying Rb1 is that it will result in effective new approaches for the diagnosis and treatment of a range of human cancer. This has not been realized as quickly as once hoped, possibly because of the large diversity of pRb function. However, with an increasing understanding of the core pRb functions, the context dependent variations on these functions, and the increasingly sophisticated tools available to study these functions, this goal remains in sight. Another key motivation for studying Rb1 is the possibility of discerning important general cancer principles from the study of a case with simple molecular etiology. From one perspective, the dizzying functional diversity of pRb renders this a false hope. From another perspective, what we have learned about Rb1 is echoed in important principles emerging in the field of cancer research. Both the developing fields of cancer stem cells and cancer epigenetics (Gupta et al., 2009; Maenhaut et al., 2010; Sharma et al., 2010) are directly relevant to Rb1. Perhaps, we are beginning to see the forest for the trees.
Figure 3. Stem/progenitor cells and sensitivity to pRb loss.

The figure summarizes the hypothesis that there exists an inverse correlation between differentiation and sensitivity to pRb loss. Stem/progenitor cells are relatively undifferentiated and have a neutral chromatin state at key lineage specifying regulatory genes. Chromatin at such genes is characterized by the simultaneous presence of protein complexes and chromatin marks characteristic of both open (shapes marked +) and closed chromatin (shapes marked −). pRb is typically associated with factors that create a closed chromatin state repressive for transcription (although there are notable exceptions). Loss of pRb in such cells is expected to have a large effect on gene expression as it will tip the balance in favor gene expression. In more differentiated cells, chromatin is more rigidly committed to an open or closed state by multiple, redundant mechanisms. Loss of pRb in such cells is expected to have less effect on the expression of stably repressed genes as there are redundant mechanisms in place to maintain the closed chromatin state.
Acknowledgments
We apologize to our colleagues whose work we were unable to cite due to constraints of space and topic. We thank Drs. Jennifer Black and Adam Karpf for critical reading of the manuscript. We thank Dr. Mike Dyer for discussion of unpublished data. We are grateful to numerous colleagues in the Rb1 field and members of the Goodrich lab for stimulating conversations. This work is supported by a grant from the National Cancer Institute (CA70292).
References
- Aggarwal BD, Calvi BR. Chromatin regulates origin activity in Drosophila follicle cells. Nature. 2004;430:372–6. doi: 10.1038/nature02694. [DOI] [PubMed] [Google Scholar]
- Ajioka I, Martins RA, Bayazitov IT, Donovan S, Johnson DA, Frase S, Cicero SA, Boyd K, Zakharenko SS, Dyer MA. Differentiated horizontal interneurons clonally expand to form metastatic retinoblastoma in mice. Cell. 2007;131:378–90. doi: 10.1016/j.cell.2007.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amato A, Lentini L, Schillaci T, Iovino F, Di Leonardo A. RNAi mediated acute depletion of retinoblastoma protein (pRb) promotes aneuploidy in human primary cells via micronuclei formation. BMC Cell Biol. 2009;10:79. doi: 10.1186/1471-2121-10-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angus SP, Mayhew CN, Solomon DA, Braden WA, Markey MP, Okuno Y, Cardoso MC, Gilbert DM, Knudsen ES. RB reversibly inhibits DNA replication via two temporally distinct mechanisms. Molecular & Cellular Biology. 2004;24:5404–20. doi: 10.1128/MCB.24.12.5404-5420.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annicotte JS, Blanchet E, Chavey C, Iankova I, Costes S, Assou S, Teyssier J, Dalle S, Sardet C, Fajas L. The CDK4-pRB-E2F1 pathway controls insulin secretion. Nat Cell Biol. 2009;11:1017–23. doi: 10.1038/ncb1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balsitis SJ, Sage J, Duensing S, Munger K, Jacks T, Lambert PF. Recapitulation of the effects of the human papillomavirus type 16 E7 oncogene on mouse epithelium by somatic Rb deletion and detection of pRb-independent effects of E7 in vivo. Molecular & Cellular Biology. 2003;23:9094–9103. doi: 10.1128/MCB.23.24.9094-9103.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baonza A, Freeman M. Control of cell proliferation in the Drosophila eye by Notch signaling. Dev Cell. 2005;8:529–39. doi: 10.1016/j.devcel.2005.01.019. [DOI] [PubMed] [Google Scholar]
- Barrientes S, Cooke C, Goodrich DW. Glutamic acid mutagenesis of retinoblastoma protein phosphorylation sites has diverse effects on function. Oncogene. 2000;19:562–70. doi: 10.1038/sj.onc.1203332. [DOI] [PubMed] [Google Scholar]
- Batsche E, Moschopoulos P, Desroches J, Bilodeau S, Drouin J. Retinoblastoma and the Related Pocket Protein p107 Act as Coactivators of NeuroD1 to Enhance Gene Transcription. Journal of Biological Chemistry. 2005;280:16088–16095. doi: 10.1074/jbc.M413427200. [DOI] [PubMed] [Google Scholar]
- Ben-Porath I, Weinberg RA, Ben-Porath I, Weinberg RA. The signals and pathways activating cellular senescence. International Journal of Biochemistry & Cell Biology. 2005;37:961–76. doi: 10.1016/j.biocel.2004.10.013. [DOI] [PubMed] [Google Scholar]
- Bender LB, Suh J, Carroll CR, Fong Y, Fingerman IM, Briggs SD, Cao R, Zhang Y, Reinke V, Strome S. MES-4: an autosome-associated histone methyltransferase that participates in silencing the X chromosomes in the C. elegans germ line. Development. 2006;133:3907–17. doi: 10.1242/dev.02584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benevolenskaya EV, Murray HL, Branton P, Young RA, Kaelin WG. Binding of pRB to the PHD Protein RBP2 Promotes Cellular Differentiation. Molecular Cell. 2005;18:623–635. doi: 10.1016/j.molcel.2005.05.012. [DOI] [PubMed] [Google Scholar]
- Berman SD, Calo E, Landman AS, Danielian PS, Miller ES, West JC, Fonhoue BD, Caron A, Bronson R, Bouxsein ML, Mukherjee S, Lees JA. Metastatic osteosarcoma induced by inactivation of Rb and p53 in the osteoblast lineage. Proc Natl Acad Sci U S A. 2008;105:11851–6. doi: 10.1073/pnas.0805462105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binne UK, Classon MK, Dick FA, Wei W, Rape M, Kaelin WG, Naar AM, Dyson NJ. Retinoblastoma protein and anaphase-promoting complex physically interact and functionally cooperate during cell-cycle exit. Nature Cell Biology. 2007;9:225–232. doi: 10.1038/ncb1532. [DOI] [PubMed] [Google Scholar]
- Borghi L, Gutzat R, Futterer J, Laizet Yh, Hennig L, Gruissem W. Arabidopsis RETINOBLASTOMA-RELATED Is Required for Stem Cell Maintenance, Cell Differentiation, and Lateral Organ Production. Plant Cell. 2010 doi: 10.1105/tpc.110.074591. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosco G, Du W, Orr-Weaver TL. DNA replication control through interaction of E2F-RB and the origin recognition complex. Nat Cell Biol. 2001;3:289–95. doi: 10.1038/35060086. [DOI] [PubMed] [Google Scholar]
- Boxem M, van den Heuvel S. lin-35 Rb and cki-1 Cip/Kip cooperate in developmental regulation of G1 progression in C. elegans. Development. 2001;128:4349–59. doi: 10.1242/dev.128.21.4349. [DOI] [PubMed] [Google Scholar]
- Boxem M, van den Heuvel S. C. elegans class B synthetic multivulva genes act in G(1) regulation. Curr Biol. 2002;12:906–11. doi: 10.1016/s0960-9822(02)00844-8. [DOI] [PubMed] [Google Scholar]
- Brehm A, Miska EA, McCance DJ, Reid JL, Bannister AJ, Kouzarides T. Retinoblastoma protein recruits histone deacetylase to repress transcription. Nature. 1998;391:597–601. doi: 10.1038/35404. [DOI] [PubMed] [Google Scholar]
