

Abstract
A long-standing question in evolutionary studies of snake venoms is the extent to which phylogenetic divergence and diet can account for between-species differences in venom composition. Here we apply phylogeny-based comparative methods to address this question. We use data on venom variation generated using proteomic techniques for all members of a small clade of rattlesnakes (Sistrurus sp.) and two outgroups for which phylogenetic and diet information is available. We first complete the characterization of venom variation for all members of this clade with a “venomic” analysis of pooled venoms from two members of this genus, S. milarius streckeri and S. m. milarius. These venoms exhibit the same general classes of proteins as those found in other Sistrurus species but differ in their relative abundances of specific protein families. We then test whether there is significant phylogenetic signal in the relative abundances of major venom proteins across species and if diet (measured as percent mammals and lizards among all prey consumed) covaries with venom composition after phylogenetic divergence is accounted for. We found no evidence for significant phylogenetic signal in venom variation: K values for seven snake venom proteins and two composite venom variables [PC 1 and 2]) were all nonsignificant and lower (mean = 0.11+0.06 sd) than mean K values (>0.35) previously reported for a wide range of morphological, life history, physiological and behavioral traits from other species. Finally, analyses based on Phylogenetic Generalized Least Squares (PGLS) methods reveal that variation in abundance of some venom proteins, most strongly CRISP is significantly related to snake diet. Our results demonstrate that venom variation in these snakes is evolutionarily a highly labile trait even among very closely-related taxa and that natural selection acting through diet variation may play a role in molding the relative abundance of specific venom proteins.
Introduction
Snake venom proteins are one of the most widely studied types of animal toxins (for general reviews see [1], [2]). These proteins largely belong to a few major protein families, including enzymes (serine proteinases, Zn2+-metalloproteinases, L-amino acid oxidase, group II PLA2s) and proteins without enzymatic activity (disintegrins, C-type lectin-like proteins, vasoactive peptides, myotoxins, CRISP, nerve and vascular endothelium growth factors, cystatin and Kunitz-type protease inhibitors) [3]–[8]. They function by interfering with the coagulation cascade, haemostatic system, and tissue repair acting to immobilize, kill and digest the prey of venomous snakes (for recent review see [9]).
Variation in venom composition at different biological levels is widespread [10], [11] and has been argued to be an adaptation that has evolved via natural selection, allowing different species to capture and digest different prey [12]–[14]. However, others have proposed that, due to the high toxicity and large doses of venom that are injected, variation in venom composition is unlikely to be subject to natural selection for lethal effects on prey, and that much of the variation in venom proteins may be a by-product of neutral evolutionary processes [15]–[17]. Under this hypothesis, interspecific differences in venom composition should be closely related to the degree of phylogenetic divergence between species.
One rarely-used approach to study the evolutionary basis of venom variation is to examine patterns of interspecific variation in venom in the context of phylogenetic relationships among a group of related species [12], [14], [18]. Specifically, phylogeny-based comparative methods [19], [20] offer ways to address key questions about venom differentiation between species. One long-standing question is the degree to which venom variation covaries with levels of phylogentic divergence between species. One way to measure such a relationship is to determine the level of phylogenetic signal in venom composition across a set of species for which a phylogeny has been established. Phylogenetic signal refers a tendency (pattern) for evolutionarily related organisms to resemble each other, with no implication as to the mechanism that might cause such resemblance (process) (20). Moderate levels of signal would support the hypothesis that neutral evolutionary mechanisms such as genetic drift are responsible for variation in venom composition across a set of related species whereas low or exceptionally high levels of signal would leave open a role for selection acting on venom composition through diet variation. If this is the case, evidence for selection could be examinedusing other comparative methods (19) to see if correlations exist between venom composition and measures of diet after taking expected similarity due to phylogeny alone into account. Here, we explore these questions using data from high resolution proteomics-based analysis of venom composition combined with phylogenetic and diet information for all members of a small clade of rattlesnakes (Sistrurus sp.).
Sistrurus rattlesnakes are a genus of New World pitvipers (Viperidae; Crotalinae) consisting of two named species: massasauga (S. catenatus) and pigmy (S. miliarius) rattlesnakes. Each species consists of three named subspecies (S. c. catenatus; S. c. tergeminus, and S. c. edwardsii, and S. m. milarius, S. m. barbouri, and S. m. streckeri ) [21]. Recently, Kubatko et al. [22] conducted a phylogenetic analysis of the genus based on multilocus data in which they generated a species tree with branch lengths based on multiple gene trees, estimated dates of divergence, and conducted tests of the genetic distinctiveness of each subspecies (Figure 1). These tests showed evidence for genetic distinctiveness of all subspecies. Further, all taxa surveyed to date show substantial differences in venom composition [23]. This is a key trait involved in prey capture and digestion and differences have functional consequences in terms of the ability to subdue ecologically-diverse prey [24]. In particular, toxicity to mammals appears to be a key axis along which venom function has evolved with whole venom from taxa showing unusually high (S. c. catenatus) and unusually low (S. m. barbouri) LD50 values for mice [24]. These data suggest that the proportion of mammals in the diets of different Sistrurus may be related to venom composition and imply a role for natural selection in generating differences in venom composition between taxa. However, the extent that levels of phylogenetic divergence alone could account for interspecific differences in venom is unknown.
Figure 1. Species tree based on multi-locus data showing phylogenetic relationships and approximate dates of divergence for all Sistrurus taxa [22] and Crotalus atrox and Agkistrodon c. contortrix outgroups [57].

Branch lengths for the in group taxa and Agkistrodon are as in Kubatko et al. [22]. Branch lengths for C. atrox were estimated based on mtDNA ATP6–8 gene sequence divergences between C. atrox [30] and S. miliarius and S. catenatus [22].
Here, we examine the association between phylogenetic divergence, diet and venom variation for Sistrurus rattlesnakes. We complete the proteomic-based analysis of the abundances of specific venom proteins (“venomics”) for all Sistrurus by analyzing the venom of the two as yet unanalyzed taxa (S. m. milarius, Carolina pigmy rattlesnake, and S. m. streckeri, Western pigmy rattlesnake). We combine this information with published data on venom composition for other Sistrurus [23] and close (Crotalus atrox) [25] and distant (Agkistrodon c. contortrix) (J Calvete et al., unpublished data) outgroups. We analyze these data in the context of a phylogeny for the group [22] to determine if there is a phylogenetic signal in shifts in venom composition. We then test whether correlations exist between venom composition and diet after taking expected similarity due to phylogenetic divergence alone into account. These analysis complement our previous work on venom variation in Sistrurus that has focused on patterns of intraspecific variation in S. c. catenatus [26] and the function of venom variation in terms of toxicity to different prey [24].
Materials and Methods
Ethics Statement
Permits for sampling snakes were obtained from the St. Louis Zoo (Permit BM 2010-06) issued by M. Duncan and from the Carolina Sandhills National Wildlife Refuge (Special Use Permit issued by L. Housh). The protocol for the collection of venom samples from individual snakes was approved by the Institutional Animal Use and Care Committee of The Ohio State University (Permit Number: 2008A0087). All efforts were made to minimize suffering of the animals while samples were being collected.
Samples of S. m. milarius and S. m. streckeri Venom
For proteomic analyses of venom, we obtained venom samples from two sources: captive adult S. m. streckeri (n = 5) held at the St. Louis Zoo which had originally been collected from the wild in SE Missouri (Barry County), and wild-caught adult S. m. milarius (n = 5) from Carolina Sandhills National Wildlife Refuge in Chesterfield County, South Carolina. Snakes were restrained in clear plastic tubes and then induced to bite the top of a glass beaker that had been covered with Parafilm. Secreted venom was immediately pipetted into a cryovial and stored in liquid nitrogen.
