

The chemistry of vanillin (1) and isovanillin (2) has been examined and reviewed extensively.1–3 A current project in our laboratory required the conversion of 5-iodoisovanillin ethers 6a–d to substituted 2,4-diaminopyrimidines followed by Heck coupling with an unsaturated ketone to generate agents displaying inhibitory activity against Bacillus anthracis, a potential bioterror threat.4 The chemistry to prepare 5-iodovanillin ethers 6a–d with variation at C-4 is straightforward5,6 starting from commercial 5-iodovanillin (3).7,8

Procedures to alter the C-3 alkoxy group, however, require demethylation of 3 to give catechol 4 followed by sequential methylation at the C-4 hydroxy group followed by O-alkylation at C-3. The synthesis of 5-iodoisovanillin ethers 6a–d, thus, requires regiocontrol in the initial methylation of 4. Although preferential alkylation of the C-4 hydroxy group of 4 would be expected due to its greater activation by the C-1 aldehyde, controlling this process to give clean monoalkylation is necessary. This has been achieved, and we now report a concise and selective method to generate 5-iodoisovanillin ethers.
Isovanillin, iodinated at C-5, has been previously reported, but the synthetic routes were lengthy9 or the procedures were unclear.10 In the first synthesis,9 the iodo group was introduced by a diazotization-replacement sequence as part of a six-step preparation from isovanillin. In the second procedure,10 the methyl ether was cleaved with aluminum chloride-pyridine and selectively alkylated at the C-4 hydroxyl group using methyl iodide and sodium bicarbonate in N,N-dimethylformamide-acetone. This approach parallels our procedure, but lacks detail about the isolation of the product from the precipitated material following decomposition of the aluminum chloride. Additionally, we had trouble controlling the subsequent alkylation using this method.
Our synthesis of 5-iodoisovanillin ethers 6a–d is outlined in Scheme 1. Demethylation of 5-iodovanillin (3) with aluminum chloride-pyridine led to 3,4-dihydroxy-5-iodobenzaldehyde (4) in 80% yield.10,11 The use of this reagent to cleave the ether was selected over anhydrous HBr in acetic acid12 and boron tribromide12 due to the simpler procedure, reduced hazard and lower cost. Following cleavage of the C-3 ether, regio-selective alkylation of the C-4 hydroxy group of 4 proceeded cleanly by treatment with 1.1 equivalent of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) in N,N-dimethylformamide (DMF) followed by addition of several portions of methyl iodide. This procedure afforded 5-iodoisovanillin 5 in 79% yield after flash chromatography. A similar alkylation using sodium bicarbonate,10 lithium carbonate13 or sodium carbonate in DMF yielded the C-4-monomethylated product in ≤30%, along with recovered starting material and the dialkylated product. The current procedure to generate 4 and 5 is superior to the previously reported syntheses in terms of yield, regiocontrol and time required. The target compounds required a final O-alkylation of the C-3 hydroxy group of 5 to give previously unknown derivatives 6a–d. Again, the use of DBU/DMF as the catalyst/solvent system for this transformation gave complete conversion in slightly more than one hour at room temperature. Purification by flash chromatography produced 6a–d in yields ranging from 78–92%. Finally, our attempts to improve the yield of 6c by carrying out the reaction in other solvents (tetrahydrofuran or dichloromethane) and with other bases (potassium carbonate or tri-ethylamine) resulted in lower yields of the desired product. Related systems 7 and 8 were synthesized similarly. Treatment of 4 with 3.5 equivalents of DBU and 3.0 equivalents of ethyl iodide produced 7 as a pale yellow solid in 80% yield. Likewise, 4 reacted with 3.5 equivalents of DBU and 1.1 equivalent of diiodomethane gave 7-iodo-1,3-benzodioxole-5-carbaldehyde (8) in 78% yield.14 Spectral and elemental analyses confirmed the structures of the final targets 6–8. Interestingly, 8 retained traces of methanol, following recrystallization from this solvent, even after extensive drying under high vacuum.
Scheme 1.
