

Abstract
We recently reported the increased oral clearance of labetalol in pregnant women. To elucidate the mechanism of the elevated oral clearance, we hypothesize that female hormones, at the high concentrations attainable during pregnancy, enhance hepatic metabolism of labetalol.
Labetalol glucuronidation, the major elimination pathway of labetalol, was characterized by screening six recombinant human UGTs (UGT1A1, 1A4, 1A6, 1A9, 2B4, and 2B7) for their capacity to catalyze labetalol glucuronidation.
The effect of female hormones (progesterone, estradiol, estriol, or estrone) on the promoter activities of relevant UDP glucuronosyltransferases (UGT) was investigated using a luciferase reporter assay in HepG2 cells. The involvement of estrogen receptor α (ERα) and pregnane X receptor (PXR) was examined by co-transfecting ERα- or PXR-constructs.
UGT1A1 and UGT2B7 were identified as the major UGT enzymes producing labetalol glucuronides (trace amount of glucuronide conjugate was formed by UGT1A9). The activities of the UGT1A1 promoter containing PXR response elements were enhanced by progesterone but not by estrogens, indicating PXR-mediated induction of UGT1A1 promoter activity by progesterone. Results from semi-quantitative real-time PCR assays are consistent with the above findings. This effect of progesterone on UGT1A1 promoter activities was concentration-dependent.
Promoter activities of UGT2B7 were not affected by either estrogens or progesterone.
These results suggest a potential role for progesterone in regulating labetalol elimination by modulating expression of UGT1A1, leading to enhanced drug metabolism during pregnancy.
Keywords: Pregnancy, labetalol glucuronidation, UGT, metabolism, female hormones, induction
Introduction
Hypertensive disorders complicate 6% to 10% of pregnancies (Brown et al. 2000; Magee 2001) and untreated hypertension during this period increases the risk of perinatal death and prematurity as well as complications associated with hypertension in the mother (Gallery 1995; Magee 2001). Labetalol, an antagonist of α-1 and β adrenoceptors, is an antihypertensive agent that serves as an important component of drug therapy in pregnancy (Magee et al. 2000; Sibai et al. 1990).
Labetalol is completely and rapidly absorbed following oral administration in both men and non-pregnant women (Donnelly and Macphee 1991; Goa et al. 1989). Systemic availability of orally administered labetalol is low due to the drug’s high hepatic clearance and first pass extraction. The mean oral bioavailability of labetalol is about 20% to 40%. Labetalol is 50% bound to plasma proteins, with alpha-1 acid glycoprotein and albumin representing the major binding proteins in plasma (Martinez-Gomez et al. 2006). In pregnant women, labetalol crosses the placenta with the ratio of fetal to maternal plasma concentrations ranging from 20% to 80% (Briggs et al. 2002; Lunell et al. 1985).
Hepatic metabolism is primarily responsible for labetalol elimination, and less than 5% of the drug is recovered unchanged in the urine (Donnelly and Macphee 1991; Goa et al. 1989). The major pathway catalyzing the biotransformation of labetalol is glucuronidation (Goa et al. 1989; Martin et al. 1976), a phase II reaction mediated by UDP-glucuronosyltransferases (UGT). Human UGTs are grouped into 2 families, UGT1 and UGT2, with the UGT1A and UGT2B subfamilies mainly responsible for hepatic glucuronidation (King et al. 2000). The UGT enzymes catalyzing labetalol glucuronidation are not known.
Recently, we reported an increased oral clearance of labetalol in women during the second and third trimesters of pregnancy as compared to post-partum women (Hardman et al. 2005). During pregnancy, the pharmacokinetic behaviour of many drugs differ from those in non-pregnant women, including changes in the oral absorption rate, plasma volume, protein binding, hepatic metabolism and renal excretion (Anderson 2005; Dawes and Chowienczyk 2001; Kaaja and Greer 2005). The increased oral clearance of labetalol suggests pregnancy-mediated changes in labetalol disposition—e.g. decreased oral absorption, enhanced hepatic metabolism, or decreased plasma protein binding. The complete oral absorption and intermediate plasma protein binding of labetalol indicate that significant changes in oral bioavailability or plasma protein binding are not likely to occur during pregnancy. Indeed, we recently showed that protein binding of labetalol is not altered during pregnancy (Choi et al. 2007). Thus, enhanced hepatic metabolism with increased first pass metabolism is most likely responsible for the altered oral clearance of labetalol during pregnancy.
One of the most noticeable features of pregnancy is the dramatic rise in levels of female hormones, i.e., estrogen and progesterone. Blood levels of estrogen and progesterone increase throughout pregnancy until peaking in the third trimester at levels over 100 times greater than the non-pregnancy baseline (Becker 2001). At these high concentrations, female hormones appear to have functions different from their conventional roles as gonadal hormones. Earlier studies have suggested that female hormones at high concentrations may regulate the hepatic expression of certain drug metabolizing enzymes and transporters mediated by pregnane X receptors (PXR) and estrogen receptor α (ERα) (Masuyama et al. 2003; Masuyama et al. 2000; Masuyama et al. 2001; Sugatani et al. 2005; Wang et al. 2006; Yamamoto et al. 2006). Thus, it seems plausible that estrogen and progesterone affect expression levels of drug-metabolizing enzymes responsible for labetalol metabolism.
In the current study, we first determined the UGT enzymes catalyzing labetalol glucuronidation and then assessed the effects of estrogen and progesterone on the expression of relevant UGT enzymes.