Isolation and Proteomic Characterization of Venom Proteins
For each taxon, separate pooled samples consisting of equal amounts of venom from five individuals were subject to proteomics analysis as described by Sanz et al. [23]. Pooled samples were used because they yield more interpretable HPLC profiles of venom composition that reflect the full range of variation of observed across multiple individual samples (27). Briefly, soluble proteins from 2 mg of crude venom were were separated on a Teknokroma Europa C18 (0.4 cm×25 cm, 5 mm particle size, 300 Å pore size) column using an Agilent LC 1100 High Pressure Gradient System equipped with DAD detector and micro-Auto-sampler. The flow-rate was set to 1 ml/min and the column was developed with a linear gradient of 0.1% TFA in water (solution A) and acetonitrile (solution B), isocratically (5% B) for 10 min, followed by 5–25% B for 20 min, 25–45% B for 120 min, and 45–70% for 20 min. Protein detection was at 215 nm and peaks were collected manually and dried in a Speed-Vac (Savant). Reverse-phase HPLC runs were consistently superimposable through their X-axes and the acetonitrile gradient profile, and thus all chromatograms of venom samples from different specimens from the same taxon were directly comparable. Given that the wavelength of absorbance for a peptide bond is 190–230 nm, protein detection at 215 nm allows to estimate the relative abundances (expressed as percentage of the total venom proteins) of the different protein families from the relation of the sum of the areas of the reverse-phase chromatographic peaks containing proteins from the same family to the total area of venom protein peaks in the reverse-phase chromatogram. In a strict sense, and according to the Lambert-Beer law, the calculated relative amounts correspond to the “% of total peptide bonds in the sample”, which is a good estimate of the % by weight (gr/100 gr) of a particular venom component.
Initially, the HPLC-isolated proteins were subjected to N-terminal sequence analysis (using a Procise instrument from Applied Biosystems (Foster City, CA, USA) following the manufacturer’s instructions). Amino acid sequence similarity searches were performed against the available databanks using the BLAST program [28] implemented in the WU-BLAST2 search engine at http://www.bork.embl-heidelberg.de. The molecular masses of the purified proteins were determined by SDS-PAGE (on 12–15% polyacrylamide gels) and by MALDI-TOF mass spectrometry using an Applied Biosystems Voyager-DE Pro mass spectrometer operated in linear mode.
Protein bands of interest were excised from Coomassie Brilliant Blue-stained SDS-PAGEs and subjected to automated reduction with DTT, alkylation with iodoacetamide, and proteolytic digestion with sequencing grade bovine pancreas trypsin (Roche). The tryptic peptide mixtures were dried, redissolved in 70% acetonitrile and 0.1% TFA and 0.65 ul of the digests spotted onto a MALDI-TOF sample holder and analyzed with an Applied Biosystems Voyager-DE Pro MALDI-TOF mass spectrometer, operated in delayed extraction and reflectror modes. For peptide sequencing, the protein digest mixture was loaded in a nanospray capilary and subjected to electrospray ionization mass spectrometric analysis using a QTRap 2000™ mass spectrometer (Applied Biosystems) equipped with a nanospray source (Proxeon Biosystems). Monoisotopic doubly- or triply-charged precursor ions were selected (within a window of ±0.5 m/z) and sequenced by CID-MS/MS using the Enhanced Product Ion mode with Q0 trapping option; Q1 was operated at unit resolution, the Q1-to-Q2 collision energy was set to 30 (for m/z ≤700) or 35 eV (for m/z >700), the Q3 entry barrier was 8 V, the LIT (linear ion trap) Q3 fill time was 250 ms, and the scan rate in Q3 was 1000 amu/s. CID spectra were interpreted manually (i.e. de novo sequenced) or using the on-line form of the MASCOT program at http://www.matrixscience.com against the SwissProt and NCBInr databases, and against a private database comprising previous assignments of Sistrurus venom proteins [23]. MS/MS mass tolerance was set to ±0.6 Da. Carbamidomethyl cysteine and oxidation of methionine were fixed and variable modifications, respectively.
Phylogeny-based Comparative Analyses
To analyze changes in venom in a phylogenetic context we used data on venom composition and phylogenetic relationships for all Sistrurus taxa and close (C. atrox) and distant (Agkistrodon c. contortix) outgroups. We chose C. atrox and A. c. contortix as representative outgroups because they are species from either the sister clade to Sistrurus (C. atrox) or the sister clade to all rattlesnakes (A. c. contortrix) [29] and because both species have haemorrhagic venoms which are the same general type of venom found in all Sistrurus [18 and JJ Calvete, unpublished data], making them good representatives of ancestral venom composition. We used the species tree topology and associated multilocus-based branch lengths from Kubatko et al. [22] as our estimate of the phylogeny for this group with the exception that we added C. atrox to the phylogeny. To estimate a value for the multilocus branch length connecting C. atrox with the Sistrurus ingroup clade, we used divergence estimates based on mtDNA ATP6–8 gene sequences from C. atrox [30] and S. miliarius and S. catenatus [22]. Venom composition for each taxa was measured as the relative percent of total venom composition made up by proteins from distinct families using data from this study and Sanz et al. [23] for Sistrurus, Calvete et al. [25] for C. atrox and Calvete et al. [unpublished data – see Supplemental Information] for A. c. contortrix. For the statstical analysis, percent values for individual proteins for each taxon were arcsin square root transformed to normalize the data and reduce the dependancy between individual values for a given taxon. We also analyzed composite variables in the form of Principle Components 1 and 2 that summarized the variation present across all venom proteins. These were calculated using the Princom subroutine in R [31].
For broad-scale estimates of diet, we used published information based on gut content analyses to estimate the percentage of each taxon’s diet made up of mammals and lizards. Diet data for six of the taxa came from studies with large samples from multiple geographic locations (S. c. catenatus [n = 139 samples]; S. c. tergeminus [n = 111]; S. c. edwardsii [n = 163] (data from 32); S. m. barbouri [n = 103] (data from T. Farrell and P. May, unpublished data); C. atrox [n = 205](data from 33)) and A. c. contortrix [n = 135] (data from 34) whereas samples for the two other taxa were more limited (S. m. streckeri [n = 14] (data from 35); S. m. miliarius [n = 12] (data from 36) which could compromise accuracy of our diet estimates for these taxa. We chose these broad categories because they represent general differences in diet that could be readily estimated from the available data that may reflect functional differences in venom toxicity among these snakes (see 24). However, we acknowledge that other prey could be important and/or that more sophisticated analyses that integrate across types of prey would be informative if better estimates of diet were available for the taxa with limited samples. Because of the high correlation between percent mammals and lizards in snake diets (see below) we also estimated a composite variable in the form of Principle Component 1 calculated as above.
We used the subroutine multiPhylosignal in the R-package Picante [37] to determine if there was evidence for phylogenetic signal in both venom and diet composition across taxa. This program calculates a K value for each trait which is an estimate of the amount of phylogenetic signal present in the trait being analyzed [20]. As described in Blomberg et al. [20] K is estimated under a generalized least squares modeling approach and is based on the calculation of mean square error of trait variation relative to its expectation under a Brownian Motion model of evolution. A Brownian Motion model assumes a linear relationship between phylogenetic distance and between taxon divergence in traits and is commonly used in population genetics to describe the evolution of characters undergoing random genetic drift. K can vary from 0 (no correspondence between phylogeny and trait variation) to 1 (evolution by Brownian motion, wherein trait differences are correlated with amount of phylogenetic divergence), to greater than 1 (closely related species have diverged in phenotype less than expected based on the levels of divergence). A statistical test of whether the observed K is greater than random expectation was then carried out by randomly assigning trait values to tree tips and calculating a “random” K value 1000 times and comparing the observed value to the distribution of random K values.
Finally, we used the subroutine PGLS in the R-package Caper [38] to examine associations between venom composition and diet independent of similarity due to phylogeny. This method implements Generalized Least Squares models which account for phylogeny by incorporating estimates of relatedness between taxa into comparisons that determine whether an independent trait (here diet) predicts values of another dependent trait (venom composition) (see ref. 19, Chapter 7 for review). It provides a more general and flexible approach to the widely-used independent contrasts methods pioneered by Felsenstein [39]for assessing correlations between traits independent of phylogenetic divergence. In our analyses we assessed whether a measure of diet (PC 1 score based on the abundance of mammals and reptiles in the diet) was significantly associated with the transformed proportions of specific venom proteins and PC 1 and 2 scores. Significance of the association was assessed using a t-test to evaluate whether the slope was significantly different from zero. The critical p-value was adjusted for the number of comparisons made using the Benjamini and Yekutieli False Discovery Rate adjustment described in [40].
