

Abstract
Acetate supplementation attenuates neuroglial activation, increases histone and non-histone protein acetylation, reduces pro-inflammatory cytokine expression, and increases IL-4 transcription in rat models of neuroinflammation and Lyme's neuroborreliosis. Because eicosanoid signaling is involved in neuroinflammation, we measured the effect acetate treatment had on phospholipase, cyclooxygenase, and prostaglandin E2 (PGE2) levels in BV-2 microglia and primary astrocytes stimulated with lipopolysaccharide (LPS). In BV-2 microglia, we found that LPS increased the phosphorylation-state of cytosolic phospholipase A2 (cPLA2), reduced the levels of phospholipase C (PLC) β1, and increased the levels of cyclooxygenase (Cox)-1 and -2. Acetate treatment returned PLCβ1 and Cox-1 levels to normal, attenuated the increase in Cox-2, but had no effect on cPLA2 phosphorylation. In primary astrocytes, LPS increased the phosphorylation of cPLA2 and increased the levels of Cox-1 and Cox-2. Acetate treatment in these cells reduced secretory PLA2 IIA and PLCβ1 levels as compared to LPS-treatment groups, reversed the increase in cPLA2 phosphorylation, and returned Cox-1 levels to normal. Acetate treatment reduced PGE2 release in astrocytes stimulated with LPS to control levels, but did not alter PGE2 levels in BV-2 microglia. The amount of acetylated H3K9 bound to the promoter regions of Cox-1, Cox-2, IL-1β and NF-κB p65 genes, but not IL-4 in were increased in BV-2 microglia treated with acetate. These data suggest that acetate treatment can disrupt eicosanoid signaling in neuroglia that may, in part, be the result of altering gene expression due chromatin remodeling as a result of increasing H3K9 acetylation.
Keywords: phospholipase, cyclooxygenase, eicosanoid, acetate, histone acetylation
Introduction
Acetate supplementation reduces tremor phenotype in a rat model of Canavan disease [1], reduces injury in an experimental model of head trauma [2], and reduces neuroglia activation in rat models of neuroinflammation [3] and Lyme neuroborreliosis [4]. Acetate supplementation further increases brain acetyl-CoA levels [3], mitochondrial acetyl-CoA metabolism [5], and alters brain histone acetylation in a time- and site-specific pattern in normal animals [6] and in rats subjected to neuroinflammation [7]. Further, long-term acetate supplementation reduces pro-inflammatory cytokine levels, and increases histone acetylation [7]. Increasing brain acetate metabolism with glyceryl triacetate also reverses LPS-induced increases in NF-κB p65 and IL-1β protein levels, and increases IL-4 protein levels in LPS-stimulated BV-2 microglia [8] and primary astrocyte cultures [9]. Acetate treatment transiently reduces LPS-induced MAPK p38 phosphorylation, and increases ERK1/2 phosphorylation in microglia cultures whereas in astrocyte cultures acetate completely attenuates LPS-induced MAPK p38 phosphorylation and reduces basal levels of phosphorylated ERK1/2. These studies suggest that acetate supplementation and increases acetyl-CoA metabolism acts to shift the brain cytokine balance toward a more anti-inflammatory state and potentially disrupts neuroglia MAPK and NF-κB signaling in a cell-type specific manner.