Materials and Methods
Chemicals and reagents
Uridine diphospho-glucuronic acid (UDPGA), alamethicin, rifampin, 17β-estradiol, estrone, estriol, progesterone, fluconazole, and labetalol were purchased from Sigma-Aldrich (St. Louis, MO). Recombinant human UGT microsomal enzymes (UGT1A1, 1A4, 1A6, 1A9, 2B4, and 2B7) prepared from baculovirus-infected insect cells, as well as human pooled hepatic microsome, were purchased from BD Gentest (Woburn, MA). Formic acid (ACS grade) and methanol (Optima grade) were purchased from Fisher Scientific (Pittsburgh, PA).
Microsomal reactions
Glucuronidation of labetalol was assayed under the following conditions. The reaction mixture consisted of 0.5 mg ml−1 UGT recombinant microsomal enzyme or human microsome, 0.025 mg ml−1 alamethicin, 2 mM UDPGA, and 10 mM MgCl2 in 50 mM Tris-Cl (pH 7.5) in a final incubation volume of 40 μl. Various concentrations of labetalol (0.0625, 0.125, 0.25, 0.5, 0.75, 1, 1.5, 2 mM) were added to characterize the concentration-dependent metabolite formation. The mixtures were incubated at 37°C for 3 h (UGT1A1 and 1A9) or 1 h (UGT2B7 and human microsome) based on preliminary data which indicated a linear increase of glucuronide metabolite production. As negative controls, microsomal reactions were also performed in the absence of UDPGA. The reaction was stopped by adding three volumes of ice-cold acetonitrile. The mixture was centrifuged, and the supernatant was injected into a liquid chromatography tandem mass spectrometer (LC/MS/MS) for quantification of labetalol and its glucuronide metabolite. Vmax and Km values were then determined for specific UGT enzymes using nonlinear regression function of GraphPad Prism 5 software (San Diego, CA).
Production of labetalol glucuronide by UGT2B7 and human microsome was further examined in the presence of fluconazole (500 μg ml−1) at a labetalol concentration of 400 μM.
LC/MS/MS Determination of labetalol glucuronides
Samples were analyzed by LC/MS/MS (Agilent 1200 HPLC interfaced with Applied Biosystems Qtrap 3200) using an electrospray ion source. The mobile phase consisted of water [0.1% (v/v) formic acid] and methanol. The following linear gradient was used: 30% methanol at time 0 increased to 85% over 9 min. Separation was performed with a Zorbax Eclipse XDB-C8 column (4.6 × 50 mm, 3.5 μm; Agilent Technologies) at a flow rate of 0.4 ml min−1. Injections were carried out at a spray voltage of 5.0 kV and a capillary temperature of 600 °C. MS detection of labetalol and glucuronide metabolites was followed in the positive ion mode by examining full MS peak at m/z 329 and 505, respectively. The glucuronide conjugate peaks were further confirmed by their disappearance in the absence of UDPGA. As the internal standard, prazosin (m/z 384) was used. Due to the unavailability of analytical standards for labetalol glucuronides, the relative amount of the glucuronide conjugates in the samples was expressed as the percentage of the largest labetalol glucuronide peak area (obtained by incubation with UGT2B7).
Plasmids and cloning
Two luciferase constructs containing different segments of the UGT1A1 promoter region, U2K and UB respectively, were provided by Dr. Masahiko Negishi (NIEHS, NC) (Sugatani et al. 2005). The U2K contains the −5.2 to −3.1 kb region (2.1 kb DNA fragment) from the translation start of the UGT1A1 promoter, and UB represents the −3 to −2 kb region (1 kb DNA fragment). A two-kb UGT1A1 promoter region proximal to the translation start site (UA, −2107/+15), was cloned into a pGL3 vector (Promega, Madison, WI) that already contains tyrosine kinase promoter (Sugatani et al. 2005). Forward and reverse primers, gcttagtagtggttctctg and ggactccacagccatgg, respectively, were used to amplify the UA promoter region, and the fragment was inserted in front of the tyrosine kinase promoter after restriction enzyme digestions (SacI and XhoI).
Human UGT2B7 promoter fragment (−1945/+14) was subcloned into pGL3-basic reporter plasmid (Promega, Madison, WI) using forward and reverse primers of atctgagtcttcagtcagtg and ccagtacagtcacctcatgac, respectively, and restriction enzymes, SacI and XhoI. This luciferase construct was named pGL3-UGT2B7.
The vector, Triplicate ERE-luciferase, containing three copies of the estrogen response element (ERE) from the Xenopus vitellogenin A2 upstream of the luciferase gene, was described before (Catherino and Jordan 1995) and used as a positive control for ERα-mediated gene regulation.
The PXR and ERα expression vectors were subcloned into pcDNA3 (Invitrogen) plasmid. The PXR expression vector was originally obtained from Dr. Bingfang Yan (University of Rhode Island) in a pCMVsport2 plasmid. The PXR gene was transferred to pcDNA3 using forward and reverse primers of actatagaaggtacgcctg and ctatgacgtcgcatgcac, and a restriction enzyme, EcoRV. The construct was named pcDNA3-PXR. Similarly, the ERα gene was transferred from pSG5-HEG0 [a gift from Dr. Pierre Chambon (Illkirch, France)] to pcDNA3 using the primers of aatacgactcactataggg and agatctggatccgaattcc, and restriction enzymes, KpnI and BamHI. The construct was named pcDNA3-ER. All sequences were confirmed by sequencing.
