

Abstract
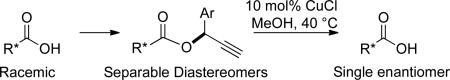
A method for the optical resolution of carboxylic acids is described. Condensation of racemic carboxylic acids with chiral terminal propargyl alcohols gave separable diastereomeric esters. Chromatographic separation followed by heating the individual diastereomers in methanol with catalytic copper (I) halide regenerated the carboxylic acids in good yields and in enantiomeric ratios of ≥94%. This method is particularly useful for the resolution of carboxylic acids that are incompatible with conventional ester hydrolysis.
Enantiomerically pure carboxylic acids are an important class of biologically active compounds. Even though great advances have been made in their asymmetric synthesis, in some cases resolution of the racemate may still be desired. The most common method of carboxylic acid resolution is still through crystallization of diastereomeric salts, although numerous other techniques exist.1 We recently wished to resolve an isoquinoline-containing carboxylic acid intermediate used in the preparation of a series of potent and selective κ opioid receptor binders.2 To accomplish this, we first attempted the low-tech approach of preparing a chiral ester from (R)-1-phenylethyl alcohol, separating the diastereomeric esters, and then regenerating the acid by basic hydrolysis (Scheme 1). Thus, the carboxylic acid shown could be converted into a pair of separable 2-phenylethyl esters, but upon saponification under basic conditions, an epimerized acid was obtained as the major product. In this Note, we present a simple means of resolving carboxylic acids that features the formation of a chiral propargylic ester from the acid substrate, separation by ordinary chromatography, and finally removal of the chiral ester under mild, neutral conditions.
Scheme 1.

An alternative to hydrolysis via acyl–oxygen bond cleavage is ester removal via alkyl–oxygen cleavage. However, such processes usually require strongly acidic or nucleophilic conditions except in cases where the alkyl carbocation that is generated is stabilized. One example of the latter is the copper catalyzed amination of propargylic esters, in which the carboxylic acid is generally viewed as a byproduct.3 In this case, the propargyl cation is stabilized by formation of a copper complex that reacts with the amine to form a propargyl amine (Scheme 2). These reactions conditions were neutral and therefore attractive for our purposes, but the literature procedure still required a stoichiometric amount of a weakly basic amine. Wishing to avoid potential complications arising from the presence of an added amine, we wondered if substituting an alcohol as the nucleophilic component would lead to an analogous reaction cleaving propargylic esters. Propargylic substitution of esters with alcohols is known, albeit under strongly Lewis acidic conditions (TiCl4).4
Scheme 2.

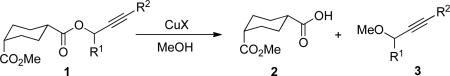
We first sought to identify conditions whereby the propargyl ester 1, containing a potentially epimerizable axial ester bystander, could be converted to acid 2 in methanol (Table 1). Copper (I) chloride was found to be better than other copper (I) halides. The reaction time was significantly shorter for ester 1b than 1a. This is probably due to better stabilization of the propargyl carbocation by the electron-rich aromatic ring. The necessity of cation stabilization by an aromatic group was evident by the poor yield obtained in the case of ester 1c, even after a long reaction time. In the case of propargyl ester 1d, with an internal alkyne, no reaction was observed under analogous conditions. Neither 1a nor 1b reacted when heated with 3 equiv of acetic acid in methanol at 40 °C, suggesting the need for Cu(I) promotion.
Table 1.
Optimization of Cu(I)-promoted propargylic substitution
| ||||
|---|---|---|---|---|
| entry | propargyl ester | CuX (10 mol%) | conditions | yield of 2 (%) |
| 1 | 1a (R1=Ph, R2=H) | CuCl | rt, 16 h | 74 |
| 2 | CuCl | 40 °C, 6 h | 86 | |
| 3 | CuBr | 40 °C, 12 h | 64 | |
| 4 | CuI | 40 °C, 12 h | <10 | |
| 5a | [CuPPh3]4Cl4 | 40 °C, 6 h | 85 | |
| 6b | - | 40 °C, 6 h | 0 | |
| 7 | 1b (R1=o-OMePh, R2=H) | CuCl | 40 °C, 2 h | 82 |
| 8 | 1c (R1=Me, R2=H) | CuCl | 40 °C, 12 h | 23 |
| 9 | 1d (R1=Ph, R2=nBu) | CuCl | 40 °C, 12 h | 0 |
2.5 mol % of catalyst was used.
AcOH (3 equiv) was added.
The requirement for a terminal alkyne is consistent with a mechanism similar to that shown in Scheme 2. The reaction took place at room temperature, however with longer time and reduced yield relative to those carried out under reflux (cf. entries 1 and 2). The major side product was propargyl ether 3; in some cases, uncharacterized diyne-containing byproducts were also observed.5 The proportion of diyne increased with decreasing yield of both 2 and 3 in the presence of air or oxygen. Thus, the reaction is air sensitive and proper degassing of the reaction with Ar was required. Significantly, no epimerization of the methyl ester to the more stable trans-1,4-cyclohexanedicarboxylic acid ester occurred under these reaction conditions.