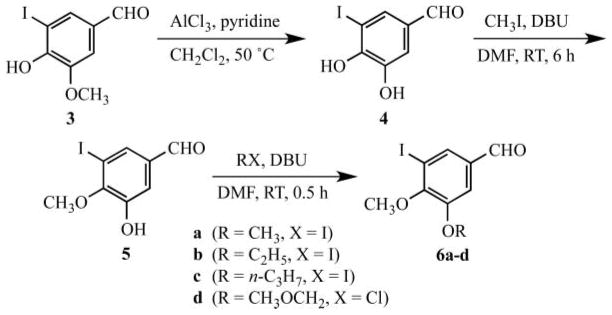

In summary, we have successfully developed a straightforward approach by which derivatives of 5-iodovanillin and 5-iodoisovanillin can be prepared. The use of aluminum chloride in pyridine proved most convenient and cost effective for the cleavage of the methyl ether in 5-iodovanillin (3) to generate 3,4-dihydroxy-5-iodobenzaldehyde (4). Regiospecific O-alkylation of 4 at the C-4 hydroxy group with methyl iodide in DBU/DMF then provided 5, which could be further alkylated at the C-5 hydroxy group using the same conditions to afford the series 6a–d. The basic procedure was further extended to the preparation of 7 and 8.
Experimental Section
All reactions were performed under dry nitrogen in oven-dried glassware. All 1H- and 13C-NMR spectra were obtained at 300 MHz and 75 MHz, respectively, using tetramethylsilane as the internal standard. All melting points are uncorrected. FT-IR spectra were taken as thin films on sodium chloride disks. Reactions were monitored by thin layer chromatography (TLC) on silica gel GF plates (Analtech No. 21521). Preparative flash chromatography15 was performed on silica gel (grade 62, 60–200 mesh) containing UV-active phosphor (Sorbent Technologies, No. UV-5) packed into quartz columns. Band elution for all chromatographic methods was monitored using a hand-held UV lamp. Pyridine was stored over potassium hydroxide pellets and distilled from calcium hydride prior to use. All other reagents and solvents were used as received from various vendors.
3,4-Dihydroxy-5-iodobenzaldehyde (4)
To a 500-mL, three-necked, round-bottomed flask, equipped with a 4.0-cm, egg-shaped stir bar and fitted with an addition funnel and a condenser (nitrogen inlet) was charged with 5-iodovanillin (3, 25.0 g, 89.9 mmol) and dichloromethane (125 mL) and stirring was begun to dissolve the solid. Aluminum chloride (13.3 g, 100 mmol, 1.1 equiv) was then added and stirring was continued for 10 min. Pyridine (31.3 g, 31.8 mL, 396 mmol, 4.4 equiv) was slowly added to the mixture via the addition funnel with stirring over a period of 20 min. [Caution: The initial addition of pyridine should be very slow]. The reaction flask was then placed in a pre-heated oil bath at 50°C and heated at reflux for 16 h. During this time, a yellow slurry formed which completely changed to a green colored solution. Near the end of the reaction a precipitate began to form. A TLC analysis of the solution using ether:hexanes (1:1) indicated the reaction was complete. After cooling for 10 min, the reaction mixture was poured into a 500-mL Erlenmeyer flask containing crushed ice (300 g). The resulting mixture was cooled in an ice bath with continuous stirring for 10 min and then 6 M hydrochloric acid (ca 50 mL) was slowly added until the pH reached 2. At this point, the reaction mixture consisted of a bottom layer of dichloromethane with a pale yellow solid suspended in the top aqueous layer. The mixture was transferred to a 1-L separatory funnel using ethyl acetate (150 mL), and water (100 mL) was slowly added. The organic layer was separated, and the remaining aqueous layer was extracted with ethyl acetate (4 × 150 mL). The combined organic extracts were washed with saturated aqueous sodium chloride (150 mL), dried (MgSO4), and concentrated using a rotary evaporator to yield a green solid. This material was dissolved with heating in ethanol (150 mL), and then water (250 mL) was slowly added. The resulting green solution was cooled to room temperature over a period of 4 h and to 0°C for 30 min to give a light brown solid, which was collected and dried under vacuum. Recrystallization from ethanol:water (3:5) yielded 4 (21.8 g, 92%) as a tan solid. A second recrystallization yielded 20.0 g (84%) and a third recrystallization gave pure 4 (18.9 g, 80%) as an off-white solid, mp. 199–200°C (lit.10 mp. 198–199°C), after drying for 12 h under high vacuum. IR: 3200, 1637 cm−1; 1H-NMR (DMSO-d6): δ 10.4 (s, 1 H, CHO), 9.69 (s, 2 H, OH), 7.75 (d, 1 H, J = 1.7 Hz), 7.26 (d, 1 H, J = 1.7 Hz); 13C-NMR (DMSO-d6): δ 190.4, 151.9, 145.1, 133.6, 130.1, 113.3, 84.3. No 13C NMR data were previously reported for 4.
