

Abstract
Induced pluripotent stem cell-derived neurons from patients promise to fill an important niche between studies in humans and model organisms in deciphering mechanisms and identifying therapeutic avenues for neurologic and psychiatric diseases. Recent work begins to tap this potential, and also highlights challenges that must be overcome for it to be fully realized.
The relative ease with which induced pluripotent cells (iPSCs) can now be generated from human somatic cells is changing the way we study human disease. Nowhere is this clearer than in the brain. Neurodevelopmental and psychiatric disorders have been challenging to study in the laboratory, and despite a large investment, few novel treatments have been developed in the last decade. One problem has been the lack of predictive preclinical models that can be used to study the pathophysiology of a disease, identify therapeutic targets, and test potential therapies. While animal models have been valuable in elucidating disease mechanisms and in providing insights into the function of specific genes, they have a poor track record when it comes to translating findings into human therapeutics (Dragunow, 2008). Post-mortem tissue can provide a window into alterations in brain structure at a cellular and molecular level, but deriving causal inferences from pathological studies of neurodevelopmental or neurodegenerative diseases has many challenges. Into this breach come iPSC-derived neurons and glial cells from human patients.
iPSCs are adult pluripotent stem cells generated from somatic cells by the introduction of reprogramming factors. Like other pluripotent stem cells, iPSCs can be coaxed to differentiate into neurons and glial cells as well as other terminally differentiated cell types by exposure to a combination of growth factors and cell culture conditions. Human iPSCs (hiPSCs) thus make it possible to study human neurons, a previously inaccessible cell type, carrying the genetic information from patients with a specific mutation or a neuropsychiatric disease.
Because hiPSCs capture the genetic diversity of the patient population, they are well suited to study how specific sets of mutations lead to disease, something that would be hard to replicate in genetically engineered animals. Most neuropsychiatric disorders including autism, schizophrenia, and depression have a strong genetic component. However, while a single mutation may cause a disease in rare cases, it is more common for disease to arise from multiple genetic events. This is evident from the fact that the mutations that have been identified for neuropsychiatric diseases are not highly penetrant and are often associated with multiple disorders, indicating that there is a contribution from other genetic, epigenetic, or environmental factors (Geschwind, 2008; Vassos et al., 2010). hiPSCs thus provide a clear advantage over animal models for studying the effect of human genetic background on disease penetrance and severity.
A further advantage of using hiPSC-derived neurons is that they offer the only practical way of studying the development and function of live human neurons. Rodents diverged from humans almost 60 million years ago, and lower vertebrates and invertebrates are even more distantly related. Therefore, there are major differences in development and structure between rodent and human brains (Clowry et al., 2010). Mice are lissencephalic, whereas the human cortex has complicated sulci and gyri. Rodents also have far less-developed prefrontal and temporal cortices, whereas the prefrontal and temporal cortex and other interconnected association areas comprise the majority of the human cortex. These regions are particularly important in the context of complex neuropsychiatric diseases. Human brain development is also quite different from the development of the rodent brain both in terms of the timing and the origin of cortical neurons. A significant amount of human neurogenesis occurs in the outer subventricular zone, an area that is significantly reduced in rodents. Specific types of neurons such as Von Economo neurons are found in humans and other large mammals and not in rodents (Allman et al., 2011), and may be key disease targets in dementia. In addition, neurons from different species, even when closely related, have different electrophysiological properties (Steffenhagen et al., 2011). These gross differences suggest that there are underlying molecular and cellular differences between humans and rodents that could impact the validity of preclinical models, a supposition that has been confirmed by genome-scale studies of transcriptional networks. Given these differences, it is not surprising that the therapies found to be effective in rodents are often not effective in patients in the clinic (Dragunow, 2008).
The advantages of hiPSC-derived neurons and the prospect of developing better preclinical models for neuropsychiatric diseases led several groups to generate hiPSCs from patients with genetic neurological disorders (Marchetto et al., 2010; Park et al., 2008; Urbach et al., 2010). Early studies focused largely on the generation of hiPSCs and their ability to generate neurons as a proof of principle. Only recently have a small number of papers reported cellular phenotypes associated with neuropsychiatric disease. This is significant because it was not clear at first that hiPSCs would be useful for modeling neuropsychiatric disorders, as many of these disorders are thought to be due to defects in the development of neural circuits that might not be picked up in cellular assays from reprogrammed somatic cells.