- Brown VD, Phillips RA, Gallie BL. Cumulative effect of phosphorylation of pRB on regulation of E2F activity. Molecular & Cellular Biology. 1999;19:3246–3256. doi: 10.1128/mcb.19.5.3246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke JR, Deshong AJ, Pelton JG, Rubin SM. Phosphorylation-induced Conformational Changes in the Retinoblastoma Protein Inhibit E2F Transactivation Domain Binding. Journal of Biological Chemistry. 2010;285:16286–16293. doi: 10.1074/jbc.M110.108167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkhart DL, Ngai LK, Roake CM, Viatour P, Thangavel C, Ho VM, Knudsen ES, Sage J. Regulation of RB Transcription In Vivo by RB Family Members. Molecular & Cellular Biology. 2010;30:1729–1745. doi: 10.1128/MCB.00952-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkhart DL, Sage J. Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nature Reviews Cancer. 2008;8:671–82. doi: 10.1038/nrc2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceol CJ, Horvitz HR. dpl-1 DP and efl-1 E2F act with lin-35 Rb to antagonize Ras signaling in C. elegans vulval development. Molecular Cell. 2001;7:461–73. doi: 10.1016/s1097-2765(01)00194-0. [DOI] [PubMed] [Google Scholar]
- Chan HM, Krstic-Demonacos M, Smith L, Demonacos C, La Thangue NB. Acetylation control of the retinoblastoma tumour-suppressor protein. Nature Cell Biology. 2001;3:667–674. doi: 10.1038/35083062. [DOI] [PubMed] [Google Scholar]
- Chau BN, Wang JY, Chau BN, Wang JYJ. Coordinated regulation of life and death by RB. Nature Reviews Cancer. 2003;3:130–8. doi: 10.1038/nrc993. [DOI] [PubMed] [Google Scholar]
- Chau CM, Deng Z, Kang H, Lieberman PM. Cell cycle association of the retinoblastoma protein Rb and the histone demethylase LSD1 with the Epstein-Barr virus latency promoter Cp. J Virol. 2008;82:3428–37. doi: 10.1128/JVI.01412-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Livne-bar I, Vanderluit JL, Slack RS, Agochiya M, Bremner R. Cell-specific effects of RB or RB/p107 loss on retinal development implicate an intrinsically death-resistant cell-of-origin in retinoblastoma. Cancer Cell. 2004;5:539–51. doi: 10.1016/j.ccr.2004.05.025. [DOI] [PubMed] [Google Scholar]
- Chen D, Opavsky R, Pacal M, Tanimoto N, Wenzel P, Seeliger MW, Leone G, Bremner R. Rb-mediated neuronal differentiation through cell-cycle-independent regulation of E2f3a. PLOS Biology. 2007;5:e179. doi: 10.1371/journal.pbio.0050179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HZ, Tsai SY, Leone G. Emerging roles of E2Fs in cancer: an exit from cell cycle control. Nature Reviews Cancer. 2009a;9:785–97. doi: 10.1038/nrc2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen PL, Riley DJ, Chen-Kiang S, Lee WH. Retinoblastoma protein directly interacts with and activates the transcription factor NF-IL6. Proceedings of the National Academy of Sciences of the United States of America. 1996a;93:465–469. doi: 10.1073/pnas.93.1.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen PL, Riley DJ, Chen Y, Lee WH. Retinoblastoma protein positively regulates terminal adipocyte differentiation through direct interaction with C/EBPs. Genes & Development. 1996b;10:2794–2804. doi: 10.1101/gad.10.21.2794. [DOI] [PubMed] [Google Scholar]
- Chen Z, Hafidh S, Poh SH, Twell D, Berger F. Proliferation and cell fate establishment during Arabidopsis male gametogenesis depends on the Retinoblastoma protein. Proc Natl Acad Sci U S A. 2009b;106:7257–62. doi: 10.1073/pnas.0810992106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chicas A, Wang X, Zhang C, McCurrach M, Zhao Z, Mert O, Dickins RA, Narita M, Zhang M, Lowe SW. Dissecting the unique role of the retinoblastoma tumor suppressor during cellular senescence. Cancer Cell. 2010;17:376–87. doi: 10.1016/j.ccr.2010.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen J, Agger K, Cloos PA, Pasini D, Rose S, Sennels L, Rappsilber J, Hansen KH, Salcini AE, Helin K. RBP2 belongs to a family of demethylases, specific for tri-and dimethylated lysine 4 on histone 3. Cell. 2007;128:1063–76. doi: 10.1016/j.cell.2007.02.003. [DOI] [PubMed] [Google Scholar]
- Clarke AR, Maandag ER, van Roon M, van der Lugt NM, van der Valk M, Hooper ML, Berns A, te Riele H. Requirement for a functional Rb-1 gene in murine development. Nature. 1992;359:328–30. doi: 10.1038/359328a0. [DOI] [PubMed] [Google Scholar]
- Classon M, Dyson N. p107 and p130: versatile proteins with interesting pockets. Experimental Cell Research. 2001;264:135–147. doi: 10.1006/excr.2000.5135. [DOI] [PubMed] [Google Scholar]
- Claudio PP, Tonini T, Giordano A. The retinoblastoma family: twins or distant cousins? Genome Biology. 2002;3:reviews3012. doi: 10.1186/gb-2002-3-9-reviews3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comings DE. A general theory of carcinogenesis. Proceedings of the National Academy of Sciences of the United States of America. 1973;70:3324–8. doi: 10.1073/pnas.70.12.3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connell-Crowley L, Harper JW, Goodrich DW. Cyclin D1/Cdk4 regulates retinoblastoma protein-mediated cell cycle arrest by site-specific phosphorylation. Molecular Biology of the Cell. 1997;8:287–301. doi: 10.1091/mbc.8.2.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coschi CH, Martens AL, Ritchie K, Francis SM, Chakrabarti S, Berube NG, Dick FA. Mitotic chromosome condensation mediated by the retinoblastoma protein is tumor-suppressive. Genes & Development. 2010;24:1351–63. doi: 10.1101/gad.1917610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtois-Cox S, Jones SL, Cichowski K. Many roads lead to oncogene-induced senescence. Oncogene. 2008;27:2801–9. doi: 10.1038/sj.onc.1210950. [DOI] [PubMed] [Google Scholar]
- Dahiya A, Wong S, Gonzalo S, Gavin M, Dean DC. Linking the Rb and Polycomb Pathways. Mol Cell. 2001;8:557–568. doi: 10.1016/s1097-2765(01)00346-x. [DOI] [PubMed] [Google Scholar]
- Dasgupta B, Milbrandt J, Dasgupta B, Milbrandt J. AMP-activated protein kinase phosphorylates retinoblastoma protein to control mammalian brain development. Developmental Cell. 2009;16:256–70. doi: 10.1016/j.devcel.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasgupta P, Sun J, Wang S, Fusaro G, Betts V, Padmanabhan J, Sebti SM, Chellappan SP. Disruption of the Rb--Raf-1 interaction inhibits tumor growth and angiogenesis. Molecular & Cellular Biology. 2004;24:9527–41. doi: 10.1128/MCB.24.21.9527-9541.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day KC, McCabe MT, Zhao X, Wang Y, Davis JN, Phillips J, Von Geldern M, Ried T, KuKuruga MA, Cunha GR, Hayward SW, Day ML. Rescue of embryonic epithelium reveals that the homozygous deletion of the retinoblastoma gene confers growth factor independence and immortality but does not influence epithelial differentiation or tissue morphogenesis. Journal of Biological Chemistry. 2002;277:44475–44484. doi: 10.1074/jbc.M205361200. [DOI] [PubMed] [Google Scholar]
- Defeo-Jones D, Huang PS, Jones RE, Haskell KM, Vuocolo GA, Hanobik MG, Huber HE, Oliff A. Cloning of cDNAs for cellular proteins that bind to the retinoblastoma gene product. Nature. 1991;352:251–4. doi: 10.1038/352251a0. [DOI] [PubMed] [Google Scholar]
- Delston RB, Harbour JW, Delston RB, Harbour JW. Rb at the interface between cell cycle and apoptotic decisions. Current Molecular Medicine. 2006;6:713–8. doi: 10.2174/1566524010606070713. [DOI] [PubMed] [Google Scholar]
- Dick FA, Dyson N. pRB contains an E2F1-specific binding domain that allows E2F1-induced apoptosis to be regulated separately from other E2F activities. Molecular Cell. 2003;12:639–649. doi: 10.1016/s1097-2765(03)00344-7. [DOI] [PubMed] [Google Scholar]
- Du W. Suppression of the rbf null mutants by a de2f1 allele that lacks transactivation domain. Development. 2000;127:367–79. doi: 10.1242/dev.127.2.367. [DOI] [PubMed] [Google Scholar]
- Du W, Dyson N. The role of RBF in the introduction of G1 regulation during Drosophila embryogenesis. EMBO Journal. 1999;18:916–925. doi: 10.1093/emboj/18.4.916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunaief JL, Strober BE, Guha S, Khavari PA, Alin K, Luban J, Begemann M, Crabtree GR, Goff SP. The retinoblastoma protein and BRG1 form a complex and cooperate to induce cell cycle arrest. Cell. 1994;79:119–30. doi: 10.1016/0092-8674(94)90405-7. [DOI] [PubMed] [Google Scholar]
- Durfee T, Becherer K, Chen PL, Yeh SH, Yang Y, Kilburn AE, Lee WH, Elledge SJ. The retinoblastoma protein associates with the protein phosphatase type 1 catalytic subunit. Genes & Development. 1993;7:555–569. doi: 10.1101/gad.7.4.555. [DOI] [PubMed] [Google Scholar]