Promoter reporter assay
HepG2 cells from ATCC were cultured in phenol red-free DMEM (MediaTech) supplemented with 10% charcoal/dextran-stripped fetal bovine serum (Gemini, Woodland, CA) for three days. The cells were then seeded in 12-well plates at a density of 2×105 cells ml−1 (day 0) and transfected on the next day (day 1) with 0.3 μg of the luciferase constructs (U2K, UB, UA, or pGL3-UGT2B7), 0.3 μg ERα expression vector (pcDNA3-ER) or PXR expression vector (pcDNA3-PXR), and 0.1 μg of a β-galactosidase expression plasmid (β-gal) using the Fugene 6 transfection reagent (Roche Applied Sciences) according to the manufacturer’s protocol. After 16–18 h (day 2), fresh media containing 1 μM of estrogen (either estradiol, estrone, or estriol), 0.5 to 500 μM of progesterone, 10 μM of rifampin, or the ethanol vehicle alone (final concentration 0.1%) was added to the transfected cells. Progesterone was not added to ERα-transfected cells because progesterone is not expected to bind to an estrogen receptor. Twenty-four h later (day 3), the cells were harvested and analyzed for both the luciferase and β-galactosidase activities using assay kits from Promega (Madison, WI). Relative luciferase activities were obtained after normalization of the luciferase activities by β-galactosidase activities. Each experiment was performed in triplicate and repeated on at least two separate occasions to confirm the findings. Statistical analysis was performed by Student’s t-test.
Hepatic RNA extraction and real-time reverse transcription-PCR (RT-PCR)
The pcDNA3-PXR or control vector (pcDNA3), 0.6 μg, was transfected into HepG2 cells using Fugene 6 and treated with 1 μM estradiol, 50 μM progesterone, 10 μM rifampin, or vehicle control (ethanol) for 72 h. The mRNA levels of UGT1A1, UGT2B7, and β-actin after drug treatment were quantified by RT-PCR. Total RNA was extracted using Trizol (Invitrogen), and cDNAs were synthesized using Superscript II (Invitrogen) according to the manufacturer’s protocol. Then, PCR was performed using an ABI PRISM 7900HT Sequence Detection System (Applied Biosystems) and SYBR Green PCR Mastermix (Applied Biosystems). The following primers were used: tagttgtcctagcacctgacgc and aaaatgctccgtctctgatgtaca for UGT1A1, attccattgtttgccgatcaa and cctggccttcatgtgagca for UGT2B7, atcctggcctcgctgtcc and ctcctgcttgctgatccacat for β-actin. The PCR conditions were as follows. After an initial denaturation at 95°C for 10 min, amplification was performed by denaturation at 95°C for 15 sec, and annealing and extension were performed at 60°C for 1 min for 40 cycles. Amplified products were monitored by measuring the increase of fluorescence intensity from the SYBR green dye that binds to double-strand DNA amplified by PCR. The dissociation curves for each reaction were examined to ensure amplication of a single PCR product in the reaction. The copy number of UGT1A1 and UGT2B7 mRNA in the cDNA samples was normalized to the β-actin expression level. Statistical analysis was performed by Student’s t-test.
Results
Microsome study
To identify UGT enzymes responsible for labetalol glucuronidation, UGT enzymes including UGT1A1, 1A4, 1A6, 1A9, 2B4, and 2B7 were screened for their capacity to catalyze labetalol glucuronidation. Each UGT enzyme as a recombinant protein was incubated with labetalol in the presence of UDPGA, a cofactor for glucuronidation reaction. No glucuronide metabolites were identified following incubation with UGT1A4, 1A6, or 2B4, whereas significant amounts of glucuronide conjugates accompanied incubation with UGT1A1 and 2B7 (data not shown). Only trace amounts of labetalol glucuronides were observed following incubation with UGT1A9. The major glucuronide conjugates produced by UGT1A1 and 2B7 eluted at different retention times–5.0 and 7.6 min–indicating that each enzyme produced a structurally distinct glucuronide. The retention time for labetalol was 7.3 min. The putative glucuronide peaks were not observed in incubations performed without UDPGA (data not shown).
The identity of the glucuronide conjugates was further confirmed by product ion scanning using tandem mass spectrometry in positive ion mode. Mass spectral analysis of protonated labetalol glucuronide at m/z 505 revealed major fragment ions at m/z 329 and 311, which corresponds to the loss of glucuronide and loss of water and glucuronide groups, respectively (Fig. 1). Previously reported mass spectra of labetalol glucuronides indicate that glucuronidation at the phenolic-OH of labetalol produces a stronger fragment ion at m/z 329 (corresponding to molecular weight of protonated labetalol) than at m/z 311, and glucuronidation at the aliphatic-OH produced a stronger m/z 311 fragment ion than m/z 329 (Niemeijer et al. 1991). Thus, UGT1A1 appears to conjugate the phenolic-OH and 2B7 the aliphatic-OH of labetalol.
Figure 1.

Mass spectrum of MS/MS product scan of labetalol glucuronide (m/z = 505) produced by UGT1A1 (A) and UGT2B7 (B) (see text for details). The glucuronide conjugates produced by UGT1A1 and UGT2B7 enzymes eluted at 5.0 and 7.6 min, respectively. Collision energy of 20 V was used to produce the product ions.
The production of distinct glucuronide conjugates of labetalol was further confirmed in human microsomes. Both early-eluting phenolic glucuronide and late-eluting aliphatic glucuronide conjugates were generated by incubation of labetalol with pooled human hepatic microsome.
Production of the aliphatic glucuronide of labetalol by UGT2B7 was further confirmed by using fluconazole, a specific UGT2B7 inhibitor (Uchaipichat et al. 2006). Fluconazole, at 500 μg ml−1 concentration, inhibited the production of late-eluting labetalol glucuronide by 30% and 28% in incubations with UGT2B7 enzyme and human microsome, respectively. The production of early-eluting glucuronide was not affected by co-incubation with fluconazole in human microsome.