One of the earliest reports of a phenotype for a neurological disease using iPSCs involved neural crest precursors from patients with familial dysautonomia (Lee et al., 2009). This disease is caused by a point mutation in the gene encoding IKBKAP and leads to loss of autonomic and sensory neurons. The authors generated iPSCs from a single patient with the disease and directed the differentiation of these cells along the neural crest lineage. They found defects in IKBKAP splicing, changes in gene expression, and defects in cell migration in patient cells that could be partially reversed by treatment with the plant hormone kinetin. This paper provided a proof of principle that iPSCs could be used to identify a cellular phenotype associated with a neurological disease, but did not provide additional insights into the pathogenesis of the disorder.
Cellular disease phenotypes were identified by two groups studying iPSC-derived neurons from patients with Rett syndrome (Cheung et al., 2011; Marchetto et al., 2010), a neurodevelopmental disorder that has been linked to mutations in the gene encoding methyl-CpG binding protein 2 (MECP2). Rett syndrome neurons were found to have small cell bodies and reduced numbers of synapses, and retained an inactive X-chromosome as observed in patients. Some of these phenotypes could be rescued by treatment with IGF-1 and large amounts of gentamycin (Marchetto et al., 2010). These studies were significant because they recapitulated observations that had been made in mouse models of Rett syndrome, demonstrating that phenotypes observed in vivo can be identified at a cellular level in iPSC-derived human neurons, and that cells developed in culture largely confirm the phenotypes of cells that develop in a mouse.
The most common mutation related to Parkinson’s disease (PD) occurs in the gene encoding Leucine-Rich Repeat Kinase-2 (LRRK2). Nguyen et al. found that iPSC-derived dopaminergic neurons from patients carrying this mutation had increased expression of oxidative stress-response genes and alpha synuclein protein (Nguyen et al., 2011). The mutant neurons were also more sensitive to caspase-3 activation and cell death caused by exposure to hydrogen peroxide, MG-132 (a proteasome inhibitor), and 6-hydroxydopamine, than control neurons. The finding of increased susceptibility to stress in patient-derived neurons provides insights into the pathogenesis of PD and a potential basis for a cellular screen.
Tackling a complex disease with less well-defined genetic determinants, Brennand et al. studied iPSC-derived neurons from four patients with idiopathic forms of schizophrenia (Brennand et al., 2011). Neurons from these patients had defects in connectivity determined by an assay that used rabies virus to label synaptically-connected neurons, providing the first report of a defect in neuronal connections in iPSC-derived neurons from neuropsychiatric patients. The synaptic connectivity phenotype was reversed by one of the five commonly used antipsychotic drugs, although the mechanism underlying this defect remains unclear. The rabies virus is transmitted retrogradely from dendrites to axons, so it is not a measure of active synapses, and neurons from schizophrenia patients did not have any intrinsic defects in excitatory or inhibitory synaptic input as measured electrophysiologically. The paper also reports changes in gene expression in the patient cells that are consistent with defects in cyclic AMP and WNT signaling. Extending iPSC technology beyond monogenic diseases to the study of complex genetic conditions could reveal common cellular features among all patients with a particular diagnosis. However, it remains to be formally addressed whether so few patient samples from a complex disease such as schizophrenia with iPSCs will yield generalizable results.
While these early papers are promising, there are clearly many issues that need to be resolved before iPSC-derived cellular models can be used more widely. These include showing reproducibility between lines from the same patient and between different patients. In this regard, recent work comparing the neural differentiation potential of 16 iPSC and six ESC lines in two different laboratories (Boulting et al., 2011) represents a step in the right direction. Given the variability in cell lines and new findings that iPSC lines may accumulate point mutations or copy number variations (CNVs) in culture (Gore et al., 2011; Hussein et al., 2011), it is also important to study more iPSC lines in greater depth. Thus, it is remarkable that genome-wide expression profiling has not been routinely performed to understand variability in this process and the cellular-molecular phenotypes from iPSC-derived neurons in a more fine-grained manner. It is also not certain that epigenetic susceptibility factors will be propogated in IPSCs derived from patients, necessitating epigenetic profiling as well. Hopefully, such analysis, such as the recent comprehensive transcriptional and epigenetic profiling in 20 iPSC and ESC lines (Bock et al., 2011) will become more standard in the field, so that transcriptional influences on molecular and physiological relevant phenotypes at a genome-wide level will be understood.