- Duronio RJ, O’Farrell PH, Xie JE, Brook A, Dyson N. The transcription factor E2F is required for S phase during Drosophila embryogenesis. Genes & Development. 1995;9:1445–55. doi: 10.1101/gad.9.12.1445. [DOI] [PubMed] [Google Scholar]
- Dynlacht BD, Flores O, Lees JA, Harlow E. Differential regulation of E2F transactivation by cyclin/cdk2 complexes. Genes & Development. 1994;8:1772–1786. doi: 10.1101/gad.8.15.1772. [DOI] [PubMed] [Google Scholar]
- Ebel C, Mariconti L, Gruissem W. Plant retinoblastoma homologues control nuclear proliferation in the female gametophyte. Nature. 2004;429:776–80. doi: 10.1038/nature02637. [DOI] [PubMed] [Google Scholar]
- Emili A, Ingles CJ. Promoter-dependent Photocross-linking of the Acidic Transcriptional Activator E2F-1 to the TATA-binding Protein. Journal of Biological Chemistry. 1995;270:13674–13680. doi: 10.1074/jbc.270.23.13674. [DOI] [PubMed] [Google Scholar]
- Fattaey AR, Helin K, Dembski MS, Dyson N, Harlow E, Vuocolo GA, Hanobik MG, Haskell KM, Oliff A, Defeo-Jones D, et al. Characterization of the retinoblastoma binding proteins RBP1 and RBP2. Oncogene. 1993;8:3149–56. [PubMed] [Google Scholar]
- Fay DS, Keenan S, Han M. fzr-1 and lin-35/Rb function redundantly to control cell proliferation in C. elegans as revealed by a nonbiased synthetic screen. Genes & Development. 2002;16:503–17. doi: 10.1101/gad.952302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong Y, Bender L, Wang W, Strome S. Regulation of the different chromatin states of autosomes and X chromosomes in the germ line of C. elegans. Science. 2002;296:2235–8. doi: 10.1126/science.1070790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friend SH, Bernards R, Rogelj S, Weinberg RA, Rapaport JM, Albert DM, Dryja TP. A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nature. 1986;323:643–6. doi: 10.1038/323643a0. [DOI] [PubMed] [Google Scholar]
- Frolov MV, Dyson NJ. Molecular mechanisms of E2F-dependent activation and pRB-mediated repression. Journal of Cell Science. 2004;117:2173–81. doi: 10.1242/jcs.01227. [DOI] [PubMed] [Google Scholar]
- Funayama R, Saito M, Tanobe H, Ishikawa F. Loss of linker histone H1 in cellular senescence. J Cell Biol. 2006;175:869–80. doi: 10.1083/jcb.200604005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung YK, Murphree AL, T’Ang A, Qian J, Hinrichs SH, Benedict WF. Structural evidence for the authenticity of the human retinoblastoma gene. Science. 1987;236:1657–61. doi: 10.1126/science.2885916. [DOI] [PubMed] [Google Scholar]
- Gabellini C, Del Bufalo D, Zupi G. Involvement of RB gene family in tumor angiogenesis. Oncogene. 2006;25:5326–32. doi: 10.1038/sj.onc.1209631. [DOI] [PubMed] [Google Scholar]
- Gladden AB, Diehl JA. The cyclin D1-dependent kinase associates with the pre-replication complex and modulates RB.MCM7 binding. Journal of Biological Chemistry. 2003;278:9754–9760. doi: 10.1074/jbc.M212088200. [DOI] [PubMed] [Google Scholar]
- Gonzalo S, Garcia-Cao M, Fraga MF, Schotta G, Peters AH, Cotter SE, Eguia R, Dean DC, Esteller M, Jenuwein T, Blasco MA. Role of the RB1 family in stabilizing histone methylation at constitutive heterochromatin. Nature Cell Biology. 2005;7:420–428. doi: 10.1038/ncb1235. [DOI] [PubMed] [Google Scholar]
- Goodrich DW. How the other half lives, the amino-terminal domain of the retinoblastoma tumor suppressor protein. Journal of Cellular Physiology. 2003;197:169–80. doi: 10.1002/jcp.10358. [DOI] [PubMed] [Google Scholar]
- Goodrich DW. The retinoblastoma tumor-suppressor gene, the exception that proves the rule. Oncogene. 2006;25:5233–5243. doi: 10.1038/sj.onc.1209616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodrich DW, Wang NP, Qian YW, Lee EY, Lee WH. The retinoblastoma gene product regulates progression through the G1 phase of the cell cycle. Cell. 1991;67:293–302. doi: 10.1016/0092-8674(91)90181-w. [DOI] [PubMed] [Google Scholar]
- Grishok A, Sharp PA. Negative regulation of nuclear divisions in Caenorhabditis elegans by retinoblastoma and RNA interference-related genes. Proc Natl Acad Sci U S A. 2005;102:17360–5. doi: 10.1073/pnas.0508989102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu W, Schneider JW, Condorelli G, Kaushal S, Mahdavi V, Nadal-Ginard B. Interaction of myogenic factors and the retinoblastoma protein mediates muscle cell commitment and differentiation. Cell. 1993;72:309–324. doi: 10.1016/0092-8674(93)90110-c. [DOI] [PubMed] [Google Scholar]
- Guo J, Longshore S, Nair R, Warner BW. Retinoblastoma protein (pRb), but not p107 or p130, is required for maintenance of enterocyte quiescence and differentiation in small intestine. Journal of Biological Chemistry. 2009;284:134–40. doi: 10.1074/jbc.M806133200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J, Sheng G, Warner BW. Epidermal growth factor-induced rapid retinoblastoma phosphorylation at Ser780 and Ser795 is mediated by ERK1/2 in small intestine epithelial cells. Journal of Biological Chemistry. 2005;280:35992–8. doi: 10.1074/jbc.M504583200. [DOI] [PubMed] [Google Scholar]
- Gupta PB, Chaffer CL, Weinberg RA. Cancer stem cells: mirage or reality? Nat Med. 2009;15:1010–2. doi: 10.1038/nm0909-1010. [DOI] [PubMed] [Google Scholar]
- Gutierrez GM, Kong E, Sabbagh Y, Brown NE, Lee JS, Demay MB, Thomas DM, Hinds PW. Impaired bone development and increased mesenchymal progenitor cells in calvaria of RB1−/− mice. Proc Natl Acad Sci U S A. 2008;105:18402–7. doi: 10.1073/pnas.0805925105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagemeier C, Cook A, Kouzarides T. The retinoblastoma protein binds E2F residues required for activation in vivo and TBP binding in vitro. Nucleic Acids Res. 1993;21:4998–5004. doi: 10.1093/nar/21.22.4998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development. Science. 2008;319:1352–5. doi: 10.1126/science.1140735. [DOI] [PubMed] [Google Scholar]
- Hallstrom TC, Nevins JR, Hallstrom TC, Nevins JR. Balancing the decision of cell proliferation and cell fate. Cell Cycle. 2009;8:532–5. doi: 10.4161/cc.8.4.7609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Hartl T, Boswell C, Orr-Weaver TL, Bosco G. Developmentally regulated histone modifications in Drosophila follicle cells: initiation of gene amplification is associated with histone H3 and H4 hyperacetylation and H1 phosphorylation. Chromosoma. 2007;116:197–214. doi: 10.1007/s00412-006-0092-2. [DOI] [PubMed] [Google Scholar]
- Hassler M, Singh S, Yue WW, Luczynski M, Lakbir R, Sanchez-Sanchez F, Bader T, Pearl LH, Mittnacht S. Crystal Structure of the Retinoblastoma Protein N Domain Provides Insight into Tumor Suppression, Ligand Interaction, and Holoprotein Architecture. Molecular Cell. 2007;28:371–385. doi: 10.1016/j.molcel.2007.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helin K, Lees JA, Vidal M, Dyson N, Harlow E, Fattaey A. A cDNA encoding a pRB-binding protein with properties of the transcription factor E2F. Cell. 1992;70:337–50. doi: 10.1016/0092-8674(92)90107-n. [DOI] [PubMed] [Google Scholar]
- Hemberger M, Dean W, Reik W. Epigenetic dynamics of stem cells and cell lineage commitment: digging Waddington’s canal. Nature Reviews Molecular Cell Biology. 2009;10:526–37. doi: 10.1038/nrm2727. [DOI] [PubMed] [Google Scholar]
- Hernando E, Nahle Z, Juan G, Diaz-Rodriguez E, Alaminos M, Hemann M, Michel L, Mittal V, Gerald W, Benezra R, Lowe SW, Cordon-Cardo C. Rb inactivation promotes genomic instability by uncoupling cell cycle progression from mitotic control. Nature. 2004;430:797–802. doi: 10.1038/nature02820. [DOI] [PubMed] [Google Scholar]
- Herrera RE, Chen F, Weinberg RA. Increased histone H1 phosphorylation and relaxed chromatin structure in Rb-deficient fibroblasts. Proc Natl Acad Sci U S A. 1996;93:11510–5. doi: 10.1073/pnas.93.21.11510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinds PW, Mittnacht S, Dulic V, Arnold A, Reed SI, Weinberg RA. Regulation of retinoblastoma protein functions by ectopic expression of human cyclins. Cell. 1992;70:993–1006. doi: 10.1016/0092-8674(92)90249-c. [DOI] [PubMed] [Google Scholar]