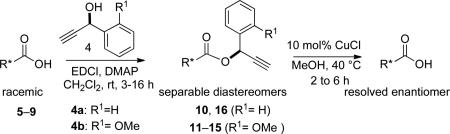
Having determined suitable conditions for ester removal, we examined the use of the propargylic alcohols as resolution agents for a series of racemic carboxylic acids (5–9). The acids were esterified with chiral propargyl alcohols 4a and 4b (Table 2).6 Propargyl alcohol 4a is commercially available and propargyl alcohol 4b was obtained by reported methods.7 The diastereomeric esters were separable using column chromatography although in most cases more than one chromatographic operation was required to ensure good yield and dr values of >95:5. The chromatographic resolving ability may depend on the specific alcohol used. For example, the Rf values of diastereomers 10a and 10b were 0.45 and 0.41 (1:3 ethyl acetate:hexane) compared to the Rf values of 0.48 and 0.42 (1:4 ethyl acetate:hexane) for diastereomers 11a and 11b.8 Some carboxylic acids did not easily afford separable diastereomers by this method, including ibuprofen and N-Boc-valine (not shown). No hydrolysis of the benzyl ester in compound 7 (entry 4) or carbamate in compound 5 (entry 1 and 2) was observed at the propargylic removal stage. Overall, highly conserved er values together with absence of any detectable diastereomeric carboxylic acids (entries 5–7) exemplify the mildness of this method. The absolute configuration of the carboxylic acids were determined by comparing reported optical rotation values of either the carboxylic acids or their derivatives, except for the acid 9 where X-ray data9 was used to determined the absolute stereochemistry.
Table 2.
Resolution of carboxylic acids
| ||||||
|---|---|---|---|---|---|---|
| entry | racemic acid | alcohol | estera | resolved enantiomers | % yield (% overall yield)b | erc,d |
| 1 |
|
4a | 10a | (S)-5 | 82 (33) | 98:2 |
| 4a | 10b | (R)-5 | 84 (33) | 98:2 | ||
| 2 | 4b | 11a | (S)-5 | 81 (36) | 96:4 | |
| 4b | 11b | (R)-5 | 82 (34) | 97:3 | ||
| 3 |
|
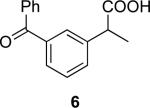
4a | 12a | (R)-6 | 84 (37) | 93:7 |
| 4a | 12b | (S)-6 | 84 (36) | 94:6 | ||
| 4e |
|
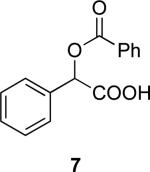
4a | 13a | (S)-7 | 82 (34) | 96:4 |
| 4a | 13b | (R)-7 | 82 (35) | 94:6 | ||
| 5f |
|
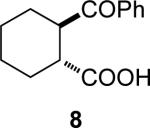
4a | 14a | (1R, 2R)-8 | 85 (35) | 94:6 |
| 4a | 14b | (1S, 2S)-8 | 82 (32) | 94:6 | ||
| 6 |
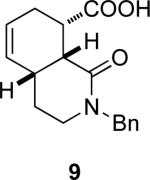
|
4a | 15a | (4aS, 8R, 8aS)-9 | 84 (35) | 96:4 |
| 4a | 15b | (4aR, 8S, 8aR)-9 | 81 (32) | 98:2 | ||
| 7 | 4b | 16a | (4aS, 8R, 8aS)-9 | 79 (38) | 98:2 | |
| 4b | 16b | (4aR, 8S, 8aR)-9 | 82 (39) | 98:2 | ||
Measured ratios for the ester diastereomers were >95:5 by 1H NMR.
“Yield” referse to the ester removal step along; whereas “overall yield” refers to the isolated yield of acid from the starting racemate.
Measured er of resolved acid by chiral analytical HPLC.
Measured er values for 4a and 4b were 99:1 and 98:2, respectively.
Ester removal performed at 50 °C.
20% CH2Cl2 in MeOH used as solvent.
In summary, we have developed a straightforward method for the resolution of carboxylic acids via diastereomeric propargylic esters under mild conditions. The method is particularly useful in chemical resolution of compounds having epimerizable centers or hydrolytically labile functional groups. Although it is not expected that the present method will supplant large-scale classical resolution methods using diastereoselective crystallization techniques, we have found it convenient for the expedient laboratory-scale separation of enantiomeric acids as it uses standard chromatography and does not require the identification of appropriate rescrystallization conditions.
Experimental Section
General Procedure for Esterification and Separation. (S)-1-tert-Butyl-2-((R)-1-(2-methoxyphenyl)prop-2-yn-1-yl)pyrrolidine-1,2-dicarboxylate (11a) and (R)-1-tert-butyl-2-((R)-1-(2-methoxyphenyl)prop-2-yn-1-yl)pyrrolidine-1,2-dicarboxylate (11b)