In pathological conditions, phospholipase activation and increases in eicosanoid signaling result in alterations in membrane permeability, the accumulation of free fatty acids and lipid peroxides, as well as the induction of many other inflammatory pathways [10, 11]. Phospholipases are a heterogeneous group of enzymes that metabolize membrane phospholipids. The physiological function of brain phospholipases include phospholipid turnover and metabolism, exocytosis, the removal of oxidized lipids, and are also involved in long-term potentiation, neural cell proliferation, and the release of neurotransmitters [12–14]. Cyclooxygenase (Cox)-1 and Cox-2 convert arachidonic acid, released by PLA2 acting at the sn-2 position of membrane phospholipids, into prostaglandin (PG) H2 which is the physiologic substrate for other terminal prostaglandin synthases' that form other biologically active prostaglandins [15]. Phospholipase A2 (PLA2) are classified into cytosolic PLA2 (cPLA2), secretory PLA2 (sPLA2), and calcium-independent PLA2 (iPLA2) depending on their requirement on calcium [13, 14]. The sPLA2 group is divided into type I (pancreatic type) and type II (inflammatory type) [16]. The most well-studied of which is type IIA sPLA2 expressed in most areas of rat brain and whose levels are elevated in inflammatory conditions like ischemia and endotoxic shock [17, 18]. Phospholipase C (PLC), based on structural characteristics are divided into PLCβ, γ, δ, and ε which cleaves the polar head group esterified at the sn-3 position of phospholipid moiety and is commonly associated with modulating intracellular calcium and protein kinase C activation [19]. The suppression of PLCβ1 by endotoxin is also associated with a disruption in purine-mediated neurotransmission that is involved in the switching macrophages from an M1 state to an M2-like phenotype [20]. Eicosanoid signaling is upregulated by lipopolysaccharide (LPS), pro-inflammatory cytokines interleukin (IL)-1β, IL-6 and tumor necrosis factor (TNF)-α, nuclear factor kappa-B (NF-κB), and mitogen-activated protein kinases (MAPK) p38, extracellular signal-regulated kinase (ERK) and c-Jun N-terminal kinases (JNK), all of which are all important mediators of neuroinflammation [11, 16, 21]. In this regard, cPLA2 is activated by MAPK phosphorylation that results in its translocation to the plasma or nuclear membranes [22, 23]. Thus understanding how these signaling pathways are altered by acetate supplementation may provide insight into the mechanism by it modulates neuroglia activation.
Histone acetylation is a common post-translational modification that is associated with changes in the gene expression due to increased accessibility of transcription machinery to chromatin [24–26], increased flexibility of the DNA associated with the end of the nucleosomes [27], and recruits proteins that facilitate downstream gene expression regulation [28]. The multitude of events and changes that take place during transcription are not due solely to changes in histone acetylation, but rather are the results of overlapping actions of several factors [29] and include the acetylation of non-histone transcription factors [30, 31]. Increases in histone acetylation is associated with anti-inflammatory outcomes in models of cerebral ischemia [32], amyotrophic lateral sclerosis [33], and reduces microglial activation in traumatic brain injury [34]. An increase in histone acetylation also reduces oxidative stress-induced apoptosis [35], polyglutamine toxicity in a mouse motor neuron–neuroblastoma fusion cell line [36] and in Drosophila models of polyglutamine diseases [37]. All of which suggests that increasing histone acetylation may provide a potential therapeutic target in to treat disorders associated with neuroinflammation [38–40].
To expand on previous studies and to begin to determine how acetate supplementation attenuates inflammatory neuroglia activation, we quantified the effect of acetate treatment had on the levels of enzymes involved in eicosanoid signaling and PGE2 release in astrocytes and BV-2 microglial cell cultures stimulated with LPS. Further, using chromatin immunoprecipitation, we measured the effect treatment had on H3K9 binding to the promoters of the genes coding for Cox-1, Cox-2, IL-1β, IL-4 and NF-κB p65 in BV-2 microglia. Acetate reduced sPLA2 IIA, PLCβ1 and cPLA2 phosphorylation and PGE2 release in astrocytes, but only Cox-2 protein levels in BV-2 microglia, while the binding affinity of acetylated H3K9 towards Cox-1 and 2, IL-1β, and NF-κB p65, but not the IL-4 promoter regions was increased in BV-2 microglia. These data suggest that acetate treatment can disrupt eicosanoid signaling in neuroglia, which may contribute to the anti-inflammatory properties found by increasing brain acetyl-CoA metabolism.