- Hou ST, Xie X, Baggley A, Park DS, Chen G, Walker T. Activation of the Rb/E2F1 pathway by the nonproliferative p38 MAPK during Fas (APO1/CD95)-mediated neuronal apoptosis. Journal of Biological Chemistry. 2002;277:48764–70. doi: 10.1074/jbc.M206336200. [DOI] [PubMed] [Google Scholar]
- Huang HJ, Yee JK, Shew JY, Chen PL, Bookstein R, Friedmann T, Lee EY, Lee WH. Suppression of the neoplastic phenotype by replacement of the RB gene in human cancer cells. Science. 1988;242:1563–1566. doi: 10.1126/science.3201247. [DOI] [PubMed] [Google Scholar]
- Humbert PO, Verona R, Trimarchi JM, Rogers C, Dandapani S, Lees JA. E2f3 is critical for normal cellular proliferation. Genes & Development. 2000;14:690–703. [PMC free article] [PubMed] [Google Scholar]
- Iavarone A, Garg P, Lasorella A, Hsu J, Israel MA. The helix-loop-helix protein Id-2 enhances cell proliferation and binds to the retinoblastoma protein. Genes & Development. 1994;8:1270–1284. doi: 10.1101/gad.8.11.1270. [DOI] [PubMed] [Google Scholar]
- Iavarone A, King ER, Dai XM, Leone G, Stanley ER, Lasorella A. Retinoblastoma promotes definitive erythropoiesis by repressing Id2 in fetal liver macrophages. Nature. 2004;432:1040–1045. doi: 10.1038/nature03068. [DOI] [PubMed] [Google Scholar]
- Ingouff M, Jullien PE, Berger F. The female gametophyte and the endosperm control cell proliferation and differentiation of the seed coat in Arabidopsis. Plant Cell. 2006;18:3491–501. doi: 10.1105/tpc.106.047266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue Y, Kitagawa M, Taya Y. Phosphorylation of pRB at Ser612 by Chk1/2 leads to a complex between pRB and E2F-1 after DNA damage. EMBO J. 2007;26:2083–93. doi: 10.1038/sj.emboj.7601652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iovino F, Lentini L, Amato A, Di Leonardo A. RB acute loss induces centrosome amplification and aneuploidy in murine primary fibroblasts. Mol Cancer. 2006;5:38. doi: 10.1186/1476-4598-5-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaac CE, Francis SM, Martens AL, Julian LM, Seifried LA, Erdmann N, Binne UK, Harrington L, Sicinski P, Berube NG, Dyson NJ, Dick FA. The retinoblastoma protein regulates pericentric heterochromatin. Molecular & Cellular Biology. 2006;26:3659–3671. doi: 10.1128/MCB.26.9.3659-3671.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida S, Huang E, Zuzan H, Spang R, Leone G, West M, Nevins JR. Role for E2F in control of both DNA replication and mitotic functions as revealed from DNA microarray analysis. Molecular & Cellular Biology. 2001;21:4684–99. doi: 10.1128/MCB.21.14.4684-4699.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwase S, Lan F, Bayliss P, de la Torre-Ubieta L, Huarte M, Qi HH, Whetstine JR, Bonni A, Roberts TM, Shi Y. The X-linked mental retardation gene SMCX/JARID1C defines a family of histone H3 lysine 4 demethylases. Cell. 2007;128:1077–88. doi: 10.1016/j.cell.2007.02.017. [DOI] [PubMed] [Google Scholar]
- Jacks T, Fazeli A, Schmitt EM, Bronson RT, Goodell MA, Weinberg RA. Effects of an Rb mutation in the mouse. Nature. 1992;359:295–300. doi: 10.1038/359295a0. [DOI] [PubMed] [Google Scholar]
- Jacques TS, Swales A, Brzozowski MJ, Henriquez NV, Linehan JM, Mirzadeh Z, COM, Naumann H, Alvarez-Buylla A, Brandner S. Combinations of genetic mutations in the adult neural stem cell compartment determine brain tumour phenotypes. Embo J. 2010;29:222–35. doi: 10.1038/emboj.2009.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji P, Jiang H, Rekhtman K, Bloom J, Ichetovkin M, Pagano M, Zhu L. An Rb-Skp2-p27 pathway mediates acute cell cycle inhibition by Rb and is retained in a partial-penetrance Rb mutant. Molecular Cell. 2004;16:47–58. doi: 10.1016/j.molcel.2004.09.029. [DOI] [PubMed] [Google Scholar]
- Jiang Z, Zacksenhaus E. Activation of retinoblastoma protein in mammary gland leads to ductal growth suppression, precocious differentiation, and adenocarcinoma. Journal of Cell Biology. 2002;156:185–198. doi: 10.1083/jcb.200106084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DA, Zhang J, Frase S, Wilson M, Rodriguez-Galindo C, Dyer MA. Neuronal differentiation and synaptogenesis in retinoblastoma. Cancer Research. 2007;67:2701–11. doi: 10.1158/0008-5472.CAN-06-3754. [DOI] [PubMed] [Google Scholar]
- Kadri Z, Shimizu R, Ohneda O, Maouche-Chretien L, Gisselbrecht S, Yamamoto M, Romeo PH, Leboulch P, Chretien S. Direct binding of pRb/E2F-2 to GATA-1 regulates maturation and terminal cell division during erythropoiesis. PLOS Biology. 2009;7:e1000123. doi: 10.1371/journal.pbio.1000123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaelin WG, Jr, Krek W, Sellers WR, DeCaprio JA, Ajchenbaum F, Fuchs CS, Chittenden T, Li Y, Farnham PJ, Blanar MA, et al. Expression cloning of a cDNA encoding a retinoblastoma-binding protein with E2F-like properties. Cell. 1992;70:351–64. doi: 10.1016/0092-8674(92)90108-o. [DOI] [PubMed] [Google Scholar]
- Ketting RF, Haverkamp TH, van Luenen HG, Plasterk RH. Mut-7 of C. elegans, required for transposon silencing and RNA interference, is a homolog of Werner syndrome helicase and RNaseD. Cell. 1999;99:133–41. doi: 10.1016/s0092-8674(00)81645-1. [DOI] [PubMed] [Google Scholar]
- Kim HY, Ahn BY, Cho Y. Structural basis for the inactivation of retinoblastoma tumor suppressor by SV40 large T antigen. EMBO Journal. 2001;20:295–304. doi: 10.1093/emboj/20.1.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HY, Cho Y. Structural similarity between the pocket region of retinoblastoma tumour suppressor and the cyclin-box. Nature Structural Biology. 1997;4:390–395. doi: 10.1038/nsb0597-390. [DOI] [PubMed] [Google Scholar]
- Kim SJ, Wagner S, Liu F, O’Reilly MA, Robbins PD, Green MR. Retinoblastoma gene product activates expression of the human TGF-beta 2 gene through transcription factor ATF-2. Nature. 1992;358:331–4. doi: 10.1038/358331a0. [DOI] [PubMed] [Google Scholar]
- Kirienko NV, Fay DS. Transcriptome profiling of the C. elegans Rb ortholog reveals diverse developmental roles. Dev Biol. 2007;305:674–84. doi: 10.1016/j.ydbio.2007.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klose RJ, Yan Q, Tothova Z, Yamane K, Erdjument-Bromage H, Tempst P, Gilliland DG, Zhang Y, Kaelin WG., Jr The retinoblastoma binding protein RBP2 is an H3K4 demethylase. Cell. 2007;128:889–900. doi: 10.1016/j.cell.2007.02.013. [DOI] [PubMed] [Google Scholar]
- Knudsen ES, Sexton CR, Mayhew CN, Knudsen ES, Sexton CR, Mayhew CN. Role of the retinoblastoma tumor suppressor in the maintenance of genome integrity. Current Molecular Medicine. 2006;6:749–57. doi: 10.2174/1566524010606070749. [DOI] [PubMed] [Google Scholar]
- Knudsen ES, Wang JYJ. Differential regulation of retinoblastoma protein function by specific Cdk phosphorylation sites. Journal of Biological Chemistry. 1996;271:8313–8320. doi: 10.1074/jbc.271.14.8313. [DOI] [PubMed] [Google Scholar]
- Knudsen ES, Wang JYJ. Dual mechanisms for the inhibition of E2F binding to RB by cyclin-dependent kinase-mediated RB phosphorylation. Molecular & Cellular Biology. 1997;17:5771–5783. doi: 10.1128/mcb.17.10.5771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudson AG., Jr Mutation and cancer: statistical study of retinoblastoma. Proceedings of the National Academy of Sciences of the United States of America. 1971;68:820–3. doi: 10.1073/pnas.68.4.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korenjak M, Brehm A, Korenjak M, Brehm A. E2F-Rb complexes regulating transcription of genes important for differentiation and development. Current Opinion in Genetics & Development. 2005;15:520–7. doi: 10.1016/j.gde.2005.07.001. [DOI] [PubMed] [Google Scholar]
- Kucherlapati MH, Yang K, Fan K, Kuraguchi M, Sonkin D, Rosulek A, Lipkin M, Bronson RT, Aronow BJ, Kucherlapati R. Loss of Rb1 in the gastrointestinal tract of Apc1638N mice promotes tumors of the cecum and proximal colon. Proc Natl Acad Sci U S A. 2008;105:15493–8. doi: 10.1073/pnas.0802933105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyritsis AP, Tsokos M, Triche TJ, Chader GJ. Retinoblastoma--origin from a primitive neuroectodermal cell? Nature. 1984;307:471–3. doi: 10.1038/307471a0. [DOI] [PubMed] [Google Scholar]