To a solution of 5 (215 mg, 1.0 mmol), EDCl (286 mg, 1.50 mmol) and DMAP (6 mg, 0.05 mmol) in 5 mL CH2Cl2 was added propargyl alcohol 4b (162 mg, 1.0 mmol) at room temperature. The reaction was stirred for 3 h at room temperature. The reaction mixture was diluted with 20 mL CH2Cl2, washed with water (5 mL), and the organic layer dried over anhydrous MgSO4. Evaporation of the solvent and chromatography (10% EtOAc in hexane) gave 11a (158 mg, 44%) as a white solid and 11b (152 mg, 42%) as colorless oil. 11a: (RF 0.32, 1:4 EtOAc:hexanes); mp 52–54; [α]D -80.8 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3; mixture of rotamers) δ 7.65 (1H, d, J = 7.5 Hz), 7.36-28 (1H, m), 7.02-6.95 (1H, m), 6.88 (1H, d, J = 8.2 Hz), 6.79 (1H, d, J = 2.0 Hz), 4.36 (0.3H, d, J = 5.9 Hz), 4.27 (0.7H, dd, J = 3.6 Hz, 8.7 Hz), 3.83 (0.9H, s), 3.82 (2.1H, s), 3.54-3.30 (2H, m), 2.58 (0.7H, d, J = 2.2 Hz), 2.55 (0.3H, d, J = 2.0 Hz), 2.28-1.72 (4H, m), 1.45 (2.7H, s), 1.37 (6.3H, s); 13C NMR (100 MHz, CDCl3) δ 171.6, 171.3, 156.7, 156.5, 154.2, 153.8, 130.5, 130.3, 128.9, 128.4, 124.4, 124.2, 120.6, 120.4, 110.7, 110.6, 80.1, 80.0, 79.9, 79.5, 74.9, 74.6, 60.6, 59.0, 58.7, 55.5, 55.4, 46.4, 46.2, 30.7, 29.5, 28.4, 28.1, 24.1, 23.3; IR (thin film) 3285, 1748, 1694 cm-1; HRMS (ESI) m/z calcd for C20H26NO5 ([M+H]+), 360.1811, found 360.1821. 11b: (RF = 0.27, 1:4 EtOAc:hexanes); [α]D -19.0 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3; mixture of rotamers) δ 7.67 (0.75H, dd, J = 1.5 Hz, 7.6 Hz), 7.62 (0.25H, d, J = 6.7 Hz), 7.38-7.27 (1H, m), 7.03-6.83 (2H, m), 6.81 (1H, d, J = 2.1 Hz), 4.38 (0.25H, dd, J = 2.8 Hz, 8.8 Hz), 4.25 (0.75H, dd, J = 3.3 Hz, 8.7 Hz), 3.82 (3H, s), 3.65-3.28 (2H, m), 2.60 (0.75H, d, J = 2.2 Hz), 2.57 (0.25H, d, J = 2.1 Hz), 2.22-1.78 (4H, m), 1.40 (2.25H, s), 1.39 (6.75H, s); 13C NMR (100 MHz, CDCl3) δ 171.7, 171.3, 156.7, 153.9, 130.6, 130.3, 128.7, 128.3, 124.8, 124.2, 120.7, 120.6, 110.7, 80.1, 80.0, 79.6, 75.1, 74.9, 60.7, 59.0, 58.9, 55.5 (2), 46.5, 46.3, 30.7, 29.8, 28.4, 28.2, 24.1, 23.5; IR (thin film) 3288, 1748, 1693cm-1; HRMS (ESI) m/z calcd for C20H26NO5 ([M+H]+), 360.1811, found 360.1817.
cis-1-Methyl 4-(1-phenylprop-2-yn-1-yl) cyclohexane-1,4-dicarboxylate (1a)
Colorless oil (574 mg, 96%). 1H NMR (400 MHz, CDCl3) δ 7.55-7.33 (5H, m), 6.46 (1H, d, J = 2.3 Hz), 3.66 (3H, s), 2.64 (1H, d, J = 2.3 Hz), 2.58-2.48 (2H, m), 1.98-1.82 (4H, m), 1.75-1.60 (4H, m); 13C NMR (100 MHz, CDCl3) δ 175.3, 173.7, 136.6, 129.0, 128.7 (2), 127.5 (2), 80.3, 75.4, 65.1, 51.6, 40.6, 40.5, 25.9 (3), 25.8; IR (thin film) 3285, 2865, 2124, 1728, 1452 cm-1; HRMS (ESI) m/z calcd for C18H21O4 ([M+H]+), 301.1440, found 301.1438.
cis-1-(1-(2-Methoxyphenyl)prop-2-yn-1-yl) 4-methyl cyclohexane-1,4-dicarboxylate (1b)
Colorless oil (296 mg, 90%). 1H NMR (400 MHz, CDCl3) δ 7.67 (1H, dd, J = 1.7 Hz, 7.6 Hz), 7.33 (1H, dt, J = 1.7 Hz, 7.6 Hz), 6.98 (1H, dt, J = 0.9 Hz, 7.5 Hz), 6.88 (1H, d, J = 8.0 Hz), 6.78 (1H, d, J = 2.2 Hz), 3.81(3H, s), 3.65 (3H, s), 2.59 (1H, d, J = 2.3 Hz), 2.55-2.38 (2H, m), 2.0-1.55 (8H, m); 13C NMR (100 MHz, CDCl3) δ 175.4, 173.5, 156.7, 130.4, 128.7, 124.5, 120.5, 110.7, 80.3, 74.8, 60.2, 55.5, 51.6, 40.8, 40.4, 26.0, 25.9 (2), 25.8; IR (thin film) 3282, 2949, 1728, 1493, 1029 cm-1; HRMS (ESI) m/z calcd for C19H23O5 ([M+H]+), 331.1545, found 331.1528.
cis-1-But-3-yn-2-yl 4-methyl cyclohexane-1,4-dicarboxylate (1c)
Colorless oil (110 mg, 92%). 1H NMR (400 MHz, CDCl3) δ 5.45 (1H, dq, J = 2.1 Hz, 6.7 Hz), 3.68 (3H, s), 2.53-2.45 (2H, m), 2.45 (1H, d, J = 2.2 Hz), 1.98-1.85 (4H, m), 1.74-1.62 (4H, m), 1.49 (3H, d, J = 6.7 Hz); 13C NMR (100 MHz, CDCl3) δ 175.2, 173.6, 82.1, 72.7, 59.7, 51.5, 40.5, 40.4, 25.9 (3), 25.8, 21.1; IR (thin film) 3273, 2867, 1728, 1451, 1031 cm-1; HRMS (ESI) m/z calcd for C13H19O4 ([M+H]+), 239.1283, found 239.1278.
cis-1-Methyl 4-(1-phenylhept-2-yn-1-yl) cyclohexane-1,4-dicarboxylate (1d)
Colorless oil (172 mg, 96%). 1H NMR (400 MHz, CDCl3) δ 7.62-7.32 (5H, m), 6.47 (1H, t, J = 2.0 Hz), 3.66 (3H, s), 5.53-2.42 (2H, m), 2.26 (2H, t, J = 7.0 Hz), 1.98-1.35 (12H, m), 0.90 (3H, t, J = 7.2 Hz); 13C NMR (100 MHz, CDCl3) δ 175.3, 173.8, 137.8, 128.5 (3), 127.5 (2), 88.2, 76.7, 65.8, 51.5, 40.6 (2), 30.4, 25.9, 25.8 (3), 21.9, 18.5, 13.5; IR (thin film) 2864, 2235, 1730, 1452, 1135 cm-1; HRMS (ESI) m/z calcd for C22H29O4 ([M+H]+), 357.2066, found 357.2061.