Glucuronidation of labetalol by UGT enzymes exhibited Michaelis-Menten kinetics (Fig 2). Vmax and Km were estimated from the metabolite production rate vs. concentration profile. Metabolite production by UGT2B7 exhibited a pattern of product inhibition at labetalol concentrations above 1 mM, thus data points at higher drug concentrations were excluded from the curve fitting. Due to the unavailability of pure labetalol glucuronide, the amount of metabolite was expressed as % of the largest glucuronide peak area. The derived Km values for UGT1A1, 1A9 and 2B7 were 956 ±14, 368 ± 14, and 386 ± 8 μM (mean ± SE, n=3), respectively, and the corresponding Vmax values were 1091 ± 10, 73 ± 2, and 4227 ± 113 % peak area min−1 mg protein−1 (mean ± SE, n=3), respectively. Relevant parameters for human microsome were not estimated as the metabolite production was linear up to 2 mM. In human microsome, the peak areas of phenolic glucuronide represented about 20% of the peak areas of late-eluting aliphatic glucuronide. Based on the above results, UGT1A1 and UGT2B7 appear to be the major UGT enzymes responsible for glucuronidation of labetalol.
Figure 2.

Production rates of labetalol glucuronide metabolites over various labetalol concentrations, expressed as % of the largest glucuronide peak area, for UGT1A1 (A), UGT2B7 (B), UGT1A9 (C; inlet figure showing the data range), human microsome (D for late- and E for early-eluting glucuronide peak). Recombinant UGT microsome was incubated in the presence of UDPGA in duplicates. Lines fitted for Mechaelis-Menten equations were shown.
Transcriptional regulation of UGT1A1 and UGT2B7
To examine potential hormonal regulation of UGT1A1 and 2B7, a luciferase reporter assay was conducted where promoter activities of UGT1A1 and 2B7 were indirectly measured by activities of luciferase. Luciferase constructs containing various regions of the UGT1A1 promoters (U2K, UB, or UA) or the UGT2B7 promoters (pGL3-UGT2B7) were co-transfected with ER (pcDNA3-ER) or PXR (pcDNA3-PXR) into HepG2 cells, and the luciferase activities were measured after treatment with estradiol, estrone, estriol, or progesterone. ERα-transfected cells were not treated with progesterone.
As shown in Fig 3, progesterone significantly induced luciferase activity of the U2K in the presence of PXR by about 7.8-fold (p = 0.006), whereas estrogen showed no induction with either co-transfected PXR or ERα. The 10-fold induction shown by rifampin, the positive control for PXR-mediated response, verified the experimental results (Fig 3). Estradiol treatment of Triplicate ERE-luciferase vector caused about 47-fold induction in the promoter activity, verifying ERα-mediated estrogen response (data not shown).
Figure 3.

Effect of female hormones on the promoter activity of various 5′-flanking fragments of UGT1A1 promoter in HepG2 cells with exogenously expressed ERα or PXR. Luciferase constructs containing −5.2 to −3.1 (U2K), −3.1 to −2.0 (UB), or −2.0 kbp (UA) regions of UGT1A1 promoter were cotransfected into HepG2 cells along with β-gal and the expression vector (empty pcDNA3, ERα or PXR). The ERα-expressed cells were treated with the vehicle (ethanol) alone or various drugs including estrogen (1 μM) or rifampin (10 μM) for 24 h, and PXR-expressed cells were treated with progesterone (10 μM) additionally. The luciferase activity was determined as described under Materials and Methods. Relative luciferase activity is expressed as ratios relative to vehicle (ethanol) control. Data presented are the mean of a triplicate experiment ± S.D. *: p<0.05 compared to vehicle-treated group.
The induction of UGT1A1 promoter activity by progesterone was concentration-dependent (Fig 4). No change in promoter activity occurred for the other UGT1A1 promoter regions, i.e. UB and UA, following treatment with rifampin, progesterone or estrogen (Fig 3).
Figure 4.

Effect of various concentrations of progesterone on the promoter activity of U2K, the UGT1A1 promoter region containing PXR response element. U2K, the luciferase construct containing −5.2 to −3.1 kbp region of UGT1A1 promoter, was cotransfected into HepG2 cells with β-gal, and the expression vector for PXR (filled circle) or pcDNA3 empty vector (empty circle). The cells were treated with progesterone (0.5, 1, 5, 10, 50, 100, or 500 μM) or vehicle control. The luciferase activity was determined as described under Materials and Methods. Relative luciferase activity is expressed as ratios relative to vehicle (ethanol) control. Data presented are the mean of a triplicate experiment ± S.D.
The promoter activities of the −2 kb region of UGT2B7 gene were not affected by rifampin or any of the female hormones tested (Fig 5).
Figure 5.
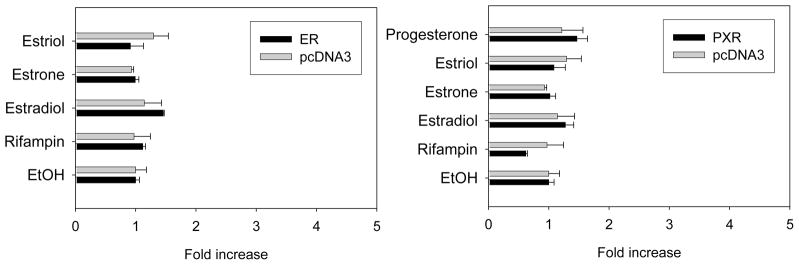
Effect of female hormones on the promoter activity of UGT2B7 promoter in HepG2 cells with exogenously expressed ER or PXR. Luciferase constructs containing −2.0 kbp region of UGT2B7 promoter were cotransfected into HepG2 cells with β-gal and the expression vector for ERα or PXR. The ERα-expressed cells were treated with the vehicle (ethanol) alone or drugs such as estrogen (1 μM) or rifampin (10 μM) for 24 h. PXR-transfected cells were treated with progesterone (10 μM) additionally. The luciferase activity was determined as described under Materials and Methods. Relative luciferase activity is expressed as ratios relative to vehicle (ethanol) control. Data presented are the mean of a triplicate experiment ± S.D.