- Lai A, Kennedy BK, Barbie DA, Bertos NR, Yang XJ, Theberge MC, Tsai SC, Seto E, Zhang Y, Kuzmichev A, Lane WS, Reinberg D, Harlow E, Branton PE. RBP1 recruits the mSIN3-histone deacetylase complex to the pocket of retinoblastoma tumor suppressor family proteins found in limited discrete regions of the nucleus at growth arrest. Molecular & Cellular Biology. 2001;21:2918–32. doi: 10.1128/MCB.21.8.2918-2932.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai A, Lee JM, Yang WM, DeCaprio JA, Kaelin WG, Jr, Seto E, Branton PE. RBP1 recruits both histone deacetylase-dependent and -independent repression activities to retinoblastoma family proteins. Molecular & Cellular Biology. 1999;19:6632–6641. doi: 10.1128/mcb.19.10.6632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landsberg RL, Sero JE, Danielian PS, Yuan TL, Lee EY, Lees JA. The role of E2F4 in adipogenesis is independent of its cell cycle regulatory activity. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:2456–2461. doi: 10.1073/pnas.0138064100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang SE, McMahon SB, Cole MD, Hearing P. E2F transcriptional activation requires TRRAP and GCN5 cofactors. Journal of Biological Chemistry. 2001;276:32627–34. doi: 10.1074/jbc.M102067200. [DOI] [PubMed] [Google Scholar]
- Laurie NA, Donovan SL, Shih CS, Zhang J, Mills N, Fuller C, Teunisse A, Lam S, Ramos Y, Mohan A, Johnson D, Wilson M, Rodriguez-Galindo C, Quarto M, Francoz S, Mendrysa SM, Guy RK, Marine JC, Jochemsen AG, Dyer MA. Inactivation of the p53 pathway in retinoblastoma. Nature. 2006;444:61–6. doi: 10.1038/nature05194. [DOI] [PubMed] [Google Scholar]
- Leduc C, Claverie P, Eymin B, Col E, Khochbin S, Brambilla E, Gazzeri S. p14ARF promotes RB accumulation through inhibition of its Tip60-dependent acetylation. Oncogene. 2006;25:4147–54. doi: 10.1038/sj.onc.1209446. [DOI] [PubMed] [Google Scholar]
- Lee CCJH. Structural basis for the recognition of the E2F transactivation domain by the retinoblastoma tumor suppressor. Genes & Development. 2002;16:3199–3212. doi: 10.1101/gad.1046102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee EY, Cam H, Ziebold U, Rayman JB, Lees JA, Dynlacht BD. E2F4 loss suppresses tumorigenesis in Rb mutant mice. Cancer Cell. 2002;2:463–472. doi: 10.1016/s1535-6108(02)00207-6. [DOI] [PubMed] [Google Scholar]
- Lee EY, Chang CY, Hu N, Wang YC, Lai CC, Herrup K, Lee WH. Mice deficient for Rb are nonviable and show defects in neurogenesis and haematopoiesis. Nature. 1992;359:288–294. doi: 10.1038/359288a0. [DOI] [PubMed] [Google Scholar]
- Lee JO, Russo AA, Pavletich NP. Structure of the retinoblastoma tumour-suppressor pocket domain bound to a peptide from HPV E7. Nature. 1998;391:859–865. doi: 10.1038/36038. [DOI] [PubMed] [Google Scholar]
- Lee WH, Bookstein R, Hong F, Young LJ, Shew JY, Lee EYHP. Human retinoblastoma susceptibility gene: cloning, identification, and sequence. Science. 1987;235:1394–1399. doi: 10.1126/science.3823889. [DOI] [PubMed] [Google Scholar]
- Leng X, Noble M, Adams PD, Qin J, Harper JW. Reversal of growth suppression by p107 via direct phosphorylation by cyclin D1/cyclin-dependent kinase 4. Molecular & Cellular Biology. 2002;22:2242–54. doi: 10.1128/MCB.22.7.2242-2254.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung JK, Berube N, Venable S, Ahmed S, Timchenko N, Pereira-Smith OM. MRG15 activates the B-myb promoter through formation of a nuclear complex with the retinoblastoma protein and the novel protein PAM14. Journal of Biological Chemistry. 2001;276:39171–8. doi: 10.1074/jbc.M103435200. [DOI] [PubMed] [Google Scholar]
- Liu H, Dibling B, Spike B, Dirlam A, Macleod K. New roles for the RB tumor suppressor protein. Current Opinion in Genetics & Development. 2004;14:55–64. doi: 10.1016/j.gde.2003.11.005. [DOI] [PubMed] [Google Scholar]
- Liu X, Marmorstein R. Structure of the retinoblastoma protein bound to adenovirus E1A reveals the molecular basis for viral oncoprotein inactivation of a tumor suppressor. Genes & Development. 2007;21:2711–6. doi: 10.1101/gad.1590607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longworth MS, Dyson NJ. pRb, a local chromatin organizer with global possibilities. Chromosoma. 2009;119:1–11. doi: 10.1007/s00412-009-0238-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longworth MS, Herr A, Ji JY, Dyson NJ. RBF1 promotes chromatin condensation through a conserved interaction with the Condensin II protein dCAP-D3. Genes & Development. 2008;22:1011–24. doi: 10.1101/gad.1631508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Danielsen M. Differential regulation of androgen and glucocorticoid receptors by retinoblastoma protein. Journal of Biological Chemistry. 1998;273:31528–33. doi: 10.1074/jbc.273.47.31528. [DOI] [PubMed] [Google Scholar]
- Ludlow JW, Glendening CL, Livingston DM, DeCaprio JA. Specific enzymatic dephosphorylation of the retinoblastoma protein. Molecular & Cellular Biology. 1993;13:367–372. doi: 10.1128/mcb.13.1.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo RX, Postigo AA, Dean DC. Rb interacts with histone deacetylase to repress transcription. Cell. 1998;92:463–73. doi: 10.1016/s0092-8674(00)80940-x. [DOI] [PubMed] [Google Scholar]
- MacLellan WR, Xiao G, Abdellatif M, Schneider MD. A Novel Rb- and p300-Binding Protein Inhibits Transactivation by MyoD. Molecular & Cellular Biology. 2000;20:8903–8915. doi: 10.1128/mcb.20.23.8903-8915.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macleod KF. The role of the RB tumour suppressor pathway in oxidative stress responses in the haematopoietic system. Nature Reviews Cancer. 2008;8:769–81. doi: 10.1038/nrc2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacPherson D, Sage J, Kim T, Ho D, McLaughlin ME, Jacks T. Cell type-specific effects of Rb deletion in the murine retina. Genes & Development. 2004;18:1681–1694. doi: 10.1101/gad.1203304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddison LA, Sutherland BW, Barrios RJ, Greenberg NM. Conditional deletion of rb causes early stage prostate cancer. Cancer Research. 2004;64:6018–6025. doi: 10.1158/0008-5472.CAN-03-2509. [DOI] [PubMed] [Google Scholar]
- Maenhaut C, Dumont JE, Roger PP, van Staveren WC. Cancer stem cells: a reality, a myth, a fuzzy concept or a misnomer? An analysis. Carcinogenesis. 2010;31:149–58. doi: 10.1093/carcin/bgp259. [DOI] [PubMed] [Google Scholar]
- Magnaghi-Jaulin L, Groisman R, Naguibneva I, Robin P, Lorain S, LeVillain JP, Troalen F, Trouche D, Harel-Bellan A. Retinoblastoma protein represses transcription by recruiting a histone deacetylase. Nature. 1998;391:601–605. doi: 10.1038/35410. [DOI] [PubMed] [Google Scholar]
- Malumbres M, Barbacid M. To cycle or not to cycle: a critical decision in cancer. Nature Reviews Cancer. 2001;1:222–231. doi: 10.1038/35106065. [DOI] [PubMed] [Google Scholar]
- Manning AL, Longworth MS, Dyson NJ. Loss of pRB causes centromere dysfunction and chromosomal instability. Genes & Development. 2010;24:1364–76. doi: 10.1101/gad.1917310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marees T, Moll AC, Imhof SM, de Boer MR, Ringens PJ, van Leeuwen FE. Risk of Second Malignancies in Survivors of Retinoblastoma: More Than 40 Years of Follow-up. J Natl Cancer Inst. 2008;100:1771–1779. doi: 10.1093/jnci/djn394. [DOI] [PubMed] [Google Scholar]
- Marino S, Hoogervoorst D, Brandner S, Berns A. Rb and p107 are required for normal cerebellar development and granule cell survival but not for Purkinje cell persistence. Development. 2003;130:3359–68. doi: 10.1242/dev.00553. [DOI] [PubMed] [Google Scholar]
- Markham D, Munro S, Soloway J, O’Connor DP, La Thangue NB. DNA-damage-responsive acetylation of pRb regulates binding to E2F-1. EMBO Rep. 2006;7:192–8. doi: 10.1038/sj.embor.7400591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Cruz AB, Santos M, Lara MF, Segrelles C, Ruiz S, Moral M, Lorz C, Garcia-Escudero R, Paramio JM. Spontaneous squamous cell carcinoma induced by the somatic inactivation of retinoblastoma and Trp53 tumor suppressors. Cancer Research. 2008;68:683–92. doi: 10.1158/0008-5472.CAN-07-3049. [DOI] [PubMed] [Google Scholar]