(S)-1-tert-Butyl-2-((R)-1-phenylprop-2-yn-1-yl)pyrrolidine-1,2-dicarboxylate (10a) and (R)-1-tert-Butyl-2-((R)-1-phenylprop-2-yn-1-yl)pyrrolidine-1,2-dicarboxylate (10b)
Isomers separated via three successive column chromatography steps using 10% EtOAc in hexane to give 10a (136 mg, 41%) and 10b (128 mg, 39%), both as colorless oils. 10a: TLC (RF 0.45, 1:4 EtOAc:hexanes); [α]D +60.7 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3; 2:1 mixture of rotamers (minor rotamer in brackets)) δ 7.55-7.46 (2H, m), 7.44-7.32 (3H, m), [6.50 (0.34H, d, J = 1.9 Hz)], 6.48 (0.66H, d, J = 2.1 Hz), [4.39 (0.34H, dd, J = 3.2 Hz, 8.7 Hz)], 4.27 (0.66H, dd, J = 3.4 Hz, 8.7 Hz), 3.60-3.25 (2H, m), 2.65 (1H, d, J = 2.0 Hz), 2.22-2.10 (1H, m), 1.98-1.72 (3H, m), [1.41 (3H, s)], 1.35 (6H, s); 13C NMR (100 MHz, CDCl3) δ 171.9, [171.5], [154.2], 153.7, [136.3], 136.1, 129.1, [128.8], 128.7 (2), [128.5], 127.5 (2), [127.4], 80.0, 79.9, [79.7], [75.7], 75.6, 65.6, 59.0, [58.9], [46.4], 46.3, 30.7, [29.7], [28.3], 28.2 (3), [24.2], 23.5; IR (thin film) 3288, 2976, 1748, 1694, 1393cm-1; HRMS (ESI) m/z calcd for C19H24NO4 ([M+H]+), 330.1705, found 330.1705. 10b: Rf 0.41 (1:4 EtOAc:hexanes); [α]D -46.3 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3; 2.3:1 mixture of rotamers by 1H NMR, minor rotamer in brackets)) δ 7.57-7.48 (2H, m), 7.43-7.32 (3H, m), 6.47 (0.7H, d, J = 2.2 Hz), [6.46 (0.3H, d, J = 2.0 Hz)], [4.37 (0.3H, dd, J = 3.4 Hz, 8.7 Hz)], 4.25 (0.7H, dd, J = 3.8 Hz, 8.7 Hz),3.58-3.47 (1H, m), 3.46-3.33 (1H, m), 2.65 (0.7H, d, J = 2.3 Hz), [2.63 (0.3H, d, J = 2.2 Hz)], 2.28-2.12 (1H, m), 2.06-1.79 (3H, m), [1.46 (2.7 H, s)], 1.30 (6.3H, s); 13C NMR (100 MHz, CDCl3) δ 171.9, [171.7], [154.4], 153.7, 136.2, [136.0], 129.2, [128.9], 128.7 (2), [128.6], 127.8 (2), [127.5], 80.0, 79.9, [79.8], 75.8, [75.5], 65.8, [65.7], 59.0, [58.7], [46.5], 46.3, 30.7, [29.6], [28.4], 28.2 (3), [24.2], 23.5; IR (thin film) 3290, 2881, 1750, 1694, 1393 cm-1; HRMS (ESI) m/z calcd for C19H24NO4 ([M+H]+), 330.1705, found 330.1706.
(S)-1-tert-Butyl-2-((R)-1-(2-methoxyphenyl)prop-2-yn-1-yl)pyrrolidine-1,2-dicarboxylate (11a) and (R)-1-tert-Butyl-2-((R)-1-(2-methoxyphenyl)prop-2-yn-1-yl)pyrrolidine-1,2-dicarboxylate (11b)
11a: White solid (158 mg, 44%); mp 52-54; Rf 0.32 (1:4 EtOAc:hexanes); [α]D -80.8 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3; 2.7:1 mixture of rotamers (minor rotamer in brackets)) δ 7.65 (1H, d, J = 7.5 Hz), 7.36-7.28 (1H, m), 7.02-6.95 (1H, m), 6.88 (1H, d, J = 8.2 Hz), 6.79 (1H, d, J = 2.0 Hz), [4.36 (0.27H, dd, J = 2.5 Hz, 8.4 Hz)], 4.27 (0.73H, dd, J = 3.6 Hz, 8.6 Hz), [3.83 (0.8H, s)], 3.82 (2.2H, s), 3.54-3.30 (2H, m), 2.58 (0.7H, d, J = 2.3 Hz), [2.55 (0.3H, d, J = 2.0 Hz)], 2.28-1.72 (4H, m), [1.45 (2.4H, s)], 1.37 (6.6H, s); 13C NMR (100 MHz, CDCl3) δ 171.6, [171.3], [156.7], 156.5, [154.2], 153.8, 130.5, [130.3], 128.9, [128.4], [124.4], 124.2, 120.6, [120.4], [110.7], 110.6, 80.1, [80.0], 79.9, [79.5], 74.9, [74.6], 60.6, 59.0, [58.7], [55.5], 55.4, [46.4], 46.2, 30.7, [29.5], [28.4], 28.1 (3), [24.1], 23.3; IR (thin film) 3285, 2976, 1748, 1694, 1393 cm-1; HRMS (ESI) m/z calcd for C20H26NO5 ([M+H]+), 360.1811, found 360.1821. 11b: Colorless oil (152 mg, 42%); Rf 0.27 (1:4 EtOAc:hexanes); [α]D -19.0 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3; 3:1 mixture of rotamers (minor rotamer in brackets)) δ 7.67 (0.75H, dd, J = 1.5 Hz, 7.6 Hz), [7.62 (0.25H, d, J = 6.7 Hz)], 7.38-7.27 (1H, m), 7.03-6.83 (2H, m), 6.81 (1H, d, J = 2.1 Hz), [4.38 (0.25H, dd, J = 2.8 Hz, 8.6 Hz)], 4.25 (0.75H, dd, J = 3.3 Hz, 8.7 Hz), 3.82 (3H, s), 3.65-3.28 (2H, m), 2.60 (0.75H, d, J = 2.2 Hz), [2.57 (0.25H, d, J = 2.2 Hz)], 2.22-1.78 (4H, m), [1.40 (2.25H, s)], 1.39 (6.75H, s); 13C NMR (100 MHz, CDCl3) δ 171.7, [171.3], 156.7, 153.9, 130.6, [130.3], 128.7, [128.3], [124.8], 124.2, 120.7, [120.6], 110.7, 80.1, 80.0, [79.6], 75.1, [74.9], 60.7, 59.0, [58.9], 55.5, [55.4], [46.5], 46.3, 30.7, [29.8], [28.4], 28.2 (3), [24.1], 23.5; IR (thin film) 3288, 2124, 1748, 1693, 1392 cm-1; HRMS (ESI) m/z calcd for C20H26NO5 ([M+H]+), 360.1811, found 360.1817.