3-Hydroxy-5-iodo-4-methoxybenzaldehyde (5)
A 250-mL, single-necked, round-bottomed flask, equipped with a 2.5-cm, egg-shaped, magnetic stir bar and fitted with an addition funnel (nitrogen inlet), was charged with 3,4-dihydroxy-5-iodobenzaldehyde (4, 15.0 g, 56.8 mmol) and dry DMF (30 mL). To the resulting solution was added dropwise 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) (9.51 g, 9.34 mL, 62.6 mmol, 1.1 equiv) with stirring over a period of 5 min. During the addition process, the color of the reaction mixture changed from brown to red. After 45 min, methyl iodide (8.00 g, 3.54 mL, 56.3 mmol) was added all at once. Thereafter, four three additional portions of methyl iodide (the same volume) were added every hour over a 5-h period to ensure maximum conversion. A TLC analysis using ether:hexanes (1:1) indicated the reaction was complete. The crude reaction mixture was acidified to pH 2 by the slow addition of 6 M hydrochloric acid and then extracted with dichloromethane (3 × 100 mL). The combined organic extracts were washed with saturated sodium chloride solution (50 mL), dried (MgSO4), and concentrated to give an off-white solid. Purification of this material by flash chromatography (30-cm × 4-cm column), eluted with dichloromethane, gave 5 (12.5 g, 79%) as a white solid, mp. 134–135°C (lit.10 mp. 134–135.8°C). IR: 3236, 1685 cm−1; 1H-NMR (DMSO-d6): δ 10.4 (s, 1 H, CHO), 9.79 (s, 1 H, OH), 7.79 (d, 1 H, J = 1.7 Hz), 7.35 (d, 1 H, J = 1.7 Hz), 3.82 (s, 3 H); 13C-NMR (DMSO-d6): δ 191.0, 152.4, 150.6, 134.0, 131.6, 116.1, 93.0, 59.8. No 13C NMR data were previously reported for 5.
3-Iodo-4,5-dimethoxybenzaldehyde (6a)
To a 100-mL, single-necked, round-bottomed flask, equipped with a 2.5-cm, egg-shaped, magnetic stir bar and fitted with a condenser (nitrogen inlet), charged with 3,4-dihydroxy-5-iodobenzaldehyde (4, 1.00 g, 3.78 mmol) and dry DMF (3 mL), was added dropwise DBU (2.01 g, 1.97 mL, 13.2 mmol, 3.5 equiv) with stirring over a period of 5 min. The reaction mixture was stirred 30 min at room temperature, methyl iodide (1.61 g, 0.71 mL, 11.4 mmol, 3.0 equiv) was added and stirring was continued for an additional 2 h. Thereafter, three additional portions of methyl iodide (the same volume) were added every hour over a 4 h-period, and the reaction mixture was stirred overnight to ensure maximum conversion. A TLC analysis using ether:hexanes (1:1) indicated the reaction was complete. The crude reaction mixture was acidified to pH 2 using 6 M hydrochloric acid and then extracted with dichloromethane (3 × 25 mL). The combined organic extracts were washed with saturated sodium chloride solution (25 mL), dried (MgSO4), and concentrated to yield a brown liquid. Purification of this material by flash column chromatography (15-cm × 2-cm column), eluted with dichloromethane, gave 6a (0.93 g, 84%) as a white solid, mp. 71–72°C (lit.16 mp. 72–73°C). IR: 2832, 2730, 1693 cm−1; 1H-NMR (CDCl3): δ 9.83 (s, 1 H), 7.85 (d, 1 H, J = 1.7 Hz), 7.41 (d, 1 H, J = 1.7 Hz), 3.93 (s, 3 H), 3.92 (s, 3 H); 13C-NMR (CDCl3): δ 189.7, 154.2, 153.0, 134.7, 133.9, 111.0, 92.1, 60.7, 56.1. Compound 6a has been previously prepared twice. One article16 gave an analysis for iodine but no experimental data, and the other5 reported a correct melting point and 1H-NMR spectrum, but no elemental analysis.