This highlights a major limitation of the current iPSC technology, namely the expensive and time-consuming nature of the research. At the moment, each iPSC line is expensive and takes months to generate and characterize. If we are to use iPSC technology to study more diverse populations of patients, we will need to invest in cell culture automation and develop new approaches for making iPSC generation and characterization more efficient and robust. A possible solution to this problem are newly emerging techniques for the direct generation of neurons from fibroblasts (Vierbuchen et al., 2010). This technology may be faster than generation of neurons from iPSCs, but is currently inefficient in humans and has the disadvantage that it does not mimic neuronal development. Thus, it may miss phenotypes that occur during early development.
A second concern is that the methods for generating neurons from iPSCs are still being developed and that the resulting populations of neurons have not been well characterized. For instance, the results of the early papers suggest that neurons derived from hiPSCs and indeed from human embryonic stem cells are not fully mature. This is apparent from the fact that only a fraction of the cells fire action potentials, many have immature synapses, and most express markers of immature neurons (Marchetto et al., 2010). Thus, phenotypes may be limited to those that can be identified in young neurons. Further optimization of culture methods and extensive electrophysiological characterization of resulting neurons will be essential for establishing useful in vitro models.
A related issue concerns the heterogeneity of the neuronal cultures generated from hiPSCs. To draw convincing conclusions about cellular phenotypes, it is essential to identify the cells that are being generated and studied. Several methods of identifying particular cell types are available, although they have not yet been used in these first iPSC papers. One approach is to use bacterial artificial chromosomes or viral reporter genes to mark specific types of cells (Lee et al., 2009). These approaches are laborious and suffer from a general lack of specificity, as the expression of a single gene is seldom enough to identify a type of neuron or glial cell. An alternative is to use antibodies to cell-specific markers to characterize different cell types. This is complicated by a dearth of good antibodies for human antigens, and the difficulty in multiplexing more than a few antibodies on a sample. An exciting new approach is to use of single cell qPCR arrays to characterize gene expression profiles in single cells (Nguyen et al., 2011). This method provides a more comprehensive characterization of single neurons, and could potentially provide insights into specific cell fate and differentiation phenotypes associated with specific diseases. The gene expression pattern of a single cell can reflect its neurotransmitter identity, cortical layer fate, predicted projections, and can even predict probable location in the cortex. This technique thus has the potential to reveal novel insights into the circuits that are altered in specific disorders from in vitro cellular models.
The ultimate question however, is whether hiPSC-derived cells will be useful for identifying new therapeutic targets and compounds? At present, hiPSC-derived neurons are difficult to generate and are not sufficiently uniform for high-throughout drug screening. On the other hand, these cells are valuable for testing small numbers of compounds for efficacy and toxicity in a specific patient or population of patients. This could be a useful method for determining which drugs or drug combinations are effective in humans or in specific patients. For this approach to be valuable, however, it is essential to validate the cellular phenotypes identified in neurons.
Validation is perhaps the largest unanswered question in hiPSC disease models, especially for psychiatric diseases. In other systems, like cardiomyocytes, it is considerably simpler to show that a cellular phenotype in the lab accounts for defects observed in patients (Yazawa et al., 2011). This is more challenging in neuropsychiatric diseases for which we have very little information about neuroanatomical and molecular phenotypes in patients.
iPSC-derived neurons are derived from patients with genetic neuropsychiatric disease and thus harbor the genetic variants that underlie human disease susceptibility. On the other hand, because they are immature and have not developed in a brain, they may not reflect the major pathology of a specific disorder. One avenue for validation is to compare the phenotypes identified in iPSC-derived neurons with those identified in postmortem samples. Finding the same phenotypes in hiPSC derived neurons and in neurons in the intact brain would provide some evidence that the cell culture model recapitulates disease specific aspects of brain development. However, post-mortem samples have many limitations and may not show neurodevelopmental phenotypes that are critical elements of the disease process. Thus, the development of other modalities, such as high-resolution human brain imaging to explore molecules and circuits at high resolution in vivo, would be of great value. The hardest level of validation to achieve for most psychiatric diseases is to show that a drug that treats the disease in patients also reverses the phenotype in the model (predictive validity). This is a tall order partly because most treatments for neuropsychiatric disease target behavioral or cognitive symptoms whose structural or physiological analogues at the cellular level are largely unknown. In addition, there are few treatments available for most neuropsychiatric disease, making predictive validation the hardest level to achieve at present.