(R)-1-(2-(R)-Methoxyphenyl)prop-2-yn-1-yl 2-(3-benzoylphenyl)propanoate (12a) and (S)-1-(2-(R)-Methoxyphenyl)prop-2-yn-1-yl 2-(3-benzoylphenyl)propanoate (12b)
Purification by three successive column chromatography steps (10% EtOAc in hexane) gave 12a (176 mg, 44%) and 12b (171 mg, 43%) as colorless oils. 12a: Rf 0.37 (1:4 EtOAc:hexanes); [α]D -27.8 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.66-7.18 (11H, m), 6.83 (1H, dt, J = 0.9 Hz, 7.5 Hz), 6.70 (1H, dd, J = 0.6 Hz, 7.6 Hz), 6.68 (1H, d, J = 2.2 Hz), 3.75 (1H, q, J = 7.2 Hz), 3.53 (3H, s), 2.53 (1H, d, J = 2.3 Hz), 1.48 (3H, d, J = 7.2 Hz); 13C NMR (100 MHz, CDCl3) δ 196.4, 172.6, 156.7, 140.5, 137.8, 137.4, 132.5, 131.6, 130.4, 130.0 (2), 129.5, 128.8, 128.5, 128.4, 128.3 (2), 124.2, 120.4, 110.5, 79.9, 75.1, 60.9, 55.3, 45.2, 18.2; IR (thin film) 3286, 2939, 2125, 1736, 1655 cm-1; HRMS (ESI) m/z calcd for C26H23O4 ([M+H]+), 399.1596, found 399.1590. 12b: Rf 0.33 (1:4 EtOAc:hexanes); [α]D -17.2 (c 0.7, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.75-7.24 (11H, m), 6.90 (1H, dt, J = 0.7 Hz, 7.6 Hz), 6.80 (1H, d, J = 8.3 Hz), 7.72 (1H, d, J = 2.2 Hz), 3.76 (1H, q, J = 7.2 Hz), 3.70 (3H, s), 2.44 (1H, d, J = 2.2 Hz), 1.46 (3H, d, J = 7.2 Hz); 13C NMR (100 MHz, CDCl3) δ 196.5, 172.7, 156.7, 140.4, 137.8, 137.5, 132.5, 131.6, 130.5, 130.1 (2), 129.4, 128.9, 128.7, 128.5, 128.3 (2), 124.3, 120.6, 110.7, 79.8, 75.0, 60.8, 55.5, 45.2, 18.5; IR (thin film) 3288, 2250, 1736, 1655, 1493 cm-1; HRMS (ESI) m/z calcd for C26H23O4 ([M+H]+), 399.1596, found 399.1587.
(S)-2-(((R)-1-(2-Methoxyphenyl)prop-2-yn-1-yl)oxy)-2-oxo-1-phenylethyl benzoate (13a) and (R)-2-(((R)-1-(2-Methoxyphenyl)prop-2-yn-1-yl)oxy)-2-oxo-1-phenylethyl benzoate (13b)
Purification by three successive column chromatography steps (10% EtOAc in hexane) gave 13a (168 mg, 42%) and 13b (172 mg, 43%) as colorless oils. 13a: Rf 0.41 (1:4 EtOAc:hexanes); [α]D +7.8 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 8.04 (2H, d, J = 7.6 Hz), 7.56-7.24 (10H, m), 6.89 (1H, t, J = 7.5 Hz6.81 (1H, d, J = 8.2 Hz), 6.78 (1H, d, J = 2.2 Hz), 6.15 (1H, s), 3.72 (3H, s), 2.44 (1H, d, J = 2.2 Hz); 13C NMR (100 MHz, CDCl3) δ 167.4, 165.6, 156.9, 133.8, 133.4, 130.7, 130.0 (2), 129.4, 129.2, 128.8, 128.7 (2), 128.4 (2), 127.9 (2), 123.7, 120.4, 110.7, 79.1, 75.4, 74.6, 61.7, 55.5; IR (thin film) 3286, 2128, 1758, 1723, 1493 cm-1; HRMS (ESI) m/z calcd for C25H21O5 ([M+H]+), 401.1389, found 401.1381. 13b: Rf 0.37 (1:4 EtOAc:hexanes); [α]D -55.0 (c 0.9, CHCl3); 1H NMR (400 MHz, CDCH3, δ 8.04 (2H, dd, J = 1.2 Hz, 8.2 Hz), 7.52-7.15 (10H, m), 6.78 (1H, dt, J = 0.8 Hz, 7.6 Hz), 6.72 (1H, d, J = 2.2 Hz), 6.64 (1H, d, J = 8.3 Hz), 6.13 (1H, m), 3.40 (3H, s), 2.59 (1H, d, J = 2.3 Hz); 13C NMR (100 MHz, CDCl3) δ 167.5, 165.7, 156.7, 133.9, 133.4, 130.5, 130.0 (2), 129.3, 129.1, 128.6 (2), 128.4 (3), 127.8 (2), 123.7, 120.3, 110.3, 79.2, 75.9, 74.7, 61.9, 55.1; IR (thin film) 1760, 1725, 1494, 1110 cm-1; HRMS (ESI) m/z calcd for C25H21O5 ([M+H]+), 401.1389, found 401.1376.