Anal. Calcd for C9H9IO3: C, 36.98; H, 3.08. Found: C, 36.76; H, 3.00.
3-Iodo-4-methoxy-5-ethoxybenzaldehyde (6b)
To a 200-mL, single-necked, round-bottomed flask, equipped with a 2.5-cm, egg-shaped, magnetic stir bar and fitted with a condenser (nitrogen inlet), charged with 3-hydroxy-5-iodo-4-methoxybenzaldehyde (5, 5.00 g, 18.0 mmol) and dry DMF (10 mL) was added dropwise DBU (4.10 g, 4.00 mL, 27.0 mmol, 1.5 equiv) with stirring over a period of 5 min. The reaction mixture was stirred for 30 min, ethyl iodide (3.08 g, 1.58 mL, 19.7 mmol, 1.1 equiv) was added, and stirring was continued for an additional 30 min. A TLC analysis using ether:hexanes (1:1) indicated the reaction was complete. The crude reaction mixture was acidified to pH 2 using 6 M hydrochloric acid and then extracted with dichloromethane (3× 50 mL). The combined dichloromethane extracts were washed with saturated sodium chloride solution (50 mL), dried (MgSO4), and concentrated to give a brown solid. The solid was dissolved with heating in ethanol (10 mL), and water (12 mL) was slowly added. The resulting solution was cooled slowly to room temperature over a period of 2 h and to 0°C for 30 min to give a solid. The crude product was collected and dried under high vacuum for 12 h to give 6b (4.95 g, 90%) as a white solid, mp. 69–70°C. IR: 1694 cm−1; 1H-NMR (CDCl3): δ 9.85 (s, 1 H), 7.92 (d, 1 H, J = 1.6 Hz), 7.51 (d, 1 H, J = 1.1 Hz), 4.16 (q, 2 H, J = 6.8 Hz), 3.83 (s, 3 H), 1.39 (t, 3 H, J = 7.1 Hz); 13C-NMR (CDCl3): δ 190.9, 153.3, 151.7, 133.9, 132.9, 112.9, 93.0, 64.4, 60.1, 14.5.
Anal. Calcd for C10H11IO3: C, 39.21; H, 3.54; I, 41.50. Found: C, 39.52; H, 3.63; I, 41.24.
3-Iodo-4-methoxy-5-propoxybenzaldehyde (6c)
To a 250-mL, single-necked, round-bottomed flask, equipped with a 2.5-cm egg-shaped magnetic stirring bar and fitted with a condenser (nitrogen inlet), charged with 3-hydroxy-5-iodo-4-methoxybenzaldehyde (5, 5.00 g, 18.0 mmol) and dry DMF (10 mL) was added dropwise DBU (4.10 g, 4.00 mL, 27.0 mmol, 1.5 equiv) with stirring over a period of 5 min. The reaction mixture was stirred for 30 min, 1-iodopropane (3.36 g, 1.93 mL, 19.8 mmol, 1.1 equiv) was added, and stirring was continued for an additional 30 min. A TLC analysis using ether:hexanes (1:1) indicated the reaction was complete. The reaction mixture was acidified to pH 2 with 6 M hydrochloric acid and then extracted with dichloromethane (3× 50 mL). The combined dichloromethane extracts were washed with saturated sodium chloride solution (50 mL), dried (MgSO4), and concentrated to yield a brown solid. This solid was dissolved with heating in ethanol (10 mL), and the resulting solution was cooled slowly to room temperature over a period of 2 h and to 0°C for 30 min to give a solid. The product was collected and dried under high vacuum for 12 h to give 6c (5.30 g, 92%) as a yellow solid, mp. 51–52°C. IR: 1695 cm−1; 1H-NMR (DMSO-d6): δ 9.85 (s, 1 H), 7.92 (d, 1 H, J = 1.6 Hz), 7.50 (d, 1 H, J = 1.1 Hz), 4.05 (t, 2 H, J = 6.5 Hz), 3.84 (s, 3 H), 1.80 (sextet, 2 H, J = 7.1 Hz), 1.03 (t, 3 H, J = 7.1 Hz); 13C-NMR (DMSO-d6): δ 190.8, 153.2, 151.9, 133.9, 132.8, 112.8, 92.9, 70.1, 60.1, 21.9, 10.5.