Results
Proteomic Analysis of S. m. streckerii and S. m. milarius Venoms
The venom proteins of S. m. streckeri and S. m. miliarius were initially separated by reverse-phase HPLC (Figs. 2 and 3). The venoms of these two subspecies yielded superimposable elution profiles and the proteins recovered in apparently equivalent chromatographic peaks were almost indistinguishable by SDS-PAGE (inserts of Figs. 2 and 3) and venomic analyses (Table 1). Only a few protein peaks differentiated the venom proteomes of the Western pigmy rattlesnake, S. m. streckeri, and the Carolina pigmy rattlesnake S. m. miliarius, eg. serine proteinase Sms-20, PI-SVMP Sms-31, CTL Sms-24, NGF Smm-14 and D49-PLA2 Smm-41 (Table 1). In addition to these qualitative differences, the venom proteomes of S. m. streckeri and S. m. miliarius also exhibit quantitative departures, most notably in their PIII-SVMP content (Table 2; Fig. 4). Notwithstanding the mentioned qualitative and quantitative differences, the Western and Carolina pigmy rattlesnakes share about 95% of their venom proteomes, which comprise 3 major toxin groups (PIII-SVMP> D49-PLA2> serine proteinase) and 6 (S. m. streckeri) or 4 (S. m. miliarius) minor (<5% of the total venom proteins) peptide/protein classes (disintegrin, C-NP, svNGF, CRISP, C-type lectin-like, PI-SVMP).
Figure 2. The venom proteome of S. m. streckeri.

The proteins from 2 mg of pooled crude venom were fractionated on a C18 column as described in the Experimental section. HPLC fractions were collected manually and analyzed by SDS-PAGE (insert; under non-reduced (upper panel) and reducing (lower paner) conditions), N-terminal sequencing, and molecular mass determination by ESI-MS or SDS-PAGE. Protein bands excised from SDS-polyacrylamide gel were identified by tryptic peptide mass fingerprinting and CID-MS/MS. The results are listed in Table 1.
Figure 3. The venom proteome of S. m. miliarius.

The proteins from 2 mg of crude venom were fractionated on a C18 column as described in as described in the Experimental section. HPLC fractions were collected manually and analyzed as in Fig. 2. The results are listed in Table 1.
Table 1. Assignment of the reverse-phase fractions from the venoms of Sistrurus miliarius streckeri (Sms) and S. m. miliarius (Smm), isolated respectively as in Figs.1 and 2, to protein families by N-terminal Edman sequencing, mass spectrometry, and collision-induced fragmentation by nESI-MS/MS of selected peptide ions from in-gel digested protein bands separated by SDS-PAGE (inserts in Figs. 1 and 2).
| HPLC Fraction | N-terminal sequence | Molecular mass | Peptide Ion | MS/MS-derived sequence | Protein family | ||
| m/z | z | ||||||
| Sms- | Smm- | ||||||
| 1 | 1 | Blocked | 624.4 | 1 | ZXPQR | Unknown | |
| 2–3 | 2–3 | Blocked | 430.1 | 1 | ZNW | SVMPi [∼P01021] | |
| 444.4 | 1 | ZBW | SVMPi [∼P01021] | ||||
| 4 | 4 | EAGEECDCGSPENPCCDAAT | 7639 | Disintegrin [P22827] (1–73) | |||
| GEECDCGSPENPCCDAAT | 7432 | Disintegrin [P22827] (3–72) | |||||
| EECDCGSPENPCCDAATCK | 7375 | Disintegrin [P22827] (4–72) | |||||
| 5 | 5 | Blocked | 569.8 | 2 | ZNWPAPHIPP | BPP | |
| 9 | 9 | GSGCFGLKLDRIGSMSGLGC | 1955.8 | C-NP [B0VXV8] 182–201 | |||
| 14 | Blocked | 16♦ kDa | 556.3 | 2 | NPNPVPTGCR | Nerve growth factor [∼BAG16511] | |
| 682.8 | 2 | AXTMEGNQASWR | |||||
| 15 | 15 | SVDFDSESPRKPEIQ | 24779 | 569.8 | 2 | SVDFDSESPR | CRISP [∼B0VXV6] |
| 533.8 | 2 | IVDLHSLR | |||||
| 16 | 16 | HLIQFETLIMKIAGR | 13997 | 686.8 | 2 | CCFVHDCCYGK | D49-PLA2 [ABY77922] |
| 490.3 | 2 | QICECDR | |||||
| 793.2 | 2 | DNIPTYDDKWR | |||||
| 753.8 | 2 | HLIQFETLIMK | |||||
| 19 | 19 | VIGGDECNINEHRFL | 36▪/38♦ kDa | 553.2 | 2 | FLVALYHSR | Serine proteinase |
| 756.7 | 2 | VIGGDECNENIHR | |||||
| 885.8 | 2 | ILCAGVLEGGIDTCHR | |||||
| 2562.6 | 1 | TFLCGGTLLNEEWVLTAAHCDR | |||||
| HTLIQFETLIMKIAGR | 16▪/♦ kDa | D49-PLA2 [ABY77929] | |||||
| 20 | 20 | VIGGDECNINEHR | 28▪/34♦ kDa | 553.2 | 2 | FLVALYHSR | Serine proteinase |
| 756.7 | 2 | VIGGDECNINEHR | |||||
| 22–23 | 22–23 | (V/I)(I/V)GGDECNINEHR(F/S)L | 28▪/34♦ kDa | 553.2 | 2 | FLVALYHSR | Serine proteinase |
| 756.7 | 2 | VIGGDECNINEHR | |||||
| 749.7 | 2 | VVGGDECNINEHR | |||||
| 763.3 | 2 | IIGGDECNINEHR | |||||
| 885.8 | 2 | ILCAGVLEGGIDTCHR | |||||
| 561.6 | 2 | AAYSSLPATSR | |||||
| 24 | 24 | VVGGDECNINEHRFL | 28▪/34♦ kDa | 749.7 | 2 | VVGGDECNINEHR | Serine proteinase |
| 594.7 | 2 | TVPNEDEQTR | |||||
| 559.6 | 2 | TLCAGILEGGK | |||||
| 24 | DCPSGWSYYEGHCYK* | 26▪/13–14♦ kDa | C-type lectin-like | ||||
| 25 | 25 | V(V/I)GGDECNINEHRFL | 28▪/33♦ kDa | 749.7 | 2 | VVGGDECNINEHR | Serine proteinase |
| 756.7 | 2 | VIGGDECNINEHR | |||||
| 594.7 | 2 | TVPNEDEQTR | |||||
| 26 | 26 | Blocked | 96▪/48♦ kDa | 606.8 | 2 | SAECIDSFQR | PIII-SVMP [B0VXU9] |
| 861.6 | 2 | MYDTVNVITPIYHR | |||||
| 617.9 | 2 | MNIHVALVGLEIWSNR | |||||