Material and Methods
Reagents
LPS (Escherichia Coli 055:B5) and proteinase K were purchased from Sigma (St. Louis, MO). Antibodies against PLCβ1, γ1, δ1, cPLA2 were from Santa Cruz Biotechnology Inc. (Santa Cruz, CA), while antibodies against Cox-1, Cox-2 and sPLA2 IIA, and prostaglandin screening EIA kits were purchased from Cayman Chemical Company (Ann Arbor, MI). A goat anti-rabbit horse radish peroxidase-linked secondary antibody was purchased from Bio-Rad Laboratories (Hercules CA), and DMEM–F-12 media, fetal bovine serum and RNAse A was from Invitrogen (Grand Island, NY). All buffering reagents and other chemicals were purchased from EMD Biosciences (Gibbstown, NJ). A chromatin immunoprecipitation assay kit, antibodies toward acetylated H3K9, and normal rabbit IgG were purchased from Millipore (Billerica, MA). A QIAquick PCR purification kit was purchased from Qiagen (Valencia, CA) and protein A and protein G magnetic beads were purchased from Invitrogen (Grand Island, NY). FastStart Universal SYBR Green Master and complete EDTA-free protease inhibitor cocktail tablets were purchased from Roche Applied Science (Indianapolis, IN) while all chromatin immunoprecipitation primers (table 1) were purchased from Integrated DNA Technologies (Coraville, IA).
Table 1.
| Gene | Primer set | Sequence | Start | End | Length | Melting Temp (° C) | Amplicon size (bp) | |
|---|---|---|---|---|---|---|---|---|
| ptgs 1 | Primer set #1 | Forward | CAA CTC CCC TCA CCT TTA ACT AT | −736 | −714 | 23 | 60.9 | 288 |
| Reverse | CTT GCT GGT CCT CGG TGT AT | −468 | −449 | 20 | 60.5 | |||
| Primer set #2 | Forward | TCA TGT CTG ACC TGG CCT CT | −393 | −374 | 20 | 60.5 | 257 | |
| Reverse | TGG GAT ATA GCA AAC TGA GGC | −157 | −137 | 21 | 59.5 | |||
| Primer set #3 | Forward | GCA TTT CTG ACA CTG TAA AAA GAT C | −129 | −105 | 25 | 60.9 | 261 | |
| Reverse | GGG AGT GGA TGG ATG TGC AA | +112 | +131 | 20 | 60.5 | |||
| Primer set #4 | Forward | ACA CCC TCG GTC CTG CTC | +296 | +313 | 18 | 60.8 | 274 | |
| Reverse | AGA ACC TGT CTC TGC TTC CC | +550 | +569 | 20 | 60.5 | |||
| Primer set #5 | Forward | TTG GGG AGG AAG CGG CAG | +668 | +685 | 18 | 60.8 | 221 | |
| Reverse | ACT TGG GGC TGT TAT CGC AC | +869 | +888 | 20 | 60.1 | |||
| ptgs2 | Primer set #1 | Forward | CTA TGT AAC AGC AGG GGG AAA AT | −720 | −698 | 23 | 60.9 | 243 |
| Reverse | GCA CAC AGC TAC CGG TTA ATT T | −499 | −478 | 22 | 60.1 | |||
| Primer set #2 | Forward | TGA GCT TTT AGG CCC CCA CT | −373 | −354 | 20 | 60.5 | 199 | |
| Reverse | AGG CTT TTA CCC ACG CAA ATG A | −196 | −175 | 22 | 60.1 | |||
| Primer set #3 | Forward | AAA GTT GGT GGG GGT TGG G | −131 | −113 | 19 | 59.5 | 264 | |
| Reverse | GCA GCG CAG AGC AGC AC | +116 | +132 | 17 | 59.8 | |||
| Primer set #4 | Forward | CTG CAA CCC ACT TTC AGG TTT | +265 | +285 | 21 | 59.5 | 270 | |
| Reverse | CGA CCT AGT GCA ATA GTC AAA ATT | +511 | +534 | 24 | 60.3 | |||
| Primer set #5 | Forward | TTA TCA TTG TAA AGT TGA CCC ATA GT | +750 | +775 | 26 | 60.1 | 244 | |
| Reverse | AGG AAG ATA CCC CAG GAA AAA CT | +971 | +993 | 23 | 60.9 | |||
| p65 | Primer set #1 | Forward | CAA CTC CCC TCA CCT TTA ACT AT | −743 | −720 | 24 | 60.3 | 209 |