These limitations highlight the fact that iPSC-derived models are only one weapon in the arsenal of tools needed to understand and treat neuropsychiatric disease. If we are going to use models to advance treatments of human neuropsychiatric disease, we must reach a systems-level understanding of disease pathophysiology, which requires leveraging and integrating multiple levels of analysis (Geschwind and Konopka, 2009). We have to integrate information derived from genetic analysis, hiPSC-derived cells, animal models, and human studies. Each level has to inform the other in a bidirectional manner (Figure 1). iPSCs have the potential to fill a critical gap in this multifaceted approach by providing live, functional human CNS cells with the complex genetic backgrounds found in patients. They thus provide an important bridge between studies in animal models and assessment of human postmortem brain and brain functioning in living patients.
Figure 1. Niches occupied by various experimental approaches.
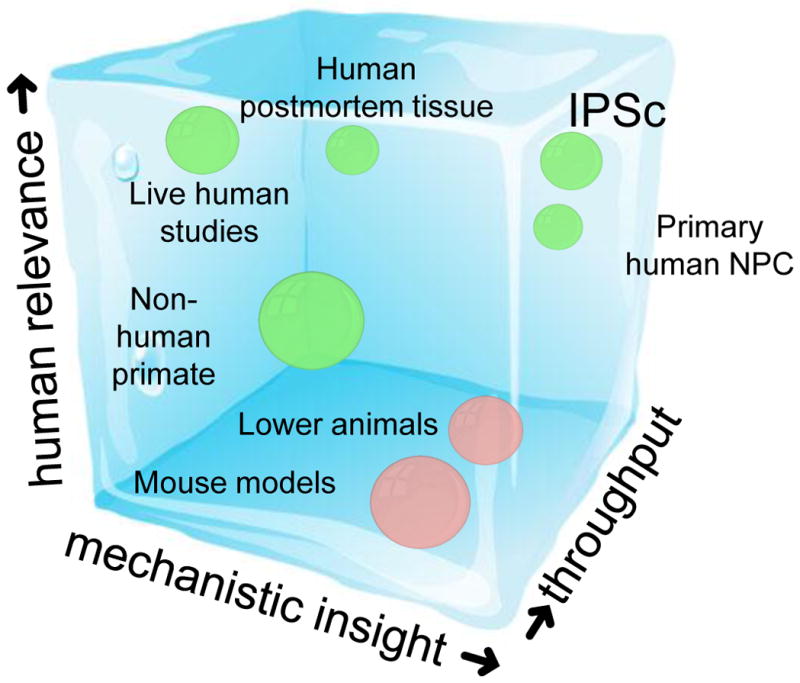
Different models and approaches are complementary and will likely all need to be pursued and integrated to achieve the level of mechanistic understanding needed to develop new therapeutic approaches. Three axes are depicted, increasing human relevance (Y), increasing molecular or mechanistic insight (X) and increasing throughput (Z). The primate (most humanoid) systems are labeled in blue and the other vertebrate models are red. Studies in living patients provide high human relevance, but low molecular insights (e.g. functional imaging) and relatively low throughput. In contrast, mouse models provide high levels of mechanistic or molecular insight at varying levels of throughput and often unknown, but assumed, high human relevance. Zebrafish provide a higher throughput model system, whereas non-human primates are more relevant to humans, but lower throughput and less genetically manipulable. iPSCs promise high human relevance, likely high molecular and mechanistic insight, and relatively low throughput currently, although there is potential for higher throughput with automation and new methods. Primary human neural cells derived from progenitors harvested from human embryonic brain are higher throughput, but because they may not have all of the genetic background characteristics of specific patients (in contrast with IPSC-derived neurons), they may have slightly diminished human relevance and molecular predictive power. Human post-mortem tissue provides human relevance at intermediate throughput, but limited causality testing and hence lower mechanistic insight than the cell or animal models.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Allman JM, Tetreault NA, Hakeem AY, Park S. Am J Hum Biol. 2011;23:5–21. doi: 10.1002/ajhb.21136. [DOI] [PubMed] [Google Scholar]