| 707.8 | 2 | DDCDMADVCTGR | |||||
| 678.3 | 2 | LYCFPNSPETK | |||||
| 604.8 | 3 | QGAQCAEGLCCDQCR | |||||
| 27 | 27 | VIGGDECNINEHRFL | 28▪/33♦ kDa | Serine proteinase | |||
| 28 | 28 | Blocked | 54▪/56♦ kDa | 777.3 | 2 | VCSNGHCVDVATAY | PIII-SVMP |
| 873.8 | 2 | VAVTMTHEXGHNXGXR | |||||
| 670.8 | 3 | XYEXVNTMNEXYXPXNXR | |||||
| 29 | 29 | ALNPEEQRYVELFIV | 26▪/♦ kDa | 631.2 | 3 | TWVHEXVNTXNVFYR | PI-SVMP |
| 30 | Blocked | 54▪/56♦ kDa | 774.6 | 2 | FALVGLEIWSNGDKI | PIII-SVMP [∼B0VXU6] | |
| 587.7 | 2 | IFPCAPQDVK | |||||
| 48▪/52♦ kDa | 664.3 | 2 | YVEXVXXWHR | PIII-SVMP | |||
| 603.3 | 2 | FTSAGNVC(gh)R | |||||
| 31, 32 | 32 | NLTPEQQAYLDAKKY | 52–54▪/♦ kDa | PIII-SVMP [∼ABG26978] | |||
| 31 | ND | 48▪ kDa | 736.8 | 3 | IYEIVNTMNEIYIPLNIR | PIII-SVMP [∼B0VXU4] | |
| 600.3 | 3 | YVEFVVVLDHGMYTK | |||||
| 875.7 | 2 | LSHQPSTQFSDCSEK | |||||
| 534.6 | 2 | GLCCDQCR | |||||
| 33 | 33 | ALNPEEQRYVELVVV | 48▪/52♦ kDa | 526.6 | 2 | GNYYGYCR | PIII-SVMP |
| 35 | 35 | ND | 39▪/♦ kDa | 657.8 | 2 | YXEXVVVADHR | PIII-SVMP |
| 37–39 | 37–39 | Heterogeneous | 66▪/♦ kDa | 583.1 | 2 | IGNYYGYCR | PIII-SVMP [∼B0VXU5] |
| 677.2 | 2 | YIELVIVADYR | |||||
| 638.1 | 3 | TWVYEIVNTLNEIYR | |||||
| 514.6 | 2 | IPCAPEDVK | |||||
| 614.4 | 3 | MCGVTQNWESYEPIK | |||||
| 2529.1 | 1 | LYCSYNFNGNQIPCVPYYTR | |||||
| 23▪/♦ kDa | 547.8 | 2 | YNSNXNTXR | PI-SVMP | |||
| 672.8 | 2 | YXEXVVVTDHR | |||||
| 631.2 | 3 | TWVHEXVNTXNVFYR | |||||
| 39 | 39 | ND | 48▪ kDa | 615.2 | 2 | XPDSEAHAVYK | PIII-SVMP [∼B0VXU5] |
| 717.1 | 2 | XQGEMYXXEPXK | |||||
| 39 | 39 | AXIYQRYIELVVVAD | 39▪/♦ kDa | 657.8 | 2 | YXEXVVVADHR | PIII-SVMP |
| 912.8 | 2 | VTXSADDTXQAFAEWR | |||||
| 26▪ kDa | 657.8 | 2 | YXEXVVVADHR | PI-SVMP | |||
| 41 | SLLQFNKMIKIMTKK | 14026 | 490.7 | 2 | QICECDR | D49-PLA2 [∼ABY77926] | |
| 581.6 | 2 | TDIYSYSWK | |||||
| 747.8 | 2 | SGVITCGEGTPCEK | |||||
| 770.6 | 2 | AAAVCFGENLPTYK | |||||
| 689.9 | 3 | NAIPSYTSYGCYCGWGGR | |||||
| 789.3 | 2 | CCFVHDCCYEK | |||||
Apparently identical proteins in Sms and Smm venoms are labelled with the same number. In MS/MS-derived sequences, X = Ile or Leu; Z, pyrrolidone carboxylic acid; B, Gln or Lys. Unless other stated, for MS/MS analyses, cysteine residues were carbamidomethylated; Molecular masses of native proteins were determined by electrospray-ionization (±0.02%) or MALDI-TOF (±0.2%) mass spectrometry. Apparent molecular masses were determined by SDS-PAGE of non-reduced (▪) and beta-mercapethanol-reduced (♦) samples. np, non-peptidic material found; ND, not determined.
Table 2. Overview of the relative occurrence of proteins of different families (in percentage of the total HPLC-separated proteins) in the venoms of Sistrurus m. streckeri (Sms) and Sistrurus m. miliarius (Smm).
| Protein Family | % of total venom proteins | |
| Sms | Smm | |
| Disintegrin | 1.4 | 1.7 |
| SVMPi | 2.9 | 2.3 |
| Vasoactive peptide | 1.3 | 0.4 |
| BPP | 0.6 | 0.3 |
| C-NP | 0.8 | <0.1 |
| svNGF | 0.7 | – |
| CRISP | 1.6 | 1.5 |
| D49-PLA2 | 21.3 | 17.9 |
| Serine proteinase | 17.9 | 12.9 |
| C-type lectin-like | 1.8 | – |
| SVMP | 51.0 | 62.4 |
| PI | 1.1 | 0.7 |
| PIII | 49.9 | 61.7 |
Figure 4. Comparison of the overall venom proteomes of S. m. streckeri and S. m. miliarius.
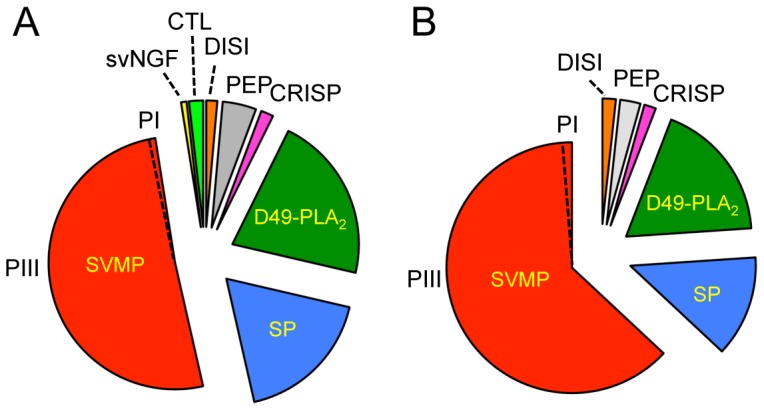
Pie charts display the relative occurrence of proteins from different toxin families in the venoms of S. m. streckeri (panel A) and S. m. miliarius (panel B). svNGF, snake venom nerve growth factor; CTL, C-type lectin-like protein; DISI, disintegrin; PEP, peptides (including tripeptide SVMP inhibitors, bradykinin-potentiating peptides (BPP), and C-natriuretic peptide, C-NP); CRISP, cysteine-rich secretory protein; D49-PLA2, D49-phospholipase A2; SP, serine proteinase; PI- and PIII-SVMP, snake venom Zn2+-metalloproteinase of class I and class III, respectively. The percentages of the different toxin families in S. m. streckeri and S. m. miliarius venoms are listed in Table 2.
Phylogenetic Signal in Venom Composition and Diet
For all subsequent analysis we focused on 7 protein families (disintegrins, L-animo acid oxidase, CRISP, PLA2s, serine proteinases, SVMP, and PEP) each of which had abundances of >1% and were present in at least 6 of 8 taxa (Table 3). To generate composite variables for both venom and diet, we calculated PC 1 scores for both venom and diet and PC 2 for venom only and used these in subsequent analyses. For venom, PC 1 and 2 together account for 81% of the total variation in protein abundance (Table 4). The highest variable loadings for PC 1 are SVMP (0.92), PLA2 (−0.88), PEP (−0.42) and CRISP (0.41) whereas for PC 2 they are LAO (0.88), SP (0.72), CRISP (0.63), PLA2 (−0.45). For diet, PC 1 accounted for 87% of total variation and had a high negative loading for % mammals (−0.95) and positive loading for % lizards (0.91).