- McClellan KA, Ruzhynsky VA, Douda DN, Vanderluit JL, Ferguson KL, Chen D, Bremner R, Park DS, Leone G, Slack RS. Unique requirement for Rb/E2F3 in neuronal migration: evidence for cell cycle-independent functions. Molecular & Cellular Biology. 2007a;27:4825–43. doi: 10.1128/MCB.02100-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClellan KA, Slack RS, McClellan KA, Slack RS. Specific in vivo roles for E2Fs in differentiation and development. Cell Cycle. 2007b;6:2917–27. doi: 10.4161/cc.6.23.4997. [DOI] [PubMed] [Google Scholar]
- Meloni AR, Smith EJ, Nevins JR. A mechanism for Rb/p130-mediated transcription repression involving recruitment of the CtBP corepressor. Proc Natl Acad Sci U S A. 1999;96:9574–9. doi: 10.1073/pnas.96.17.9574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meuwissen R, Linn SC, Linnoila RI, Zevenhoven J, Mooi WJ, Berns A. Induction of small cell lung cancer by somatic inactivation of both Trp53 and Rb1 in a conditional mouse model. Cancer Cell. 2003;4:181–9. doi: 10.1016/s1535-6108(03)00220-4. [DOI] [PubMed] [Google Scholar]
- Miccadei S, Provenzano C, Mojzisek M, Giorgio Natali P, Civitareale D. Retinoblastoma protein acts as Pax 8 transcriptional coactivator. Oncogene. 2005;24:6993–7001. doi: 10.1038/sj.onc.1208861. [DOI] [PubMed] [Google Scholar]
- Miyake S, Sellers WR, Safran M, Li X, Zhao W, Grossman SR, Gan J, DeCaprio JA, Adams PD, Kaelin WG., Jr Cells degrade a novel inhibitor of differentiation with E1A-like properties upon exiting the cell cycle. Molecular & Cellular Biology. 2000;20:8889–8902. doi: 10.1128/mcb.20.23.8889-8902.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohn F, Schubeler D. Genetics and epigenetics: stability and plasticity during cellular differentiation. Trends Genet. 2009;25:129–36. doi: 10.1016/j.tig.2008.12.005. [DOI] [PubMed] [Google Scholar]
- Moreira AL, Gonen M, Rekhtman N, Downey RJ. Progenitor stem cell marker expression by pulmonary carcinomas. Mod Pathol. 2010;23:889–95. doi: 10.1038/modpathol.2010.68. [DOI] [PubMed] [Google Scholar]
- Morris EJ, Dyson NJ. Retinoblastoma protein partners. Advances in Cancer Research. 2001;82:1–54. doi: 10.1016/s0065-230x(01)82001-7. [DOI] [PubMed] [Google Scholar]
- Mukherjee P, Winter SL, Alexandrow MG, Mukherjee P, Winter SL, Alexandrow MG. Cell cycle arrest by transforming growth factor beta1 near G1/S is mediated by acute abrogation of prereplication complex activation involving an Rb-MCM interaction. Molecular & Cellular Biology. 2010;30:845–56. doi: 10.1128/MCB.01152-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L. A global double-fluorescent Cre reporter mouse. Genesis. 2007;45:593–605. doi: 10.1002/dvg.20335. [DOI] [PubMed] [Google Scholar]
- Nair JS, Ho AL, Tse AN, Coward J, Cheema H, Ambrosini G, Keen N, Schwartz GK, Nair JS, Ho AL, Tse AN, Coward J, Cheema H, Ambrosini G, Keen N, Schwartz GK. Aurora B kinase regulates the postmitotic endoreduplication checkpoint via phosphorylation of the retinoblastoma protein at serine 780. Molecular Biology of the Cell. 2009;20:2218–28. doi: 10.1091/mbc.E08-08-0885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narita M, Narita M, Krizhanovsky V, Nunez S, Chicas A, Hearn SA, Myers MP, Lowe SW. A novel role for high-mobility group a proteins in cellular senescence and heterochromatin formation. Cell. 2006;126:503–14. doi: 10.1016/j.cell.2006.05.052. [DOI] [PubMed] [Google Scholar]
- Narita M, Nunez S, Heard E, Narita M, Lin AW, Hearn SA, Spector DL, Hannon GJ, Lowe SW. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell. 2003;113:703–716. doi: 10.1016/s0092-8674(03)00401-x. [DOI] [PubMed] [Google Scholar]
- Nath N, Wang S, Betts V, Knudsen E, Chellappan S. Apoptotic and mitogenic stimuli inactivate Rb by differential utilization of p38 and cyclin-dependent kinases. Oncogene. 2003;22:5986–94. doi: 10.1038/sj.onc.1206843. [DOI] [PubMed] [Google Scholar]
- Nguyen DX, McCance DJ. Role of the retinoblastoma tumor suppressor protein in cellular differentiation. Journal of Cellular Biochemistry. 2005;94:870–879. doi: 10.1002/jcb.20375. [DOI] [PubMed] [Google Scholar]
- Nielsen SJ, Schneider R, Bauer UM, Bannister AJ, Morrison A, O’Carroll D, Firestein R, Cleary M, Jenuwein T, Herrera RE, Kouzarides T. Rb targets histone H3 methylation and HP1 to promoters. Nature. 2001;412:561–565. doi: 10.1038/35087620. [DOI] [PubMed] [Google Scholar]
- Novitch BG, Mulligan GJ, Jacks T, Lassar AB. Skeletal muscle cells lacking the retinoblastoma protein display defects in muscle gene expression and accumulate in S and G2 phases of the cell cycle. Journal of Cell Biology. 1996;135:441–456. doi: 10.1083/jcb.135.2.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novitch BG, Spicer DB, Kim PS, Cheung WL, Lassar AB. pRb is required for MEF2-dependent gene expression as well as cell-cycle arrest during skeletal muscle differentiation. Current Biology. 1999;9:449–459. doi: 10.1016/s0960-9822(99)80210-3. [DOI] [PubMed] [Google Scholar]
- Ouellet J, Roy R. The lin-35/Rb and RNAi pathways cooperate to regulate a key cell cycle transition in C. elegans. BMC Dev Biol. 2007;7:38. doi: 10.1186/1471-213X-7-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page BD, Guedes S, Waring D, Priess JR. The C. elegans E2F- and DP-related proteins are required for embryonic asymmetry and negatively regulate Ras/MAPK signaling. Molecular Cell. 2001;7:451–60. doi: 10.1016/s1097-2765(01)00193-9. [DOI] [PubMed] [Google Scholar]
- Papadimou E, Menard C, Grey C, Puceat M. Interplay between the retinoblastoma protein and LEK1 specifies stem cells toward the cardiac lineage. EMBO Journal. 2005;24:1750–1761. doi: 10.1038/sj.emboj.7600652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park M, Krause MW. Regulation of postembryonic G(1) cell cycle progression in Caenorhabditis elegans by a cyclin D/CDK-like complex. Development. 1999;126:4849–60. doi: 10.1242/dev.126.21.4849. [DOI] [PubMed] [Google Scholar]
- Pearson A, Greenblatt J. Modular organization of the E2F1 activation domain and its interaction with general transcription factors TBP and TFIIH. Oncogene. 1997;15:2643–58. doi: 10.1038/sj.onc.1201451. [DOI] [PubMed] [Google Scholar]
- Pennaneach V, Salles-Passador I, Munshi A, Brickner H, Regazzoni K, Dick F, Dyson N, Chen TT, Wang JY, Fotedar R, Fotedar A. The large subunit of replication factor C promotes cell survival after DNA damage in an LxCxE motif- and Rb-dependent manner. Molecular Cell. 2001;7:715–27. doi: 10.1016/s1097-2765(01)00217-9. [DOI] [PubMed] [Google Scholar]
- Polager S, Ofir M, Ginsberg D. E2F1 regulates autophagy and the transcription of autophagy genes. Oncogene. 2008;27:4860–4. doi: 10.1038/onc.2008.117. [DOI] [PubMed] [Google Scholar]
- Prieur A, Peeper DS. Cellular senescence in vivo: a barrier to tumorigenesis. Curr Opin Cell Biol. 2008;20:150–5. doi: 10.1016/j.ceb.2008.01.007. [DOI] [PubMed] [Google Scholar]
- Qian YW, Lee EY. Dual retinoblastoma-binding proteins with properties related to a negative regulator of ras in yeast. Journal of Biological Chemistry. 1995;270:25507–13. doi: 10.1074/jbc.270.43.25507. [DOI] [PubMed] [Google Scholar]
- Qian YW, Wang YC, Hollingsworth RE, Jr, Jones D, Ling N, Lee EY. A retinoblastoma-binding protein related to a negative regulator of Ras in yeast. Nature. 1993;364:648–52. doi: 10.1038/364648a0. [DOI] [PubMed] [Google Scholar]
- Ren B, Cam H, Takahashi Y, Volkert T, Terragni J, Young RA, Dynlacht BD. E2F integrates cell cycle progression with DNA repair, replication, and G(2)/M checkpoints. Genes & Development. 2002;16:245–256. doi: 10.1101/gad.949802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson KD, Ait-Si-Ali S, Yokochi T, Wade PA, Jones PL, Wolffe AP. DNMT1 forms a complex with Rb, E2F1 and HDAC1 and represses transcription from E2F-responsive promoters. Nature Genetics. 2000;25:338–342. doi: 10.1038/77124. [DOI] [PubMed] [Google Scholar]
- Royzman I, Austin RJ, Bosco G, Bell SP, Orr-Weaver TL. ORC localization in Drosophila follicle cells and the effects of mutations in dE2F and dDP. Genes & Development. 1999;13:827–40. doi: 10.1101/gad.13.7.827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin SM, Gall AL, Zheng N, Pavletich NP. Structure of the Rb C-Terminal Domain Bound to E2F1-DP1: A Mechanism for Phosphorylation-Induced E2F Release. Cell. 2005;123:1093–1106. doi: 10.1016/j.cell.2005.09.044. [DOI] [PubMed] [Google Scholar]