- Bock C, Kiskinis E, Verstappen G, Gu H, Boulting G, Smith ZD, Ziller M, Croft GF, Amoroso MW, Oakley DH, et al. Cell. 2011;144:439–452. doi: 10.1016/j.cell.2010.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulting GL, Kiskinis E, Croft GF, Amoroso MW, Oakley DH, Wainger BJ, Williams DJ, Kahler DJ, Yamaki M, Davidow L, et al. Nat Biotechnol. 2011;29:279–286. doi: 10.1038/nbt.1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennand KJ, Simone A, Jou J, Gelboin-Burkhart C, Tran N, Sangar S, Li Y, Mu Y, Chen G, Yu D, et al. Nature. 2011 doi: 10.1038/nature09915. epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung AY, Horvath LM, Grafodatskaya D, Pasceri P, Weksberg R, Hotta A, Carrel L, Ellis J. Hum Mol Genet. 2011 doi: 10.1093/hmg/ddr093. epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clowry G, Molnár Z, Rakic P. J Anat. 2010;217:276–288. doi: 10.1111/j.1469-7580.2010.01281.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dragunow M. Nat Rev Drug Discov. 2008;7:659–666. doi: 10.1038/nrd2617. [DOI] [PubMed] [Google Scholar]
- Geschwind DH. Cell. 2008;135:391–395. doi: 10.1016/j.cell.2008.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geschwind DH, Konopka G. Nature. 2009;461:908–915. doi: 10.1038/nature08537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gore A, Li Z, Fung HL, Young JE, Agarwal S, Antosiewicz-Bourget J, Canto I, Giorgetti A, Israel MA, Kiskinis E, et al. Nature. 2011;471:63–67. doi: 10.1038/nature09805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussein SM, Batada NN, Vuoristo S, Ching RW, Autio R, Närvä E, Ng S, Sourour M, Hämäläinen R, Olsson C, et al. Nature. 2011;471:58–62. doi: 10.1038/nature09871. [DOI] [PubMed] [Google Scholar]
- Lee G, Papapetrou EP, Kim H, Chambers SM, Tomishima MJ, Fasano CA, Ganat YM, Menon J, Shimizu F, Viale A, et al. Nature. 2009;461:402–406. doi: 10.1038/nature08320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchetto MC, Carromeu C, Acab A, Yu D, Yeo GW, Mu Y, Chen G, Gage FH, Muotri AR. Cell. 2010;143:527–539. doi: 10.1016/j.cell.2010.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen HN, Byers B, Cord B, Shcheglovitov A, Byrne J, Gujar P, Kee K, Schle B, Dolmetsch RE, Langston W, et al. Cell Stem Cell. 2011;8:267–280. doi: 10.1016/j.stem.2011.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park IH, Arora N, Huo H, Maherali N, Ahfeldt T, Shimamura A, Lensch MW, Cowan C, Hochedlinger K, Daley GQ. Cell. 2008;134:877–886. doi: 10.1016/j.cell.2008.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffenhagen C, Kraus S, Dechant FX, Kandasamy M, Lehner B, Poehler AM, Furtner T, Siebzehnrubl FA, Couillard-Despres S, Strauss O, et al. Stem Cell Rev. 2011 doi: 10.1007/s12015-011-9251-9. epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Urbach A, Bar-Nur O, Daley GQ, Benvenisty N. Cell stem cell. 2010;6:407–411. doi: 10.1016/j.stem.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassos E, Collier DA, Holden S, Patch C, Rujescu D, St Clair D, Lewis CM. Hum Mol Genet. 2010;19:3477–3481. doi: 10.1093/hmg/ddq259. [DOI] [PubMed] [Google Scholar]
- Vierbuchen T, Ostermeier A, Pang ZP, Kokubu Y, Sudhof TC, Wernig M. Nature. 2010;463:1035–1041. doi: 10.1038/nature08797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazawa M, Hsueh B, Jia X, Pasca AM, Bernstein JA, Hallmayer J, Dolmetsch RE. Nature. 2011;471:230–234. doi: 10.1038/nature09855. [DOI] [PMC free article] [PubMed] [Google Scholar]