Table 3. Summary of proteomics and diet information used for comparative evolutionary analysis of rattlesnake venom composition.
| Protein family (% total venom proteins) | Diet | |||||||||||||
| Taxon | DISI | LAO | CRISP | PLA2 | SP | SVMP | svNGF | CTL | MYO | DC | PEP | Kunitz | % mam | % liz |
| Sms | 1.4 | 0 | 1.6 | 21.3 | 17.8 | 51.1 | 0.7 | 1.8 | 0 | 0 | 4.3 | 0 | 0.20 | 0.20 |
| Smm | 1.8 | 0 | 1.6 | 18.1 | 13.1 | 62.7 | 0 | 0 | 0 | 0 | 2.7 | 0 | 0.17 | 0.50 |
| Smb | 7.5 | 2.0 | 2.8 | 31.8 | 16.8 | 35.3 | 0.1 | 0.1 | 0 | 1.3 | 2.1 | 0.1 | 0.17 | 0.32 |
| Sca | 2.4 | 4.0 | 0.6 | 28.9 | 17.5 | 42.2 | 0.1 | 0.1 | 0.4 | 0.1 | 3.7 | 0 | 0.85 | 0.00 |
| Sct | 4.1 | 1.4 | 1.2 | 30.3 | 19.6 | 38.9 | 0.1 | 0.1 | 0.1 | 0.1 | 4.1 | 0 | 0.73 | 0.14 |
| Sce | 0.8 | 2.4 | 10.1 | 13.1 | 23.3 | 45.6 | 0.1 | 0.1 | 0 | 0.1 | 4.2 | 0.1 | 0.32 | 0.60 |
| Catrox | 6.2 | 8.0 | 4.3 | 7.3 | 19.8 | 49.8 | 0.0 | 1.6 | 0.0 | 0.0 | 3.0 | 0.0 | 0.87 | 0.05 |
| Agc | 1.3 | 0.4 | 3.7 | 31.7 | 14.3 | 32.5 | 0.3 | 2.4 | 0.0 | 0.0 | 0.7 | 0.0 | 0.37 | 0.06 |
Values represent % total venom proteome made up by a given protein as determined by HPLC analyses (see Methods). Abbreviations for names of protein families are as follows: DIS – Disintegrin; LAO – L-amino acid oxidase; CRISP – cysteine rich secretory protein; PLA2– phospholipase A2; SP – serine proteinase; SVMP – snake venom metalloproteinase; svNGF – snake venom nerve growth factor; CTL - C-type lectin-like; MYO – myotoxin; DC – Disintegrin-like/cysteine-rich SVMP C-terminal fragment; PEP - vasoactive peptides; Kunitz - Kunitz-type inhibitor. Diet measures: % mam – proportion of mammals in diet; % liz: proportion of lizards in diet. Taxon names: Sms – Sistrurus m. streckerii; Smm – S. m. milarius; Smb – S. m. barbouri; Sca – S. c. catenatus; Sct – S. c. tergenimus; Sce – S. c. edwardsii; Catrox – Crotalus atrox; Agc – Agkstrodon c. contortrix. Sources for proteomics and diet information are given in the Materials and Methods section.
Table 4. Measures of phylogenetic signal (estimated as K values) for venom and diet variation across the 8 taxa shown in Fig. 1.
| Trait - venom | K-value | P-value |
| DISI | 0.052 | 0.70 |
| LAO | 0.26 | 0.06 |
| CRISP | 0.04 | 0.83 |
| PLA2 | 0.08 | 0.37 |
| SP | 0.07 | 0.50 |
| SVMP | 0.11 | 0.37 |
| PEP | 0.15 | 0.20 |
| PC 1 - venom | 0.09 | 0.37 |
| PC 2 - venom | 0.12 | 0.23 |
| Trait - diet | ||
| % mam | 0.16 | 0.11 |
| % liz | 0.08 | 0.44 |
| PC 1 - diet | 0.11 | 0.22 |
P-value represents the probably that the observed value is greater then the expected value if trait values were randomly assigned to tip taxa. Trait names are as in Table 3 with the exception that PC 1 and 2– venom are the PC scores for composite venom variable and PC 1– diet are PC 1 scores for the composite diet variable. For venom, PC 1 and 2 explained 49% and 31% of total variation respectively, and had the following variable loadings (PC1 - DISI (−0.11); LAO (−0.36); CRISP (0.41); PLA2 (−0.88); SP (−0.12); SVMP (0.92); PEP (−0.42); PC2 - DISI (0.23); LAO (0.87); CRISP (0.64); PLA2 (−0.44); SP (0.72); SVMP (−0.33); PEP (0.12). For diet, PC 1 explained 87% of total variation and had the following loadings (% mammals (−0.95); % lizards (0.91).
Phylogeny-based comparative analysis of evolutionary change in venom compositon as well as diet show no evidence for a significant effect of phylogenetic signal in either set of variables. K values for individual venom components as well as PC 1 and 2 are mostly small (≤0.15) and none are significantly different from random (Table 4). The highest K value was observed for LAO (0.26) suggesting that of all the venom components analyzed it shows the strongest evidence for an assocation with phylogeny. However, this K value is also not signficant albeit marginally so (P = 0.06). Similar to venom, diet variation shows no association with phylogeny: K values for both % mammals and % lizards as well as PC 1 scores are also small (≤0.16) and not significantly different from random expectations. Overall both venom and diet are evolutionarily highly labile traits in these snakes.
Associations between Diet and Venom Variation
The PGLS analyses allow us to look for evidence of correlated evolution between diet and venom composition independent of modest phylogenetic signal. Because % mammals and % lizards were inversely correlated with each other (r = −0.68) we focused on the diet PC 1 scores as overall measures of diet. Analyses based on % mammals and % lizards separately showed similar patterns to those based on diet PC 1 described below (results not shown). We initially found significant (P<0.05) correlations between diet PC 1 and CRISP (r = 0.26), PLA2 (−0.26) and venom PC1 scores (0.31) (Table 5); however, after adjusting the critical P-value for the number of tests performed, only the association between diet and CRISP protein variation remained significant, with snakes with high CRISP abundance having a greater abundance of lizards relative to mammals in their diets. Despite the small simple size, these analyses are consistent with a role for natural selection in molding variation in the abundance of some venom proteins in these snakes through variation in diet.
Table 5. Phylogenetic General Least Squares analyses for associations between diet and individual and composite venom traits for the 8 taxa shown in Fig. 1.
| Trait - venom | Slope | P-value |
| DISI | −0.12 | 0.16 |
| LAO | 0.01 | 0.84 |
| CRISP | 0.26 | 0.003* |
| PLA2 | −0.26 | 0.026 |
| SP | 0.03 | 0.50 |
| SVMP | 0.12 | 0.32 |
| PEP | −0.01 | 0.67 |
| PC 1 - venom | 0.31 | 0.047 |
| PC 2 - venom | 0.18 | 0.12 |
Abbreviations for venom traits are given in Table 3. Diet was measured as composite trait PC 1 based on % mammals and % lizards in diet and has a high negative loading for % mammals and high positive loading for % lizards (see above).
represents a p-value that remained significant after a FDR-based adjustment of critical p-value to 0.017 based on number of comparisons.
Discussion
Proteomic Analysis of Sistrurus Venom Composition
The proteomic characterization of S. m. streckeri and S. m. miliarius completes the venomic overview of genus Sistrurus initiated by Juárez et al [41] and Sanz et al. [23], and provides hints for understanding the toxinological profile and natural history of Sistrurus venoms.
The high abundance of extracellular matrix-disrupting Zn2+-dependent metalloproteinases (PIII-SVMP) [42], [43], cytolytic PLA2 molecules [44], thrombin-like serine proteinases affecting the blood coagulation cascade and platelet aggregation [3], and, to a lesser extent, hypotensive bradykinin-potentiating peptides (BPPs) [45]–[47] in both S. miliarius subspecies investigated suggests that the major effects of these venoms may involve local tissue damage, hemorrhage, coagulation disturbances, and vascular shock. Further, low abundant venom toxins such as disintegrins, C-type lectin-like molecules, and PI-SVMPs, which also interact with components of the human hemostatic system, may synergistically potentiate the activity of PIII-SVMPs and serine proteinases, resulting in increased incidence of bleeding and systemic disseminated coagulopathy (see discussion in Rodrigues et al. [48]).
Notwithstanding qualitative and quantitative differences, the large conservation (95%) in their venom proteomes underscores the close phylogenetic kinship of the two pigmy rattlesnake subspecies, which had a common ancestor as early as 0.49 Myr (Fig.1). In contrast, S. m. streckeri and S. m. miliarius share only a few venom proteins (disintegrin barbourin 4 [P22827], BPP/C-NP precursor 9 [B0VXV8], PLA2 18 [ABY77929], NGF 14 [∼BAG16511], serine proteinase 23, PI-SVMP 29) with S. m. barbouri, indicating that only ∼24% of their venom proteomes has been preserved since their split 0.77 Mbp. These data highlight the evolutionary plasticity of pygmy rattlesnake venoms and also indicate two very different divergence rates across the S. miliarius clade: rapid differenciation of the venom proteomes of Smb and the common ancestor of Sms and Smm (∼9% divergence per 100,000 years) and a slow evolutionary rate (∼1% divergence per 100,000 years) for the venom proteomes of S. m. streckeri and S. m. miliarius. The high degree of differentiation among recently evolved subspecies points to a strong role for adaptive diversification via natural selection as a cause of this distinctiveness.