Anal. Calcd for C11H13IO3: C, 41.26; H, 4.06, I, 39.66. Found: C, 41.47; H, 4.17; I, 39.88.
3-Iodo-4-methoxy-5-(methoxymethyl)benzaldehyde (6d)
Using the above procedure, alkylation was carried out on 5 (5.00 g, 18.0 mmol) using chloromethyl methyl ether (MOMCl) (1.59 g, 1.50 mL, 19.7 mmol, 1.1 equiv) and DBU (4.10 g, 4.00 mL, 27.0 mmol, 1.5 equiv) in 10 mL of dry DMF to give 6d (5.21 g, 90%) as a white solid, mp. 51–52°C (EtOH). IR: 1698 cm−1; 1H-NMR (CDCl3): δ 9.83 (s, 1 H), 7.93 (d, 1 H, J = 1.7 Hz), 7.64 (d, 1 H, J = 1.1 Hz), 5.29 (s, 2 H), 3.96 (s, 3 H), 3.53 (s, 3 H); 13C-NMR (CDCl3): δ 189.4, 154.5, 150.3, 134.6, 133.9, 116.3, 95.0, 92.5, 60.7, 56.5.
Anal. Calcd for C10H11IO4: C, 37.26; H, 3.41; I, 39.44. Found: C, 37.56; H, 3.41; I, 39.35.
3-Iodo-4,5-diethoxybenzaldehyde (7)
Using the above procedure, alkylation was carried out on 3,4-dihydroxy-5-iodobenzaldehyde (4, 1.00 g, 3.79 mmol) using ethyl iodide (1.77 g, 0.91 mL, 11.3 mmol, 3.0 equiv) and DBU (2.01 g, 1.97 mL, 13.2 mmol, 3.5 equiv) in 3 mL of dry DMF to give 7 (0.97 g, 80%) as a pale yellow solid, mp. 45–46°C (EtOH). IR: 1694 cm−1; 1H-NMR (CDCl3): δ 9.81 (s, 1 H), 7.83 (d, 1 H, J = 1.7), 7.37 (d, 1 H, J = 1.7 Hz), 4.20 (q, 2 H, J = 7.1 Hz), 4.12 (q, 2 H, J = 7.0 Hz), 1.48 (t, 3 H, J = 7.0 Hz), 1.47 (t, 3 H, J = 7.0 Hz); 13C-NMR (CDCl3): δ 189.7, 153.6, 152.1, 134.5, 133.6, 111.8, 92.8, 69.4, 64.6, 15.8, 14.6.
Anal. Calcd for C11H13IO3: C, 41.25; H, 4.06; I, 39.68. Found: C, 41.44; H, 4.04; I, 39.82.