To further confirm the findings from the luciferase assays, mRNA levels of UGT1A1 and UGT2B7 were determined by semi-quantitative RT-PCR in HepG2 cells after estradiol, progesterone, or rifampin treatment. Upon co-transfection with PXR, progesterone and rifampin caused a 3.0- and 6.5-fold increase in UGT1A1 mRNA levels, respectively (p < 0.05 for both), whereas UGT2B7 expression was not affected by either compound (Fig 6). Estradiol treatment caused no change in expression of UGT1A1 or UGT2B7. mRNA level of pS2, a known estrogen-responsive gene (Barkhem et al. 2002), increased by 5-fold upon estradiol treatment (data not shown).
Figure 6.

Effect of female hormones on the mRNA levels of UGT1A1 and UGT2B7. The pcDNA3-PXR or control vector (pcDNA3) was transfected into HepG2 cells, and the transfected cells were treated with 1 μM estradiol (E), 50 μM progesterone (P), 10 μM rifampin (R), or vehicle control (V, ethanol) for 72 h. The mRNA levels were semi-quantitatively determined by real-time PCR. Expression levels of UGT1A1 and UGT2B7 were normalized by those of β-actin. Relative expression levels were expressed as ratios relative to vehicle control. Data presented are the mean of a triplicate experiment ± S.D. *: p<0.05 compared to vehicle-treated group.
Discussion
The pharmacokinetics of a drug provides essential information to guide dosage recommendations. During pregnancy, various physiological changes can alter the pharmacokinetic profiles of drugs, requiring dosage regimens different from those in non-pregnant women and men (Anderson 2005; Dawes and Chowienczyk 2001; Kaaja and Greer 2005). Elucidation of the mechanisms for the altered pharmacokinetics is important in guiding the optimal drug therapy in pregnancy.
We have previously reported that the oral clearance of labetalol is increased in pregnant women as compared to post-partum controls (Hardman et al. 2005) and that the extent of labetalol protein binding is not influenced by pregnancy (Choi et al. 2007). An altered hepatic metabolism of labetalol is a potential mechanism for the increased oral clearance seen in pregnancy.
In the current study, we hypothesized that female hormones, the levels of which rise dramatically during pregnancy, regulate expression of UGTs that catalyze labetalol glucuronidation. UGT enzymes that are responsible for labetalol glucuronidation were identified using 6 recombinant enzymes, UGT1A1, 1A4, 1A6, 1A9, 2B4, and 2B7. The effects of female hormones on expression of certain UGT enzymes were further examined.
Although contribution of other hepatic UGT enzymes, i.e., UGT1A3, 2B10, 2B11, 2B15, and 2B17, to labetalol glucuronidation cannot be excluded, the results from the microsomal reactions using recombinant UGT indicate that UGT1A1 and UGT2B7 are major UGT enzymes catalyzing labetalol glucuronidation. Also, results from promoter reporter assays and RT-PCR analysis indicate that the expression of UGT1A1 is up-regulated by high concentrations of progesterone via a PXR-mediated mechanism. Taken together, our results indicate progesterone-mediated regulation of UGT1A1 via a nuclear receptor, PXR, and suggest induction of UGT1A1 as a potential mechanism for altered labetalol metabolism in pregnancy. Consistent with the above findings, elimination of another UGT1A1 substrate, acetaminophen, is faster in pregnant women as compared with non-pregnant control; the mean half-life was 20% faster in the first trimester, with a corresponding increase in oral clearance (Beaulac-Baillargeon and Rocheleau 1994). A separate study reported an increase in oral clearance of 58% during pregnancy, which was due to a 75% increase in glucuronidation capacity (Miners et al. 1986).
In the current study, the promoter activities of the −2 kb region of the UGT2B7 promoter were not affected by the female hormones or rifampin (a PXR ligand). The results obtained from RT-PCR also correspond to the results obtained from promoter reporter assay, showing no induction in mRNA levels of UGT2B7 with rifampin or progesterone treatment. These results are consistent with a previous report where the pharmacokinetic profiles of zidovudine, a UGT2B7 probe drug, were not altered in pregnancy as compared to non-pregnant controls (O’Sullivan et al. 1993). However, plasma clearance of another UGT2B7 probe drug, morphine, was reported to be 70% higher in pregnant women than in the non-pregnant controls (Gerdin et al. 1990). UGT2B7 has been often suggested as a PXR-responsive gene based on clinical reports, such as drug-drug interactions with rifampin. Glucuronidation of zidovudine was increased by co-administration of rifampin in HIV patients (Gallicano et al. 1999), and production of the acyl glucuronide metabolite of mycophenolate, a reaction mediated by UGT2B7, was increased 2-fold after rifampin co-administration (Naesens et al. 2006). However, rifampin was also capable of decreasing, rather than increasing, UGT2B7-mediated morphine glucuronidation in healthy volunteers (Fromm et al. 1997). These conflicting data appear to indicate complex regulatory mechanisms of UGT2B7 expression. So far, no PXR response elements have been identified in the UGT2B7 promoter region although relevant binding sites for PXR have been well-characterized in the promoter region of another UGT gene, UGT1A1 (Sugatani et al. 2005).
Fetal livers express UGT2B7 at 10–20% of adult levels whereas UGT1A1 is almost undetectable during fetal life (de Wildt et al. 1999). Because labetalol crosses placenta (Briggs et al. 2002; Lunell et al. 1985), additional glucuronidation of the drug by fetal hepatic UGT2B7 may partially contribute to theenhanced labetalol elimination in pregnant women. Further investigation is needed to determine the extent of contribution of fetal UGT enzymes to the labetalol glucuronidation in pregnant women.