(1R,2R)-(R)-1-(2-Methoxyphenyl)prop-2-yn-1-yl-2-benzoylcyclohexanecarboxylate (14a) and (1S,2S)-(R)-1-(2-Methoxyphenyl)prop-2-yn-1-yl-2-benzoylcyclohexanecarboxylate (14b)
Purification by three successive column chromatography steps (10% EtOAc in hexane) gave 14a (154 mg, 41%) and 14b (141 mg, 38%) as colorless oils. 14a: Rf 0.41 (1:4 EtOAc:hexanes); [α]D -15.2 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 8.02-7.95 (2H, m), 7.62-7.41 (4H, m), 7.32 (1H, dt, J = 1.7 Hz, 8.2 Hz, 6.95 (1H, dt, J = 0.8 Hz, 7.6 Hz), 6.86 (1H, d, J = 8.3 Hz), 6.68 (1H, d, J = 2.2 Hz), 3.79 (3H, s), 3.70-3.60 (1H, m), 3.02-2.92 (1H, m), 2.51 (1H, d, J = 2.2 Hz), 2.34-1.20 (8H, m); 13C NMR (100 MHz, CDCl3) δ 202.8, 173.9, 156.8, 136.4, 132.8, 130.3, 128.5 (3), 128.4 (2), 124.3, 120.4, 110.7, 80.2, 74.5, 60.4, 55.5, 46.7, 44.3, 29.9, 28.9, 25.6, 25.5; IR (thin film) 3285, 1732, 1679, 1493, 1167 cm-1; HRMS (ESI) m/z calcd for C24H25O4 ([M+H]+), 377.1753, found 377.1747. 14b: Rf 0.35 (1:4 EtOAc:hexanes); [α]D -30.5 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.90-7.85 (2H, m), 7.57 (1H, dd, J = 1.7 Hz, 7.6 Hz), 7.55-7.23 (4H, m), 6.90 (1H, dt, J = 0.9 Hz, 7.6 Hz), 6.71 (1H, d, J = 8.2 Hz), 6.68 (1H, d, J = 2.2 Hz), 3.67 (3H, s), 3.63-3.55 (1H, m), 3.03-2.94 (1H, m), 2.54 (1H, d, J = 2.3 Hz), 2.22-1.15 (8H, m); 13C NMR (100 MHz, CDCl3) δ 202.6, 173.6, 156.6, 136.3, 132.7, 130.2, 128.5 (2), 128.4, 128.3 (2), 124.5, 120.4, 110.4, 80.0, 74.9, 60.4, 55.3, 46.7, 44.4, 29.9, 29.0, 25.6, 25.5; IR (thin film) 3285, 1732, 1678 cm-1; HRMS (ESI) m/z calcd for C24H25O4 ([M+H]+), 377.1753, found 377.1739.
(4aS,8R,8aS)-(R)-1-Phenylprop-2-yn-1-yl-2-benzyl-1-oxo-1,2,3,4,4a,7,8,8a-octahydroisoquinoline-8-carboxylate (15a) and (4aR,8S,8aR)-(R)-1-(2-Methoxyphenyl)prop-2-yn-1-yl-2-benzyl-1-oxo-1,2,3,4,4a,7,8,8a-octahydroisoquinoline-8-carboxylate (15b)
Purification by three successive column chromatography steps (20% EtOAc in hexane) gave 15a (181 mg, 42%) as colorless oil and 15b (168 mg, 39%) as a white solid. 15a: Rf 0.34 (1:3 EtOAc:hexanes); [α]D -20.5 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.68 (1H, dd, J = 1.7 Hz, 7.6 Hz), 7.36-7.10 (6H, m), 6.99 (1H, td, J = 0.9 Hz, 7.6 Hz), 6.88 (1H, d, J = 0.7 Hz), 6.87 (1H, d, J = 2.2 Hz), 5.86-5.80 (1H, m), 5.51 (1H, d, J = 10.0 Hz), 4.53 (1H, d, J = 14.7), 4.42 (1H, d,J = 14.7 Hz), 3.81 (3H, s), 3.39-3.34 (1H, m), 3.05-2.98 (2H, m), 2.85-2.76 (1H, m), 2.74-2.64 (1H, m), 2.57 (1H, d, J = 2.3 Hz), 2.51-2.29 (2H, m), 2.01-1.89 (1H, m), 1.83-1.72 (1H, m); 13C NMR (100 MHz, CDCl3) δ 172.4, 168.9, 156.7, 137.4, 130.0, 129.1, 128.5, 128.4 (2), 127.9 (2), 127.7, 127.1, 125.3, 120.4, 110.8, 81.1, 74.0, 60.3, 55.6, 49.9, 43.8, 42.6, 41.4, 34.4, 27.3, 23.7; IR (thin film) 3285, 1737, 1635, 1493, 1251 cm-1; HRMS (ESI) m/z calcd for C27H28NO4 ([M+H]+), 430.2018, found 430.2011. 