Phylogentic Comparative Analyses of Venom Composition
Evidence for a strong association between venom composition and phylogenetic relationships of species comes from observations that broad-scale patterns of venom composition mirror deep phylogenetic relationships among venomous snakes (for review see [11]). For example, snakes in the family Elapidae tend to have smaller toxins such as three-finger toxins and PLA2s, whereas species in the family Viperidae have more high molecular weight toxins such as SVMPs. However, there are a number of examples where in more closely related, and well-defined lineages, patterns of venom composition do not closely reflect phylogenetic affinities. Specifically, rattlesnakes (Crotalus and Sistrurus), show variation in the presence of Type I (high levels of metalloprotease and low toxicity) versus Type II (low metalloprotease, high toxicity) venoms that shows no strong association with phylogeny [11].
Our analysis of the Sisturus venoms reported here is an example of the second pattern but with several differences. First, we compare venom composition among snakes that are exceptionally closely-related to each other phylogenetically, and probably represent a clade in the earliest stages of an adaptive radiation [22]. One implication is that, given the close relationships among the taxa analyzed, divergence effects should be especially strong and yet we observe little evidence of phylogenetic constraint operating on venom evolution, suggesting that phylogenetic effects are weak at best.
Second, our analyses are the first to apply phylogeny-based comparative methods to venom data generated using proteomics methods. These methods allow the characterization of venom composition to an unprecedented level of detail and as a result we are able to assess whether there is evidence for phylogenetic signal in the abundance of individual venom proteins across all taxa. As a result we are able to identify a large number of shifts in the composition of individual proteins that are statistically greater than expectations based on phylogenetic relationships among taxa. We found no evidence for a significant effect of signal on phylogeny-wide variation in any protein. In fact, K values for indivdual proteins are substantially lower than those observed for a wide range of traits in other species. Specifically, Blomberg et al [20] calculated K values for 119 traits that were associated with 34 different phylogenies from between six to 254 species. In contrast to our results, 92% of all traits in their analysis showed evidence for significant signal and mean K values for broad clases of traits (body size (mean K = 0.83), other morphological traits (0.70), life history traits (0.63), physiology (0.53), and behavior (0.35) were all greater than values observed for individual venom proteins and PC 1 and 2 scores). The small number of species in our analysis could have resulted in low statistical power in our ability to detect a significant K value [20] but the low observed K values demonstrate that even if they were significant the effects of phylogenetic divergence would be low. Overall, our result argues that the abundance of individual venom proteins are exceptionally evolutionarily labile traits and that phylogenetic divergence plays an extremely limited role in shifts in venom composition in terms of the abundance of specific types of proteins. This result leaves open the possiblity that another evolutionary force, possibly natural selection in relation to diet, plays a role in molding the venom composition of these snakes.
Our analysis makes a number of important assumptions. First, we use a single estimate of venom composition for each taxon based on a pooled sample to represent the venom profile of a taxon. In particular, samples from several taxa (e.g. S. m. streckeri) come from a single geographic location. This is of concern because geographic variation in venom composition certainly occurs in Sistrurus (for example, see 25). What is different between the proteomic data used in this study and other work in these snakes and others is that in general the venom variation is usually assessed at much more fine-scale (e.g. individual protein bands on 1-D PAGE gels) than was done here where we pooled venom proteins into broad categories of major protein families.
To assess whether there is significant geographic variation in venom composition characterized in this way, we reanalyzed proteomics-based venom profiles generated for individual snakes from three distinct geographic regions that span the western and central range of this species (Illinois, Ohio, and Ontario) as reported by Gibbs et al. [49]. For each snake, we generated values for % total venom made up by 5 major types of venom proteins (disintegrins, metalloprotenases, PLA2s, serine proteinases, and CRISP – for technical issues these were the only proteins that we were able to analyze) and compared proportions among individuals in each of the populations. The results show that only 2 of 15 population-by-protein comparisons were significant (p≤0.05) based on Mann-Whitney U tests. Our conclusion is that there is some possibility that geographic variation could be underestimated in our data but that this effect is limited, at least based on this analysis of data from this taxa.
We also assume that the relative percent composition made up by each venom protein has a genetic basis. Previous work has shown that venom composition based on HPLC profiles varies among individuals and that some of this variation is caused by differences in gene expression among individuals [26], [49]. We assume that the use of a pooled sample accounts for individual variation that may be present but the extent to which this is true is unknown. In terms of the genetic basis of protein variation, at least for one major protein family studied here (PLA2), Gibbs and Rossiter [50] showed a significant link between genomic and proteomic variation, supporting the assumption.
Finally an additional concern is that our use of proportions to measure the relative abundance of different proteins introduces a dependency among the values for individual proteins across taxa. Our transformation of the raw data using an arcsin square root transformation reduced this dependency but to an unknown level. This issue has been discussed for proportional data derived from HPLC analyses in chemical ecology and the use of a centered log ratio transformation has been proposed [51]. We chose not to use this transformation because of the difficulty of interpreting the meaning of parametric-based composite variables based on Principle Component analyses derived from centered log ratio transformed data. However, we suggest it would be fruitful to explore the use of this and possibly other transformations that minimize the covariation between indivdual values in future analyses.
Causes of Venom Variation
Our results clearly demonstrate the both venom composition and diet are evolutionarily variable traits in these snakes which leaves open the possibility that adaptation by natural selection acting through diet variation could play a key role in molding venom variation. In support of a role for diet-mediated selection we found a strong positive correlation between CRISP abundance and snake diets that were high in lizards and low in mammals and weaker correlations between PLA2 abundance and a measure of overall venom composition in the form of PC 1 scores. These results focus attention on the CRISP venom protein as possibly playing a key role in mediating the effectiveness of rattlesnake venom in killing and/or digesting mammals and lizards and are consistent with recent molecular evolutionary analyses that found strong evidence for positive selection on the coding regions of CRISP venom genes in snakes [52]. Future work involving prey-specific toxicity tests which evaluated the effect of this protein and possibly others on mammals and lizards as described in [24] would be valuable in evaluating whether selection is a viable hypothesis to explain interspecific variation in these proteins.
Nevertheless, the fact that our independant contrast analyses failed to show correlations between diet and other major venom proteins which are major components of rattlesnake venom (e.g. snake venom metaloproteinases) is sobering. It is unclear whether this is due to shortcomings of our analyses in terms of the accuracy of our estimates of diet and venom composition of specific taxa, limited simple size or the primacy of other traits such as snake body size over venom in determining diet (see 32). A broad-based approach to developing evolutionary explanations for variation in major snake venom proteins should be an important focus of future studies of venom evolution in these rattlesnakes. However, our results make it clear that phylogenetic relatedness among species accounts for little of the observed variation and that causal explanations must involve other evolutionary and ecological mechanisms.
Concluding Remarks
There has been a recent explosion of proteomic-based data on venom composition and this promises to continue in the future as the technological approaches become more refined and comprehensive genomic and/or transcriptomic databases become available [53]. At the same time, phylogenetic analysis of the relationships of the snakes producing these venoms has also increased dramatically [54], [55]. We feel that a joint analysis of these data sets using phylogenetic comparative methods like those described here offers an unprecedented opportunity to study venom evolution at a variety of time-scales to better understand the evolutionary forces that have shaped this variation in venomous snakes and will complement the handful of other studies that adopted the same general approach to studying the evolutionary basis for interspecific variation in venom composition [14], [56].