The hormone concentrations used in this study were determined based on the range of blood levels reported at term and the projected accumulation of female hormones in the liver. During pregnancy, blood levels of these hormones rise gradually until they peak during the third trimester, at levels ~100 times higher than the baseline maintained during a regular menstrual cycle. Blood concentrations of estradiol and progesterone at term reach 0.1 and 1 μM, respectively (Becker 2001). Accordingly, the hormone concentrations in the liver, an organ receiving ~30% of cardiac output, are expected to be high. Because the liver is the major elimination organ for the hormones, intra-hepatic concentrations of progesterone and estrogen may be even higher as compared to the hormone levels in the blood. In fact, progesterone concentrations in the liver are reported to be about 10-fold higher than blood levels in liver perfusion models (Carlson et al. 1988). Also, 3-fold higher hepatic levels of estrogen, as compared to the levels in blood, was reported after intravenous administration of 17β-estradiol to rats (Schleicher et al. 1998). Thus, the concentrations of 1 μM for estrogen and 10 μM for progesterone should provide a realistic representation of the concentrations attainable in the livers of pregnant women.
Female hormones play a well-established role as gonadal hormones at nM blood concentrations. The concentration-dependent effect of progesterone on UGT1A1 promoter activities, shown in this study, suggests differential roles of female hormones at concentrations attainable during pregnancy. Consistent with this notion, a recent study demonstrated that supra-therapeutic doses of estrogen (10 mg kg−1 ethinyl estradiol subcutaneously in mice) down-regulate multiple hepatic transporters for bile acid and cholesterol elimination (Yamamoto et al. 2006). Another study in pregnant mice showed that expression of a major hepatic drug transporter, breast cancer resistance protein (Bcrp1/Abcg2), correlates with hepatic ERα expression (Wang et al. 2006), suggesting estrogen-mediated regulation of hepatic transporters at high hormonal concentrations. This concentration-dependent effect of female hormones also seems evident in the pharmacokinetic drug-drug interactions caused by oral contraceptives. For many drugs, use of oral contraceptives produces changes in hepatic drug elimination, which is qualitatively similar to those observed in pregnancy. For example, in both pregnant women and oral contraceptive users, hepatic metabolism of CYP2D6, UGT1A1 substrates is increased (Abernethy et al. 1982; Walle et al. 1996) and hepatic elimination of CYP2C19 or CYP1A2 substrates is decreased (Balogh et al. 1995; Laine et al. 2000) as compared to controls. This similarity between pregnant women and oral contraceptive users may be because hormone levels in the liver of oral contraceptive users are significantly higher than blood levels due to hepatic first-pass effect. In fact, 50-fold higher hormonal levels in the liver as compared to blood have been reported after administration of oral contraceptives in rats (Eisenfeld and Aten 1987; Schleicher et al. 1998). These results strongly support the notion that increased levels of female hormones influence expression levels of drug metabolizing enzymes and transporters in the liver, ultimately resulting in altered hepatic metabolism of drugs.
In the current study, we investigated the effects of estrogen and progesterone on the expression of drug metabolizing enzymes responsible for labetalol metabolism, in an effort to understand the mechanism for altered oral clearance of the drug in pregnant women. Both UGT1A1 and UGT2B7 were identified as major UGT enzymes mediating the glucuronidation of labetalol, and UGT1A1 showed enhanced promoter activities with progesterone treatment mediated by PXR. The results obtained in this study suggest that the induction of UGT1A1 expression by rising progesterone levels in pregnant women may be responsible for the increased oral clearance of labetalol.
Acknowledgments
This investigation was conducted in a facility constructed with support from grant C06RR15482 from the NCRR NIH.
References
- Abernethy DR, Divoll M, Ochs HR, Ameer B, Greenblatt DJ. Increased metabolic clearance of acetaminophen with oral contraceptive use. Obstet Gynecol. 1982;60(3):338–41. [PubMed] [Google Scholar]
- Anderson GD. Pregnancy-induced changes in pharmacokinetics: a mechanistic-based approach. Clin Pharmacokinet. 2005;44(10):989–1008. doi: 10.2165/00003088-200544100-00001. [DOI] [PubMed] [Google Scholar]
- Balogh A, Klinger G, Henschel L, Borner A, Vollanth R, Kuhnz W. Influence of ethinylestradiol-containing combination oral contraceptives with gestodene or levonorgestrel on caffeine elimination. Eur J Clin Pharmacol. 1995;48(2):161–6. doi: 10.1007/BF00192743. [DOI] [PubMed] [Google Scholar]