15b: Rf 0.31 (1:3 EtOAc:hexanes); mp 149-151 °C; [α]D - 30.6 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.69 (1H, dd, J = 1.7 Hz, 7.6 Hz), 7.36-7.14 (6H, m), 6.99 (1H, dt, J = 0.9 Hz, 7.5 Hz), 6.91 (1H, d, J = 2.2 Hz), 6.89 (1H, d, J = 8.2 Hz), 5.84-5.80 (1H, m), 5.49 (1H, d, J = 10.0 Hz), 4.79 (1H, d, J = 14.7), 4.31 (1H, d, J = 14.7), 3.84 (3H, s), 3.47-3.42 (1H, m), 3.07-2.62 (4H, m), 2.58 (1H, d, J = 2.3 Hz), 2.50-1.65 (4H, m); 13C NMR (100 MHz, CDCl3) δ 172.2, 169.0, 156.7, 137.3, 130.0, 129.0, 128.5, 128.4 (2), 127.9 (2), 127.8, 127.1, 125.6, 120.5, 110.8, 80.7, 74.2, 60.3, 55.7, 50.2, 43.9, 42.6, 41.3, 34.3, 27.3, 23.8; IR (thin film) 3285, 1736, 1634, 1492, 1250 cm-1; HRMS (ESI) m/z calcd for C27H28NO4 ([M+H]+), 430.2018, found 430.2005.
(4aS,8R,8aS)-1-((R)-Phenylprop-2-yn-1-yl)-2-benzyl-1-oxo-1,2,3,4,4a,7,8,8a octahydroisoquinol-ine-8-carboxylate (16a) and (4aR,8S,8aR)-1-((R)-Phenylprop-2-yn-1-yl)-2-benzyl-1-oxo-1,2,3,4,4a,7,8,8a-octahydroisoquinoline-8-carboxylate (16b)
Purification by three successive column chromatography steps (10% EtOAc in hexane) gave 16a (190 mg, 48%) and 16b (192 mg, 48%) as white foams. 16a: Rf 0.58 (1:3 EtOAc:hexanes); [α]D -37.0 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.70-7.60 (2H, m), 7.40-7.10 (8H, m), 6.63 (1H, d, J = 2.2 Hz), 5.90-5.83 (1H, m), 5.51 (1H, dd, J = 10.0 Hz, 1 Hz), 4.64 (1H, d, J = 14.7 Hz), 4.38 (1H, d, J = 14.7 Hz), 3.34 (1H, dd, J = 5.4 Hz, 3.3 Hz), 3.10-2.98 (2H, m), 2.85- 2.76 (1H, m), 2.74-2.65 (1H, m), 2.69 (1H, d, J = 2.2 Hz), 2.46-2.38 (2H, m), 1.98-1.86 (1H, m), 1.84-1.74 (1H, m); 13C NMR (100 MHz, CDCl3) δ 172.6, 169.0, 137.3, 136.5, 129.1, 128.6, 128.5 (2), 128.4 (2), 127.9 (2), 127.8 (2), 127.6, 127.2, 80.5, 75.5, 65.0, 50.1, 43.8, 42.5, 41.1, 34.2, 27.3, 23.6; IR (thin film) 2122, 1737, 1630, 1356 cm-1; HRMS (ESI) m/z calcd for C26H26NO3 ([M+H]+), 400.1913, found 400.1902. 16b: Rf 0.52 (1:3 EtOAc:hexanes); [α]D -15.0 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.70-7.60 (2H, m), 7.40-7.15 (8H, m), 6.62 (1H, d, J = 2.2 Hz), 5.85-5.78 (1H, m), 5.50 (1H, dd, J = 10.0 Hz, 1.6 Hz), 4.75 (1H, d, J = 14.7 Hz), 4.36 (1H, d, J = 14.7 Hz), 3.47 (1H, dd, J = 5.4 Hz, 3.2 Hz), 3.10-3.00 (2H, m), 2.85- 2.60 (2H, m), 2.65 (1H, d, J = 2.2 Hz), 2.48-2.28 (2H, m), 2.05-1.95 (1H, m), 1.82-1.77 (1H, m); 13C NMR (100 MHz, CDCl3) δ 172.6, 169.1, 137.2, 137.0, 129.0, 128.7, 128.5 (2), 128.4 (2), 127.9 (2), 127.8, 127.7 (2), 127.2, 80.5, 75.3, 65.3, 50.3, 43.8, 42.8, 41.4, 34.3, 27.3, 23.6; IR (thin film) 2123, 1734, 1630, 1356 cm-1; HRMS (ESI) m/z calcd for C26H26NO3 ([M+H]+), 400.1913, found 400.1908.
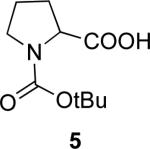
General Procedure for Removal of Propargylic Esters. N-Boc-D-Proline ((R)-5
To the propargyl ester 11b (108 mg, 0.30 mmol) and CuCl (3 mg, 0.03 mmol) in a 10 mL flask was added 3.0 mL MeOH (degassed with Ar). The reaction solution was degassed with Ar for 5 min and then heated for 2 h at 40 °C with constant stirring. The reaction was cooled, concentrated, and chromatographed with 20–100% EtOAc in hexane to give (R)-5 (53 mg, 82% yield) as a white solid. The er (97:3) of (R)-5 was determined by converting it into phenyl amide (EDCl, DMAP–mediated coupling with aniline in CH2Cl2) and then using analytical HPLC (Chiralcel OD-H, 0.5 mL/min, 10% i-PrOH/hexane).