7-Iodo-1,3-benzodioxole-5-carbaldehyde (8)
Using the above procedure, alkylation was carried out on 3,4-dihydroxy-5-iodobenzaldehyde (4, 1.00 g, 3.79 mmol) using diiodomethane (1.11 g, 0.33 mL, 4.14 mmol, 1.1 equiv) and DBU (2.01 g, 1.97 mL, 13.2 mmol, 3.5 equiv) in 3 mL of dry DMF to give 8 (0.82 g, 78%) as a white solid, mp. 138–139°C (MeOH). A second recrystallization of this solid gave mp. 139–140°C (MeOH). IR: 1683 cm−1; 1H NMR (CDCl3): δ 9.75 (s, 1 H), 7.69 (d, 1 H, J = 1.4 Hz), 7.27 (d, 1 H, J = 1.4 Hz), 6.15 (s, 2 H); 13C NMR (CDCl3): δ 189.0, 154.5, 147.4, 136.5, 133.3, 106.8, 101.7, 70.1. A small amount of methanol was retained in the solid.
Anal. Calcd for C8H5IO3: C, 34.78; H, 1.81; I, 46.01. Found: C, 35.36; H, 2.03; I, 45.85. Found: C8H5IO3·0.3 CH3OH: C, 34.93; H, 2.23; I, 45.85.
Acknowledgments
We gratefully acknowledge support of this work by the National Institute of Allergy and Infectious Diseases [1-R01-AI 090685-01] of the NIH to WWB. We also are pleased to acknowledge funding for the Statewide NMR facility by the National Science Foundation (BIR-9512269), the Oklahoma State Regents for Higher Education, the W. M. Keck Foundation, and Conoco, Inc. RAB and KDB gratefully acknowledge support by the Department of Chemistry at Oklahoma State University.
References
- 1.Priefert H, Rabenhorst J, Steinbuchel A. Appl Microbiol Biotech. 2001;56:296. doi: 10.1007/s002530100687. [DOI] [PubMed] [Google Scholar]
- 2.Walton NJ, Mayer MJ, Narbad A. Phytochemistry. 2003;63:505. doi: 10.1016/s0031-9422(03)00149-3. [DOI] [PubMed] [Google Scholar]
- 3.Sinha AK, Sharma UK, Sharma N. Int J Food Sci Nutr. 2008;59:299. doi: 10.1080/09687630701539350. [DOI] [PubMed] [Google Scholar]
- 4.Bourne CR, Bunce RA, Bourne PC, Berlin KD, Barrow EW, Barrow WW. Antimicrob Agents Chemother. 2009;53:3065. doi: 10.1128/AAC.01666-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nimgirawath S. Australian J Chem. 1994;47:957. [Google Scholar]
- 6.Joshua AV, Sharma SK, Abrams DN. Synth Commun. 2008;38:434. [Google Scholar]
- 7.Thorn GD, Purves CB. Can J Chem. 1954;32:373. [Google Scholar]
- 8.Gordon JT, Kirk KL, Dratman MB. Org Prep Proced Int. 1983;15:49. The sample used in our work was obtained from Alfa Aesar in 99% purity with mp. 181–182 °C. [Google Scholar]
- 9.Borchardt RT, Huber JA, Houston M. J Med Chem. 1982;25:258. doi: 10.1021/jm00345a012. [DOI] [PubMed] [Google Scholar]
- 10.Anhoury ML, Crooy P, De Neys R, Eliaers J. J Chem Soc, Perkin Trans. 1974;I:1015. [Google Scholar]
- 11.The use of aluminum chloride-pyridine to cleave the methyl ether of vanillin was first reported by Lange RG. J Org Chem. 1962;27:2037.
- 12.Jacks TE, Belmont DT, Briggs CA, Horne NM, Kanter GD, Karrick GL, Krikke JJ, McCabe RJ, Mustakis JG, Nanniga TN, Risedorph GS, Seamans RE, Skeean R, Winkle DD, Zennie TM. Org Proc Res Dev. 2004;8:201. [Google Scholar]
- 13.Wymann WE, Davis R, Patterson JW, Jr, Pfister JR. Synth Commun. 1988;18:1379. [Google Scholar]
- 14.To date, the only known iodinated piperonal is 2-iodopiperonal, see Young WB, Masters JJ, Danishefsky S. J Am Chem Soc. 1995;117:5228.
- 15.Still WC, Kahn M, Mitra A. J Org Chem. 1978;43:2923. [Google Scholar]
- 16.Raiford LC, Perry RP. J Org Chem. 1942;7:354. [Google Scholar]