- Barkhem T, Haldosen LA, Gustafsson JA, Nilsson S. pS2 Gene expression in HepG2 cells: complex regulation through crosstalk between the estrogen receptor alpha, an estrogen-responsive element, and the activator protein 1 response element. Mol Pharmacol. 2002;61(6):1273–83. doi: 10.1124/mol.61.6.1273. [DOI] [PubMed] [Google Scholar]
- Beaulac-Baillargeon L, Rocheleau S. Paracetamol pharmacokinetics during the first trimester of human pregnancy. Eur J Clin Pharmacol. 1994;46(5):451–4. doi: 10.1007/BF00191910. [DOI] [PubMed] [Google Scholar]
- Becker KL. Principles and practice of endocrinology and metabolism. xxxiv. Philadelphia: Lippincott Williams & Wilkins Co; 2001. p. 2477. [Google Scholar]
- Briggs GG, Freeman RK, Yaffe SJ. Drugs in pregnancy and lactation: a reference guide to fetal and neonatal risk. xxv. Philadelphia, Pa. London: Lippincott Williams & Wilkins; 2002. p. 1595. [Google Scholar]
- Brown MA, Hague WM, Higgins J, Lowe S, McCowan L, Oats J, Peek MJ, Rowan JA, Walters BN. The detection, investigation and management of hypertension in pregnancy: executive summary. Aust N Z J Obstet Gynaecol. 2000;40(2):133–8. doi: 10.1111/j.1479-828x.2000.tb01136.x. [DOI] [PubMed] [Google Scholar]
- Carlson KE, Brandes SJ, Pomper MG, Katzenellenbogen JA. Uptake of three [3H]progestins by target tissues in vivo: implications for the design of diagnostic imaging agents. Int J Rad Appl Instrum B. 1988;15(4):403–8. doi: 10.1016/0883-2897(88)90010-4. [DOI] [PubMed] [Google Scholar]
- Catherino WH, Jordan VC. Increasing the number of tandem estrogen response elements increases the estrogenic activity of a tamoxifen analogue. Cancer Lett. 1995;92(1):39–47. doi: 10.1016/0304-3835(95)03755-l. [DOI] [PubMed] [Google Scholar]
- Choi S, Jeong H, Deyo K, Fischer J. Protein binding of labetalol in pregnancy. Clin Pharm Ther. 2007;81(S1):S79. [Google Scholar]
- Dawes M, Chowienczyk PJ. Drugs in pregnancy. Pharmacokinetics in pregnancy. Best Pract Res Clin Obstet Gynaecol. 2001;15(6):819–26. doi: 10.1053/beog.2001.0231. [DOI] [PubMed] [Google Scholar]
- de Wildt SN, Kearns GL, Leeder JS, van den Anker JN. Glucuronidation in humans. Pharmacogenetic and developmental aspects. Clin Pharmacokinet. 1999;36(6):439–52. doi: 10.2165/00003088-199936060-00005. [DOI] [PubMed] [Google Scholar]
- Donnelly R, Macphee GJ. Clinical pharmacokinetics and kinetic-dynamic relationships of dilevalol and labetalol. Clin Pharmacokinet. 1991;21(2):95–109. doi: 10.2165/00003088-199121020-00002. [DOI] [PubMed] [Google Scholar]
- Eisenfeld AJ, Aten RF. Estrogen receptors and androgen receptors in the mammalian liver. J Steroid Biochem. 1987;27(4–6):1109–18. doi: 10.1016/0022-4731(87)90197-x. [DOI] [PubMed] [Google Scholar]
- Fromm MF, Eckhardt K, Li S, Schanzle G, Hofmann U, Mikus G, Eichelbaum M. Loss of analgesic effect of morphine due to coadministration of rifampin. Pain. 1997;72(1–2):261–7. doi: 10.1016/s0304-3959(97)00044-4. [DOI] [PubMed] [Google Scholar]
- Gallery ED. Hypertension in pregnancy. Practical management recommendations. Drugs. 1995;49(4):555–62. doi: 10.2165/00003495-199549040-00006. [DOI] [PubMed] [Google Scholar]
- Gallicano KD, Sahai J, Shukla VK, Seguin I, Pakuts A, Kwok D, Foster BC, Cameron DW. Induction of zidovudine glucuronidation and amination pathways by rifampicin in HIV-infected patients. Br J Clin Pharmacol. 1999;48(2):168–79. doi: 10.1046/j.1365-2125.1999.00987.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdin E, Salmonson T, Lindberg B, Rane A. Maternal kinetics of morphine during labour. J Perinat Med. 1990;18(6):479–87. doi: 10.1515/jpme.1990.18.6.479. [DOI] [PubMed] [Google Scholar]
- Goa KL, Benfield P, Sorkin EM. Labetalol. A reappraisal of its pharmacology, pharmacokinetics and therapeutic use in hypertension and ischaemic heart disease. Drugs. 1989;37(5):583–627. doi: 10.2165/00003495-198937050-00002. [DOI] [PubMed] [Google Scholar]
- Hardman J, Endres L, Fischer P, Fischer J. Pharmacokinetics of labetalol in pregnancy. Pharmacotherapy. 2005;25(10):1493. [Google Scholar]
- Kaaja RJ, Greer IA. Manifestations of chronic disease during pregnancy. Jama. 2005;294(21):2751–7. doi: 10.1001/jama.294.21.2751. [DOI] [PubMed] [Google Scholar]
- King CD, Rios GR, Green MD, Tephly TR. UDP-glucuronosyltransferases. Curr Drug Metab. 2000;1(2):143–61. doi: 10.2174/1389200003339171. [DOI] [PubMed] [Google Scholar]
- Laine K, Tybring G, Bertilsson L. No sex-related differences but significant inhibition by oral contraceptives of CYP2C19 activity as measured by the probe drugs mephenytoin and omeprazole in healthy Swedish white subjects. Clin Pharmacol Ther. 2000;68(2):151–9. doi: 10.1067/mcp.2000.108949. [DOI] [PubMed] [Google Scholar]
- Lunell NO, Kulas J, Rane A. Transfer of labetalol into amniotic fluid and breast milk in lactating women. Eur J Clin Pharmacol. 1985;28(5):597–9. doi: 10.1007/BF00544073. [DOI] [PubMed] [Google Scholar]
- Magee LA. Treating hypertension in women of child-bearing age and during pregnancy. Drug Saf. 2001;24(6):457–74. doi: 10.2165/00002018-200124060-00004. [DOI] [PubMed] [Google Scholar]