- Ruiz S, Santos M, Segrelles C, Leis H, Jorcano JL, Berns A, Paramio JM, Vooijs M. Unique and overlapping functions of pRb and p107 in the control of proliferation and differentiation in epidermis. Development. 2004;131:2737–2748. doi: 10.1242/dev.01148. [DOI] [PubMed] [Google Scholar]
- Sage C, Huang M, Karimi K, Gutierrez G, Vollrath MA, Zhang DS, Garcia-Anoveros J, Hinds PW, Corwin JT, Corey DP, Chen ZY. Proliferation of functional hair cells in vivo in the absence of the retinoblastoma protein. Science. 2005;307:1114–8. doi: 10.1126/science.1106642. [DOI] [PubMed] [Google Scholar]
- Sage J, Miller AL, Perez-Mancera PA, Wysocki JM, Jacks T. Acute mutation of retinoblastoma gene function is sufficient for cell cycle re-entry. Nature. 2003;424:223–228. doi: 10.1038/nature01764. [DOI] [PubMed] [Google Scholar]
- Sage J, Mulligan GJ, Attardi LD, Miller A, Chen S, Williams B, Theodorou E, Jacks T. Targeted disruption of the three Rb-related genes leads to loss of G(1) control and immortalization. Genes & Development. 2000;14:3037–50. doi: 10.1101/gad.843200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saijo M, Sakai Y, Kishino T, Niikawa N, Matsuura Y, Morino K, Tamai K, Taya Y. Molecular cloning of a human protein that binds to the retinoblastoma protein and chromosomal mapping. Genomics. 1995;27:511–9. doi: 10.1006/geno.1995.1084. [DOI] [PubMed] [Google Scholar]
- de Sanchez ML, Caro E, Desvoyes B, Ramirez-Parra E, Gutierrez C. Chromatin dynamics during the plant cell cycle. Semin Cell Dev Biol. 2008;19:537–46. doi: 10.1016/j.semcdb.2008.07.014. [DOI] [PubMed] [Google Scholar]
- Sang Y, Wu MF, Wagner D. The stem cell--chromatin connection. Semin Cell Dev Biol. 2009;20:1143–8. doi: 10.1016/j.semcdb.2009.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider JW, Gu W, Zhu L, Mahdavi V, Nadal-Ginard B. Reversal of terminal differentiation mediated by p107 in Rb−/− muscle cells. Science. 1994;264:1467–1471. doi: 10.1126/science.8197461. [DOI] [PubMed] [Google Scholar]
- Shan B, Zhu X, Chen PL, Durfee T, Yang Y, Sharp D, Lee WH. Molecular cloning of cellular genes encoding retinoblastoma- associated proteins: identification of a gene with properties of the transcription factor E2F. Molecular & Cellular Biology. 1992;12:5620–5631. doi: 10.1128/mcb.12.12.5620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Kelly TK, Jones PA. Epigenetics in cancer. Carcinogenesis. 2010;31:27–36. doi: 10.1093/carcin/bgp220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen WH. The plant E2F-Rb pathway and epigenetic control. Trends Plant Sci. 2002;7:505–11. doi: 10.1016/s1360-1385(02)02351-8. [DOI] [PubMed] [Google Scholar]
- Sherr CJ, McCormick F. The RB and p53 pathways in cancer. Cancer Cell. 2002;2:103–12. doi: 10.1016/s1535-6108(02)00102-2. [DOI] [PubMed] [Google Scholar]
- Siddiqui H, Fox SR, Gunawardena RW, Knudsen ES. Loss of RB compromises specific heterochromatin modifications and modulates HP1alpha dynamics. J Cell Physiol. 2007;211:131–7. doi: 10.1002/jcp.20913. [DOI] [PubMed] [Google Scholar]
- Sijen T, Plasterk RH. Transposon silencing in the Caenorhabditis elegans germ line by natural RNAi. Nature. 2003;426:310–4. doi: 10.1038/nature02107. [DOI] [PubMed] [Google Scholar]
- Singh P, Coe J, Hong W. A role for retinoblastoma protein in potentiating transcriptional activation by the glucocorticoid receptor. Nature. 1995;374:562–5. doi: 10.1038/374562a0. [DOI] [PubMed] [Google Scholar]
- Sotillo R, Hernando E, Diaz-Rodriguez E, Teruya-Feldstein J, Cordon-Cardo C, Lowe SW, Benezra R. Mad2 overexpression promotes aneuploidy and tumorigenesis in mice. Cancer Cell. 2007;11:9–23. doi: 10.1016/j.ccr.2006.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spike BT, Dirlam A, Dibling BC, Marvin J, Williams BO, Jacks T, Macleod KF. The Rb tumor suppressor is required for stress erythropoiesis. EMBO Journal. 2004;23:4319–4329. doi: 10.1038/sj.emboj.7600432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterner JM, Dew-Knight S, Musahl C, Kornbluth S, Horowitz JM. Negative regulation of DNA replication by the retinoblastoma protein is mediated by its association with MCM7. Molecular & Cellular Biology. 1998;18:2748–2757. doi: 10.1128/mcb.18.5.2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strober BE, Dunaief JL, Guha, Goff SP. Functional interactions between the hBRM/hBRG1 transcriptional activators and the pRB family of proteins. Molecular & Cellular Biology. 1996;16:1576–83. doi: 10.1128/mcb.16.4.1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuma K, Takahara N, Suzuma I, Isshiki K, Ueki K, Leitges M, Aiello LP, King GL. Characterization of protein kinase C beta isoform’s action on retinoblastoma protein phosphorylation, vascular endothelial growth factor-induced endothelial cell proliferation, and retinal neovascularization. 2002. pp. 721–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaki T, Fukasawa K, Suzuki-Takahashi I, Semba K, Kitagawa M, Taya Y, Hirai H. Preferences for phosphorylation sites in the retinoblastoma protein of D-type cyclin-dependent kinases, Cdk4 and Cdk6, in vitro. J Biochem. 2005;137:381–6. doi: 10.1093/jb/mvi050. [DOI] [PubMed] [Google Scholar]
- Takemoto A, Maeshima K, Ikehara T, Yamaguchi K, Murayama A, Imamura S, Imamoto N, Yokoyama S, Hirano T, Watanabe Y, Hanaoka F, Yanagisawa J, Kimura K. The chromosomal association of condensin II is regulated by a noncatalytic function of PP2A. Nat Struct Mol Biol. 2009;16:1302–8. doi: 10.1038/nsmb.1708. [DOI] [PubMed] [Google Scholar]
- Templeton DJ, Park SH, Lanier L, Weinberg RA. Nonfunctional mutants of the retinoblastoma protein are characterized by defects in phosphorylation, viral oncoprotein association, and nuclear tethering. Proceedings of the National Academy of Sciences of the United States of America. 1991;88:3033–3037. doi: 10.1073/pnas.88.8.3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas DM, Carty SA, Piscopo DM, Lee JS, Wang WF, Forrester WC, Hinds PW. The retinoblastoma protein acts as a transcriptional coactivator required for osteogenic differentiation. Molecular Cell. 2001;8:303–316. doi: 10.1016/s1097-2765(01)00327-6. [DOI] [PubMed] [Google Scholar]
- Tracy K, Dibling BC, Spike BT, Knabb JR, Schumacker P, Macleod KF. BNIP3 is an RB/E2F target gene required for hypoxia-induced autophagy. Molecular & Cellular Biology. 2007;27:6229–42. doi: 10.1128/MCB.02246-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trojer P, Li G, Sims RJ, 3rd, Vaquero A, Kalakonda N, Boccuni P, Lee D, Erdjument-Bromage H, Tempst P, Nimer SD, Wang YH, Reinberg D. L3MBTL1, a histone-methylation-dependent chromatin lock. Cell. 2007;129:915–28. doi: 10.1016/j.cell.2007.03.048. [DOI] [PubMed] [Google Scholar]
- Trouche D, Le Chalony C, Muchardt C, Yaniv M, Kouzarides T. RB and hbrm cooperate to repress the activation functions of E2F1. Proc Natl Acad Sci U S A. 1997;94:11268–73. doi: 10.1073/pnas.94.21.11268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai KY, Hu Y, Macleod KF, Crowley D, Yamasaki L, Jacks T. Mutation of E2f-1 suppresses apoptosis and inappropriate S phase entry and extends survival of Rb-deficient mouse embryos. Molecular Cell. 1998;2:293–304. doi: 10.1016/s1097-2765(00)80274-9. [DOI] [PubMed] [Google Scholar]
- van den Heuvel S, Dyson NJ. Conserved functions of the pRB and E2F families. Nature Reviews Molecular Cell Biology. 2008;9:713–24. doi: 10.1038/nrm2469. [DOI] [PubMed] [Google Scholar]