Supporting Information
Reverse-phase separation of Agkistrodon contortrix contortrix venom. Chromatographic fractions were characterized by SDS-PAGE and assigned to protein families by de novo sequencing of in-gel trypsin digested protein bands (Table 1). Relative abundances of each protein family were computed from the sum of the relative chromatographic peak areas. SVMP, snake venom metalloproteinase; SP, serine proteinase; PEP, peptides; LAO, L-amino acid oxidase; DIS, disintegrin; CTL, C-type lectin-like; UNK, unknown; CRISP, cysteine-rich secretory protein; PLA2, phospholipase A2 (source: J. Calvete et al. unpublished ms.).
(TIF)
De novo assignment of RP-HPLC isolated fractions of Agkistrodon contortrix contortrix venom to protein families by MALDI-TOF-TOF or nESI-MS-MS (confidence ≥99%) of selected peptide ions from in-gel trypsin-digested protein bands. Cysteine residues are carbamidomethylated; X: Leu/Ile; B: Lys/Gln. Confidence values were calculated by the Paragon algorithm of ProteinPilot® (source: J. Calvete et al. unpublished ms.).
(DOCX)
Acknowledgments
We thank James Chiucchi, Jeff Ettling (St. Louis Zoo), and Kevin Messenger for help with collecting venom samples, Ron DeBry, Emilia Martins, Darin Rokyta, John Wenzel for discussion, Trevor Price for advice and suggesting the use of Picante and Caper R-packages for analyses, and Anita Malhotra, Wolfgang Wuster and two anonymous reviewers for helpful comments.
Funding Statement
This work was supported by funds from the Ohio State University and grants BFU2010-17373 (from the Ministerio de Economía y Competitividad, Madrid, Spain), Programa de Cooperacion Interuniversitaria e Investigacion Cientifica between CSIC and PROMETEO/2010/005 from the Generalitat Valenciana (Valencia, Spain). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Meier J, Stocker KF (1995) Biology and distribution of venomous snakes of medical importance and the composition of snake venoms. In Handbook of Clinical Toxicology of Animal Venoms and Poisons (Meier J, White J, eds), CRC Press Inc, Boca Raton, Florida, USA, p.367–412.
- 2.Ménez A (ed) (2002) Perspectives in Molecular Toxinology. J Wiley and Sons, UK.
- 3. Markland FS (1998) Snake venoms and the hemostatic system. Toxicon 36: 1749–1800. [DOI] [PubMed] [Google Scholar]
- 4.Calvete JJ (2010) Snake venomics, antivenomics, and venom phenotyping: The Ménage à Trois of proteomic tools aimed at understanding the biodiversity of venoms. In Toxins and Hemostasis: from Bench to Bedside (Kini RM, Markland F, McLane MA, Morita T, eds.), Springer, Dordrecht (The Netherlands), p.45–72.
- 5. Fry BG, Wüster W (2004) Assembling an arsenal: origin and evolution of the snake venom proteome inferred from phylogenetic analysis of toxin sequences. Mol. Biol. Evol. 21: 870–83. [DOI] [PubMed] [Google Scholar]
- 6. Fry BG (2005) From genome to ‘venome’: Molecular origin and evolution of the snake venom proteome inferred from phylogenetic analysis of toxin sequences and related body proteins. Genome Res. 15: 403–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Serrano SM, Shannon JD, Wang D, Camargo AC, Fox JW (2005) A multifaceted analysis of viperid snake venoms by two-dimensional gel electrophoresis: an approach to understanding venom proteomics. Proteomics 5: 501–10. [DOI] [PubMed] [Google Scholar]
- 8. Calvete JJ, Juárez P, Sanz L (2007) Snake venomics. Strategy and applications. J. Mass. Spectrom. 42: 1405–14. [DOI] [PubMed] [Google Scholar]
- 9.Mackessy SP, ed (2010) Handbook of Venoms and Toxins of Reptiles. CRC Press, Taylor & Francis Group, Boca Raton, FL.
- 10. Chippaux J-P, Williams V, White J (1991) Snake venom variability: methods of study, results and interpretation. Toxicon 29: 1279–303. [DOI] [PubMed] [Google Scholar]
- 11.Mackessy SP (2010)The field of reptile toxinology: snakes, lizards and their venoms. In Handbook of Venoms and Toxins of Reptiles (Mackessy SP, ed). CRC Press, Taylor & Francis Group, Boca Raton, FL, P.3–23.
- 12. Daltry JC, Wüster W, Thorpe RS (1996) Diet and snake venom evolution. Nature 379: 537–40. [DOI] [PubMed] [Google Scholar]
- 13. Wüster W, Daltry JC, Thorpe RS (1999) Can diet explain intraspecific venom variation? Reply to Sasa. Toxicon 37: 253–8. [DOI] [PubMed] [Google Scholar]
- 14. Barlow A, Pook CE, Harrison RA, Wüster W (2009) Co-evolution of diet and prey-specific venom activity supports the role of selection in snake venom evolution. Proceedings of the Royal Society B 276: 2443–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Williams V, White J, Schwaner TD, Sparrow A (1998) Variation in venom proteins from isolated populations of tiger snakes (Notechis ater niger, N. scutatus) in South Australia. Toxicon 26: 1067–75. [DOI] [PubMed] [Google Scholar]
- 16. Sasa M (1999) Diet and snake venom evolution: can local selection alone explain intraspecific vemon variation? Toxicon 32: 249–52. [DOI] [PubMed] [Google Scholar]
- 17. Mebs D. Toxicity in animals (2001) Trends in evolution? Toxicon 39: 87–96. [DOI] [PubMed] [Google Scholar]
- 18.Mackessy SP (2008) Venom composition in rattlesnakes: trends and biological significance. In The Biology of Rattlesnakes (Hayes WK, Beaman KR, Cardwell MD, Bush SP (eds). Loma Linda University Press, Loma Linda, California, p.495–510.
- 19.Nunn CL (2011) The comparative approach in evolutionary anthropology and Biology. University of Chicago Press, Chicago.
- 20. Blomberg SP, Garland T Jr, Ives AR (2003) Testing for phylogenetic signal in comparative data: behavioral traits are more laible. Evolution 57: 717–745. [DOI] [PubMed] [Google Scholar]
- 21.Campbell JA, Lamar W (2004) The Venomous Reptiles of the Western Hemisphere, Cornell University Press, Ithaca, New York.
- 22. Kubatko LS, Gibbs HL, Bloomquist EW (2011) Inferring species-level phylogenies and taxonomic distinctiveness using multilocus data in Sistrurus rattlesnakes. Syst. Biol. 60: 393–409. [DOI] [PubMed] [Google Scholar]
- 23. Sanz L, Gibbs HL, Mackessy SP, Calvete JJ (2006) Venom proteomes of closely related Sistrurus rattlesnakes with divergent diets. J. Proteome Res. 5: 2098–112. [DOI] [PubMed] [Google Scholar]
- 24. Gibbs HL, Mackessy SP (2006) Functional basis of a molecular adaptation: Prey-specic toxic effects of venom from Sistrurus rattlesnakes. Toxicon 53: 672–9. [DOI] [PubMed] [Google Scholar]
- 25. Calvete JJ, Fasoli E, Sanz L, Boschetti E, Righetti PG (2009) Exploring the venom proteome of the western diamondback rattlesnake, Crotalus atrox, via snake venomics and combinatorial peptide ligand library approaches.J. Proteome Res. 8: 3055–67. [DOI] [PubMed] [Google Scholar]
- 26. Gibbs HL, Chiucchi JE (2011) Deconstructing a complex molecular phenotype: population- level variation in individual venom proteins in eastern massasauga rattlesnakes (Sistrurus c. catenatus). Journal of Molecular Evolution 72: 383–397. [DOI] [PubMed] [Google Scholar]
- 27. Gibbs HL, Sanz L, Chiucchi JE, Farrell TM, Calvete JJ (2011) Proteomic analysis of ontogenetic and diet-related changes in venom composition of juvenile and adult Dusky Pigmy rattlesnakes (Sistrurus miliarius barbouri). J Proteomics 74: 2169–79. [DOI] [PubMed] [Google Scholar]
- 28. Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipmann DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25: 3389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Murphy RW, Fu J, Lathrop A, Feltham JV, Kovak V (2002) Phylogeny of the rattlesnakes (Crotalus and Sistrurus) inferred from sequences of five mitochondrial DNA genes. In Biology of the Vipers (Schuett GW, Hoggren M, Douglas ME, Greene HW, eds), Eagle Mountain Publishing, LC, Eagle Mountain, Utah, p.69–92.