- Magee LA, Elran E, Bull SB, Logan A, Koren G. Risks and benefits of beta-receptor blockers for pregnancy hypertension: overview of the randomized trials. Eur J Obstet Gynecol Reprod Biol. 2000;88(1):15–26. doi: 10.1016/s0301-2115(99)00113-x. [DOI] [PubMed] [Google Scholar]
- Martin LE, Hopkins R, Bland R. Metabolism of labetalol by animals and man. Br J Clin Pharmacol. 1976;3(4 Suppl 3):695–710. [PubMed] [Google Scholar]
- Martinez-Gomez MA, Sagrado S, Villanueva-Camanas RM, Medina-Hernandez MJ. Characterization of basic drug-human serum protein interactions by capillary electrophoresis. Electrophoresis. 2006;27(17):3410–9. doi: 10.1002/elps.200600102. [DOI] [PubMed] [Google Scholar]
- Masuyama H, Hiramatsu Y, Kodama J, Kudo T. Expression and potential roles of pregnane X receptor in endometrial cancer. J Clin Endocrinol Metab. 2003;88(9):4446–54. doi: 10.1210/jc.2003-030203. [DOI] [PubMed] [Google Scholar]
- Masuyama H, Hiramatsu Y, Kunitomi M, Kudo T, MacDonald PN. Endocrine disrupting chemicals, phthalic acid and nonylphenol, activate Pregnane X receptor-mediated transcription. Mol Endocrinol. 2000;14(3):421–8. doi: 10.1210/mend.14.3.0424. [DOI] [PubMed] [Google Scholar]
- Masuyama H, Hiramatsu Y, Mizutani Y, Inoshita H, Kudo T. The expression of pregnane X receptor and its target gene, cytochrome P450 3A1, in perinatal mouse. Mol Cell Endocrinol. 2001;172(1–2):47–56. doi: 10.1016/s0303-7207(00)00395-6. [DOI] [PubMed] [Google Scholar]
- Miners JO, Robson RA, Birkett DJ. Paracetamol metabolism in pregnancy. Br J Clin Pharmacol. 1986;22(3):359–62. doi: 10.1111/j.1365-2125.1986.tb02901.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naesens M, Kuypers DR, Streit F, Armstrong VW, Oellerich M, Verbeke K, Vanrenterghem Y. Rifampin induces alterations in mycophenolic acid glucuronidation and elimination: implications for drug exposure in renal allograft recipients. Clin Pharmacol Ther. 2006;80(5):509–21. doi: 10.1016/j.clpt.2006.08.002. [DOI] [PubMed] [Google Scholar]
- Niemeijer NR, Gerding TK, De Zeeuw RA. Glucuronidation of labetalol at the two hydroxy positions by bovine liver microsomes. Isolation, purification, and structure elucidation of the glucuronides of labetalol. Drug Metab Dispos. 1991;19(1):20–3. [PubMed] [Google Scholar]
- O’Sullivan MJ, Boyer PJ, Scott GB, Parks WP, Weller S, Blum MR, Balsley J, Bryson YJ. The pharmacokinetics and safety of zidovudine in the third trimester of pregnancy for women infected with human immunodeficiency virus and their infants: phase I acquired immunodeficiency syndrome clinical trials group study (protocol 082). Zidovudine Collaborative Working Group. Am J Obstet Gynecol. 1993;168(5):1510–6. doi: 10.1016/s0002-9378(11)90791-1. [DOI] [PubMed] [Google Scholar]
- Schleicher F, Tauber U, Louton T, Schunack W. Tissue distribution of sex steroids: concentration of 17beta-oestradiol and cyproterone acetate in selected organs of female Wistar rats. Pharmacol Toxicol. 1998;82(1):34–9. doi: 10.1111/j.1600-0773.1998.tb01395.x. [DOI] [PubMed] [Google Scholar]
- Sibai BM, Mabie WC, Shamsa F, Villar MA, Anderson GD. A comparison of no medication versus methyldopa or labetalol in chronic hypertension during pregnancy. Am J Obstet Gynecol. 1990;162(4):960–6. doi: 10.1016/0002-9378(90)91297-p. discussion 966–7. [DOI] [PubMed] [Google Scholar]
- Sugatani J, Nishitani S, Yamakawa K, Yoshinari K, Sueyoshi T, Negishi M, Miwa M. Transcriptional regulation of human UGT1A1 gene expression: activated glucocorticoid receptor enhances constitutive androstane receptor/pregnane X receptor-mediated UDP-glucuronosyltransferase 1A1 regulation with glucocorticoid receptor-interacting protein 1. Mol Pharmacol. 2005;67(3):845–55. doi: 10.1124/mol.104.007161. [DOI] [PubMed] [Google Scholar]
- Uchaipichat V, Winner LK, Mackenzie PI, Elliot DJ, Williams JA, Miners JO. Quantitative prediction of in vivo inhibitory interactions involving glucuronidated drugs from in vitro data: the effect of fluconazole on zidovudine glucuronidation. Br J Clin Pharmacol. 2006;61(4):427–39. doi: 10.1111/j.1365-2125.2006.02588.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walle T, Fagan TC, Walle UK, Topmiller MJ. Stimulatory as well as inhibitory effects of ethinyloestradiol on the metabolic clearances of propranolol in young women. Br J Clin Pharmacol. 1996;41(4):305–9. doi: 10.1046/j.1365-2125.1996.03097.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Wu X, Hudkins K, Mikheev A, Zhang H, Gupta A, Unadkat JD, Mao Q. Expression of the breast cancer resistance protein (Bcrp1/Abcg2) in tissues from pregnant mice: effects of pregnancy and correlations with nuclear receptors. Am J Physiol Endocrinol Metab. 2006;291(6):E1295–304. doi: 10.1152/ajpendo.00193.2006. [DOI] [PubMed] [Google Scholar]
- Yamamoto Y, Moore R, Hess HA, Guo GL, Gonzalez FJ, Korach KS, Maronpot RR, Negishi M. Estrogen receptor alpha mediates 17alpha-ethynylestradiol causing hepatotoxicity. J Biol Chem. 2006;281(24):16625–31. doi: 10.1074/jbc.M602723200. [DOI] [PubMed] [Google Scholar]