- van Harn T, Foijer F, van Vugt M, Banerjee R, Yang F, Oostra A, Joenje H, te Riele H. Loss of Rb proteins causes genomic instability in the absence of mitogenic signaling. Genes & Development. 2010;24:1377–88. doi: 10.1101/gad.580710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance KW, Shaw HM, Rodriguez M, Ott S, Goding CR. The Retinoblastoma Protein Modulates Tbx2 Functional Specificity. Molecular Biology of the Cell. 2010 doi: 10.1091/mbc.E09-12-1029. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandel L, Nicolas E, Vaute O, Ferreira R, Ait-Si-Ali S, Trouche D. Transcriptional repression by the retinoblastoma protein through the recruitment of a histone methyltransferase. Molecular & Cellular Biology. 2001;21:6484–94. doi: 10.1128/MCB.21.19.6484-6494.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderluit JL, Ferguson KL, Nikoletopoulou V, Parker M, Ruzhynsky V, Alexson T, McNamara SM, Park DS, Rudnicki M, Slack RS. p107 regulates neural precursor cells in the mammalian brain. Journal of Cell Biology. 2004;166:853–863. doi: 10.1083/jcb.200403156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaute O, Nicolas E, Vandel L, Trouche D. Functional and physical interaction between the histone methyl transferase Suv39H1 and histone deacetylases. Nucleic Acids Res. 2002;30:475–81. doi: 10.1093/nar/30.2.475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verschure PJ, van der Kraan I, de Leeuw W, van der Vlag J, Carpenter AE, Belmont AS, van Driel R. In vivo HP1 targeting causes large-scale chromatin condensation and enhanced histone lysine methylation. Molecular & Cellular Biology. 2005;25:4552–64. doi: 10.1128/MCB.25.11.4552-4564.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vooijs M, van dV, te RH, Berns A. Flp-mediated tissue-specific inactivation of the retinoblastoma tumor suppressor gene in the mouse. Oncogene. 1998;17:1–12. doi: 10.1038/sj.onc.1202169. [DOI] [PubMed] [Google Scholar]
- Wang D, Kennedy S, Conte D, Jr, Kim JK, Gabel HW, Kamath RS, Mello CC, Ruvkun G. Somatic misexpression of germline P granules and enhanced RNA interference in retinoblastoma pathway mutants. Nature. 2005;436:593–7. doi: 10.1038/nature04010. [DOI] [PubMed] [Google Scholar]
- Wang H, Bauzon F, Ji P, Xu X, Sun D, Locker J, Sellers RS, Nakayama K, Nakayama KI, Cobrinik D, Zhu L. Skp2 is required for survival of aberrantly proliferating Rb1-deficient cells and for tumorigenesis in Rb1+/− mice. Nature Genetics. 2010;42:83–88. doi: 10.1038/ng.498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg RA. The retinoblastoma protein and cell cycle control. Cell. 1995;81:323–330. doi: 10.1016/0092-8674(95)90385-2. [DOI] [PubMed] [Google Scholar]
- Welch PJ, Wang JYJ. A c-terminal protein-binding domain in the retinoblastoma protein regulates nuclear c-Abl tyrosine kinase in the cell cycle. Cell. 1993;75:779–790. doi: 10.1016/0092-8674(93)90497-e. [DOI] [PubMed] [Google Scholar]
- Wenzel PL, Wu L, de Bruin A, Chong JL, Chen WY, Dureska G, Sites E, Pan T, Sharma A, Huang K, Ridgway R, Mosaliganti K, Sharp R, Machiraju R, Saltz J, Yamamoto H, Cross JC, Robinson ML, Leone G. Rb is critical in a mammalian tissue stem cell population. Genes & Development. 2007;21:85–97. doi: 10.1101/gad.1485307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wikenheiser-Brokamp KA. Rb family proteins differentially regulate distinct cell lineages during epithelial development. Development. 2004;131:4299–4310. doi: 10.1242/dev.01232. [DOI] [PubMed] [Google Scholar]
- Wildwater M, Campilho A, Perez-Perez JM, Heidstra R, Blilou I, Korthout H, Chatterjee J, Mariconti L, Gruissem W, Scheres B. The RETINOBLASTOMA-RELATED Gene Regulates Stem Cell Maintenance in Arabidopsis Roots. Cell. 2005;123:1337–1349. doi: 10.1016/j.cell.2005.09.042. [DOI] [PubMed] [Google Scholar]
- Williams JP, Stewart T, Li B, Mulloy R, Dimova D, Classon M, Williams JP, Stewart T, Li B, Mulloy R, Dimova D, Classon M. The retinoblastoma protein is required for Ras-induced oncogenic transformation. Molecular & Cellular Biology. 2006;26:1170–1182. doi: 10.1128/MCB.26.4.1170-1182.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wintersteiner H. Das Neuroepithelioma Retinae. Franz Deuticke; Vienna: 1897. [Google Scholar]
- Wistuba II, Gazdar AF, Minna JD. Molecular genetics of small cell lung carcinoma. Semin Oncol. 2001;28:3–13. [PubMed] [Google Scholar]
- Wong S, Weber JD. Deacetylation of the retinoblastoma tumour suppressor protein by SIRT1. Biochem J. 2007;407:451–60. doi: 10.1042/BJ20070151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, de Bruin A, Saavedra HI, Starovic M, Trimboli A, Yang Y, Opavska J, Wilson P, Thompson JC, Ostrowski MC, Rosol TJ, Woollett LA, Weinstein M, Cross JC, Robinson ML, Leone G. Extra-embryonic function of Rb is essential for embryonic development and viability. Nature. 2003;421:942–947. doi: 10.1038/nature01417. [DOI] [PubMed] [Google Scholar]
- Wu L, Timmers C, Maiti B, Saavedra HI, Sang L, Chong GT, Nuckolls F, Giangrande P, Wright FA, Field SJ, Greenberg ME, Orkin S, Nevins JR, Robinson ML, Leone G. The E2F1–3 transcription factors are essential for cellular proliferation. Nature. 2001;414:457–462. doi: 10.1038/35106593. [DOI] [PubMed] [Google Scholar]
- Xia X, Cheng A, Lessor T, Zhang Y, Hamburger AW. Ebp1, an ErbB-3 binding protein, interacts with Rb and affects Rb transcriptional regulation. J Cell Physiol. 2001;187:209–17. doi: 10.1002/jcp.1075. [DOI] [PubMed] [Google Scholar]
- Xiao B, Spencer J, Clements A, Ali-Khan N, Mittnacht S, Broceno C, Burghammer M, Perrakis A, Marmorstein R, Gamblin SJ. Crystal structure of the retinoblastoma tumor suppressor protein bound to E2F and the molecular basis of its regulation. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:2363–2368. doi: 10.1073/pnas.0436813100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu XL, Fang Y, Lee TC, Forrest D, Gregory-Evans C, Almeida D, Liu A, Jhanwar SC, Abramson DH, Cobrinik D. Retinoblastoma Has Properties of a Cone Precursor Tumor and Depends Upon Cone-Specific MDM2 Signaling. Cell. 2009;137:1018–1031. doi: 10.1016/j.cell.2009.03.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao G, Lee TJ, Mori S, Nevins JR, You L, Yao G, Lee TJ, Mori S, Nevins JR, You L. A bistable Rb-E2F switch underlies the restriction point. Nature Cell Biology. 2008;10:476–82. doi: 10.1038/ncb1711. [DOI] [PubMed] [Google Scholar]
- Yu BD, Becker-Hapak M, Snyder EL, Vooijs M, Denicourt C, Dowdy SF. Distinct and nonoverlapping roles for pRB and cyclin D:cyclin-dependent kinases 4/6 activity in melanocyte survival. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:14881–14886. doi: 10.1073/pnas.2431391100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zacksenhaus E, Jiang Z, Chung D, Marth JD, Phillips RA, Gallie BL. pRb controls proliferation, differentiation, and death of skeletal muscle cells and other lineages during embryogenesis. Genes & Development. 1996;10:3051–3064. doi: 10.1101/gad.10.23.3051. [DOI] [PubMed] [Google Scholar]
- Zarkowska T, Mittnacht S. Differential phosphorylation of the retinoblastoma protein by G1/S cyclin-dependent kinases. Journal of Biological Chemistry. 1997;272:12738–12746. doi: 10.1074/jbc.272.19.12738. [DOI] [PubMed] [Google Scholar]
- Zhang J, Gray J, Wu L, Leone G, Rowan S, Cepko CL, Zhu X, Craft CM, Dyer MA. Rb regulates proliferation and rod photoreceptor development in the mouse retina. Nature Genetics. 2004;36:351–360. doi: 10.1038/ng1318. [DOI] [PubMed] [Google Scholar]
- Zhang R, Poustovoitov MV, Ye X, Santos HA, Chen W, Daganzo SM, Erzberger JP, Serebriiskii IG, Canutescu AA, Dunbrack RL, Pehrson JR, Berger JM, Kaufman PD, Adams PD. Formation of MacroH2A-containing senescence-associated heterochromatin foci and senescence driven by ASF1a and HIRA. Dev Cell. 2005;8:19–30. doi: 10.1016/j.devcel.2004.10.019. [DOI] [PubMed] [Google Scholar]
- Zheng L, Flesken-Nikitin A, Chen PL, Lee WH. Deficiency of Retinoblastoma gene in mouse embryonic stem cells leads to genetic instability. Cancer Research. 2002;62:2498–2502. [PubMed] [Google Scholar]
- Zhou Z, Flesken-Nikitin A, Corney DC, Wang W, Goodrich DW, Roy-Burman P, Nikitin AY. Synergy of p53 and Rb Deficiency in a Conditional Mouse Model for Metastatic Prostate Cancer. Cancer Research. 2006;66:7889–98. doi: 10.1158/0008-5472.CAN-06-0486. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Flesken-Nikitin A, Nikitin AY. Prostate Cancer Associated with p53 and Rb Deficiency Arises from the Stem/Progenitor Cell-Enriched Proximal Region of Prostatic Ducts. Cancer Research. 2007;67:5683–5690. doi: 10.1158/0008-5472.CAN-07-0768. [DOI] [PubMed] [Google Scholar]