- 30. Bryson RW Jr, Murphy RW, Graham MR, Lathrop A, Lazcano-Villareal D (2011) Ephemeral Pleistocene woodlands connect the dots for highland rattlesnakes of the Crotalus intermedius group. J. Biogeograph. 38: 2299–310. [Google Scholar]
- 31.R Development Core Team (2005). R: A language and environment for statistical computing, reference index version 2.2.1. R Foundation for Statistical Computing, Vienna, Austria.
- 32. Holycross AT, Mackessy SP (2002) Variation in the diet of Sistrurus catenatus (massasauga) with emphasis on Sistrurus catenatus edwardsii (desert massasauga). Journal of Herpetology 36: 454–464. [Google Scholar]
- 33. Beavers RA (1976) Food habits of the western diamond rattlesnake, Crotalus atrox, in Texas (Viperidae) Southwest. Nat. 20: 503–15. [Google Scholar]
- 34.Gloyd HK, Conant R (1990) Snakes of the Agkistrodon Complex: A Monographic Review. Society for the Study of Amphibians and Reptiles. 614 pp.
- 35.Holder TL (1988) Movement and life history aspects of the pigmy rattlesnake in southwest Missouri. Unpublished M.S. thesis. Southwest Missouri State University.
- 36. Hamilton WJ, Pollack JA (1955) The food of some crotalid snakes from Fort Benning, Georgia. Nat. Hist. Misc. 140: 1–4. [Google Scholar]
- 37. Kembel SW, Cowan PD, Helmus MR, Cornwell WK, Morlon H, Ackerly DD, Blomberg SP, Webb CO (2010) Picante: R tools for integrating phylogenies and ecology. Bioinformatics 26: 1463–1464. [DOI] [PubMed] [Google Scholar]
- 38.Orme D (2012) The Caper package: comparative analysis of phylogenetics and evolution in R. Available: http://cran.r-project.org/web/packages/caper
- 39. Felsenstein J (1995) Phylogenies and the comparative method. Am. Nat. 125: 1–15. [Google Scholar]
- 40. Narum SR (2006) Beyond Bonferroni: Less conservative analyses for conservation genetics. Conservation Genetics 7: 783–787. [Google Scholar]
- 41. Juárez P, Sanz L, Calvete JJ (2004) Snake venomics: characterization of protein families in Sistrurus barbouri venom by cysteine mapping, N-terminal sequencing, and tandem mass spectrometry analysis. Proteomics 4: 327–38. [DOI] [PubMed] [Google Scholar]
- 42. Lomonte B, Angulo Y, Sasa M, Gutiérrez JM (2009) The phospholipase A2 homologues of snake venoms: biological activities and their possible adaptive roles. Protein Pept Lett. 16: 860–76. [DOI] [PubMed] [Google Scholar]
- 43. Escalante T, Rucavado A, Fox JW, Gutiérrez JM (2011) Key events in microvascular damage induced by snake venom hemorrhagic metalloproteinases. J. Proteomics 74: 1781–94. [DOI] [PubMed] [Google Scholar]
- 44. Gutiérrez JM, Lomonte B (1995) Phospholipase A2 myotoxins from Bothrops snake venoms. Toxicon 33: 1405–24. [DOI] [PubMed] [Google Scholar]
- 45. Ferreira SH, Bartelt DC, Greene LJ (1970) Isolation of bradykinin-potentiating peptides from Bothrops jararaca venom. Biochemistry 9: 2583–93. [DOI] [PubMed] [Google Scholar]
- 46. Greene L-J, Camargo AC, Krieger EM, Stewart JM, Ferreira SH (1972) Inhibition of the conversion of angiotensin I to II and potentiation of bradykinin by small peptides present in Bothrops jararaca venom. Circ. Res. 31 (suppl 2)62–71. [PubMed] [Google Scholar]
- 47. Luft FC (2008) The Bothrops legacy: Vasoactive peptides from Brazil. Renin Rep. 10: 57–64. [DOI] [PubMed] [Google Scholar]
- 48. Rodrigues RS, Boldrini-França J, Fonseca FP, de la Torre P, Henrique-Silva F, Sanz L, Calvete JJ, Rodrigues VM (2012) Combined snake venomics and venom gland transcriptomic analysis of Bothropoides pauloensis. J. Proteomics 75: 2707–20. [DOI] [PubMed] [Google Scholar]
- 49. Gibbs HL, Sanz L, Calvete JJ (2009) Snake population venomics: proteomics-based analyses of individual variation reveals significant gene regulation effects on venom protein expression in Sistrurus rattlesnakes. J. Mol. Evol. 68: 113–25. [DOI] [PubMed] [Google Scholar]
- 50. Gibbs HL, Rossiter W (2008) Rapid evolution by positive selection and gene gain and loss: PLA2 venom genes in closely related Sistrurus rattlesnakes with divergent diets. J. Mol. Evol. 66: 151–66. [DOI] [PubMed] [Google Scholar]
- 51. Ranganathan Y, Borges RM (2011) To transform or not to transform: that is the dilemma in the statistical analysis of plant volatiles. Plant Signaling & Behaviour 6: 113–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sunagar K, Johnson WE, O’Brien SJ, Vasconcelos V, Antunes A (2012) Evolution of CRISPs associated with toxicoferan-reptilian venom and mammalian reproduction. Molecular Biology and Evolution 29: 1807–22. [DOI] [PubMed] [Google Scholar]
- 53. Calvete JJ (2011) Proteomic tools against the neglected pathology of snake bite envenoming. Expert Rev. Proteomics 8: 739–58. [DOI] [PubMed] [Google Scholar]
- 54. Vidal N, Hedges SB (2002) Higher-level relationships of snakes inferred from four nuclear and mitochondrial genes. C. R. Biologies 325: 977–85. [DOI] [PubMed] [Google Scholar]
- 55. Pyron RA, Burbrink FT, Colli GR, de Oca AN, Vitt LJ, Kuczynski CA, Wiens JJ (2011) The phylogeny of advanced snakes (Colubroidea), with discovery of a new subfamily and comparison of support methods for likelihood trees. Molecular Phylogenetics and Evolution 58: 329–42. [DOI] [PubMed] [Google Scholar]
- 56.Casewell NR, Harrison RA, Wüster W, Wagstaff SC (2009) Comparative venom gland transcriptome surveys of the saw-scaled vipers (Viperidae: Echis) reveal substantial intra-family gene diversity and novel venom transcripts. BMC Genomics 10, 564. [DOI] [PMC free article] [PubMed]
- 57.Douglas ME, Douglas MR, Schuett Gw, Porra LW (2006) Evolution of rattlesnakes (Viperidae: Crotalus) in the warm deserts of western North America shaped by Neogen vicariance and Quaternary climate change. Molecular Ecology 15, 3353–3374. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Reverse-phase separation of Agkistrodon contortrix contortrix venom. Chromatographic fractions were characterized by SDS-PAGE and assigned to protein families by de novo sequencing of in-gel trypsin digested protein bands (Table 1). Relative abundances of each protein family were computed from the sum of the relative chromatographic peak areas. SVMP, snake venom metalloproteinase; SP, serine proteinase; PEP, peptides; LAO, L-amino acid oxidase; DIS, disintegrin; CTL, C-type lectin-like; UNK, unknown; CRISP, cysteine-rich secretory protein; PLA2, phospholipase A2 (source: J. Calvete et al. unpublished ms.).
(TIF)
De novo assignment of RP-HPLC isolated fractions of Agkistrodon contortrix contortrix venom to protein families by MALDI-TOF-TOF or nESI-MS-MS (confidence ≥99%) of selected peptide ions from in-gel trypsin-digested protein bands. Cysteine residues are carbamidomethylated; X: Leu/Ile; B: Lys/Gln. Confidence values were calculated by the Paragon algorithm of ProteinPilot® (source: J. Calvete et al. unpublished ms.).
(DOCX)