(R)-Ketoprofen ((R)-6) and (S)-ketoprofen ((S)-6)
The er values of (R)- and (S)-6 were determined by converting them into phenyl amides (EDCl, DMAP mediated coupling with aniline in CH2Cl2) and then using analytical HPLC (Chiralcel OD-H, 0.5 mL/min, 5% i-PrOH/hexane).
(S)-O-Benzoyl-mandelic acid [(S)-7] and (R)-O-benzoyl-mandelic acid [(R)-7]
The er values of (R)- and (S)-7 were determined by converting them into phenyl amides (EDCl, DMAP mediated coupling with aniline in CH2Cl2) and then using analytical HPLC (Chiralcel OD-H, 1.0 mL/min, 1% i-PrOH/hexane). (R)-7: [α]D -47.1 (c 1.0, CHCl3); [lit.10 [α]D -48.28 (c 1.0, CHCl3).
(1R,2R)-2-Benzoylcyclohexanecarboxylic acid [(1R,2R)-8] and (1S,2S)-2-Benzoylcyclohexanecarboxylic acid [(1S,2S)-8]
The er values of (1R,2R)-8 and (1S,2S)-8 were determined by first converting them into the methyl ester (EDCl, DMAP mediated coupling with methanol in CH2Cl2) and then using analytical HPLC (Chiralcel OD-H, 0.5 mL/min, 5% i-PrOH/hexane). (1R,2R)-8: Optical rotation for the methyl ester: [α]D +8.5 (c 0.8, hexane); [lit.11 [α]D +11.1 (c 1.9, iso-octane)].
(4aS,8R,8aS)-2-Benzyl-1-oxo-1,2,3,4,4a,7,8,8a octahydroisoquinoline-8-carboxylic acid ((4aS, 8R, 8aS)-9) and (4aS, 8R, 8aS)-2-Benzyl-1-oxo-1,2,3,4,4a,7,8,8a octahydroisoquinoline-8-carboxylic acid ((4aS, 8R, 8aS))-9
The er values of (4aS, 8R, 8aS)-9 and (4aR, 8S, 8aR)-9 were determined by first converting them into 4-chloro-3-trifluoromethylphenyl amides (EDCl, DMAP mediated coupling with 4-chloro-3-trifluoromethyl aniline in CH2Cl2) and then using analytical HPLC (Chiralcel OD-H, 0.5 mL/min, 10% i-PrOH/hexane).
Supplementary Material
ACKNOWLEDGEMENT
We thank Dr. Victor Day for the X-ray analysis and Stephen Slausen, Chan Woo Huh, and Kevin Frankowski for editorial assistance. This work was supported by the National Institutes of General Medical Sciences (2P50GM069663).
Footnotes
Supporting Information Available: Copies of 1H and 13C NMR spectra for new compounds and chiral HPLC data and X-ray data for the derivative of compound 9. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Reviews: Jacques J, Collet A, Wilen SH. Enantiomers, Racemates and Resolutions. Wiley–Interscience; New York, NY: 1981. Porter W.H. Pure Appl. Chem. 1991;63:1119–1122.Sekhon BS. Int. J. PharmTech Res. 2010;2:1584–1594.
- 2.a Frankowski KJ, Hirt EE, Zeng Y, Neuenswander B, Fowler D, Schoenen F, Aubé J. J. Comb. Chem. 2007;9:1188–1192. doi: 10.1021/cc700127f. [DOI] [PubMed] [Google Scholar]; b Frankowski KJ, Ghosh P, Setola V, Tran TB, Roth BL, Aubé J. ACS Med. Chem. Lett. 2010;1:189–193. doi: 10.1021/ml100040t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a Yamada Y, Yuasa M, Nakamura I, Murahashi S. J. Org. Chem. 1994;59:2282–2284. [Google Scholar]; b Detz RJ, Delville MME, Hiemstra H, van Maarseveen JH. Angew. Chem. Int. Ed. 2008;47:3777–3780. doi: 10.1002/anie.200705264. [DOI] [PubMed] [Google Scholar]
- 4.Mahrwald R, Quint S. Tetrahedron. 2000;56:7463–7468. [Google Scholar]
- 5.For a review of the Glaser coupling of alkynes, see: Gribble GW. In: Name Reactions for Homologation, Part 1. Pt. 1. Li JJ, editor. John Wiley & Sons, Inc.; Hoboken: 2009. pp. 236–257.
- 6.Carboxylic acids 5-8 are commercially available and 9 is known (reference 2b).
- 7.a Waldinger C, Schneider M, Botta M, Corelli F, Summa V. Tetrahedron: Asymmetry. 1996;7:1485–1488. [Google Scholar]; b Baldoli C, Buttero PD, Licandro E, Maiorana S, Papagni A, Torchio M. Tetrahedron Lett. 1993;34:7943–7946. [Google Scholar]
- 8.Authentic samples were prepared by reacting racemic 1-(2-methoxyphenyl)prop-2-yn-1-ol with L-Bocproline, diastereomer separation, and hydrolysis with LiOH.
- 9.Carboxylic acid (4aS,8S,8aR)-9 was converted to the analogous 4-bromo-2-(trifluoromethyl)aniline amide, which was subjected to anomalous X-ray diffraction to determine the absolute stereochemistry.
- 10.Kumar PS, Banerjee A, Baskaran S. Angew. Chem. Int. Ed. 2010;49:804–807. doi: 10.1002/anie.200905952. [DOI] [PubMed] [Google Scholar]
- 11.Hagishita S, Kuriyama K. J. Chem. Soc. Perkin. Trans. 2. 1974;6:686–696. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.