| Reverse | CTT GCT GGT CCT CGG TGT AT | −557 | −535 | 23 | 60.9 | |||
| Primer set #2 | Forward | TCA TGT CTG ACC TGG CCT CT | −518 | −499 | 20 | 60.5 | 201 | |
| Reverse | TGG GAT ATA GCA AAC TGA GGC | −336 | −318 | 19 | 61.6 | |||
| Primer set #3 | Forward | GCA TTT CTG ACA CTG TAA AAA GAT C | −93 | −71 | 23 | 60.9 | 186 | |
| Reverse | GGG AGT GGA TGG ATG TGC AA | +75 | +92 | 18 | 60.8 | |||
| Primer set #4 | Forward | ACA CCC TCG GTC CTG CTC | +146 | +163 | 18 | 60.8 | 148 | |
| Reverse | AGA ACC TGT CTC TGC TTC CC | +272 | +293 | 22 | 60.1 | |||
| Primer set #5 | Forward | TTG GGG AGG AAG CGG CAG | +538 | +555 | 18 | 60.8 | 255 | |
| Reverse | ACT TGG GGC TGT TAT CGC AC | +773 | +792 | 20 | 60.5 | |||
| il1 β | Primer set #1 | Forward | CAA CTC CCC TCA CCT TTA ACT AT | −749 | −730 | 20 | 60.5 | 253 |
| Reverse | CTT GCT GGT CCT CGG TGT AT | −516 | −496 | 20 | 60.5 | |||
| Primer set #2 | Forward | TCA TGT CTG ACC TGG CCT CT | −450 | −429 | 22 | 60.1 | 241 | |
| Reverse | TGG GAT ATA GCA AAC TGA GGC | −229 | −210 | 20 | 60.5 | |||
| Primer set #3 | Forward | GCA TTT CTG ACA CTG TAA AAA GAT C | −125 | −103 | 23 | 60.9 | 237 | |
| Reverse | GGG AGT GGA TGG ATG TGC AA | +89 | +111 | 23 | 60.9 | |||
| Primer set #4 | Forward | TTG GGG AGG AAG CGG CAG | +550 | +575 | 26 | 60.1 | 266 | |
| Reverse | ACT TGG GGC TGT TAT CGC AC | +794 | +815 | 22 | 60.1 | |||
| il4 | Primer set #1 | Forward | CAA CTC CCC TCA CCT TTA ACT AT | −748 | −729 | 20 | 60.5 | 251 |
| Reverse | CTT GCT GGT CCT CGG TGT AT | −517 | −498 | 20 | 60.5 | |||
| Primer set #2 | Forward | TCA TGT CTG ACC TGG CCT CT | −449 | −427 | 23 | 60.9 | 236 | |
| Reverse | TGG GAT ATA GCA AAC TGA GGC | −239 | −214 | 26 | 60.1 | |||
| Primer set #3 | Forward | GCA TTT CTG ACA CTG TAA AAA GAT C | −113 | −91 | 23 | 60.9 | 218 | |
| Reverse | GGG AGT GGA TGG ATG TGC AA | +82 | +104 | 23 | 60.9 | |||
| Primer set #4 | Forward | ACA CCC TCG GTC CTG CTC | +151 | +173 | 23 | 60.9 | 278 | |
| Reverse | AGA ACC TGT CTC TGC TTC CC | +404 | +428 | 25 | 60.9 | |||
| Primer set #5 | Forward | TTG GGG AGG AAG CGG CAG | +602 | +624 | 23 | 60.9 | 134 | |
| Reverse | ACT TGG GGC TGT TAT CGC AC | +713 | +735 | 23 | 60.9 |
Cell Cultures
Primary astrocyte cultures were prepared from C57BL/6 mouse brains as described previously [41] and BV-2 cells were cultures as previously described [8]. All procedures involving the use of mice were conducted in accordance with an animal care and protocol approved by the UND ACUC (Protocol # 1004-1). Prior to stimulating the cells (3 hr), the culture media was changed to serum-free media. At this time, plates were divided into 4 different groups; group one was treated with 12 mM sodium chloride (NaCl) as a control group, group two was treated with 12 mM sodium acetate, group three was treated with both 6.25 ng/ml LPS and 12 mM NaCl, and the fourth group was treated with both 6.25 ng/ml LPS and 12 mM sodium acetate (n = 6 per group). The sodium acetate concentration used in this study is the same as that previous reported in vitro, which does not result in cell death over a 24 hr exposure period, maximizes cellular levels of acetyl-CoA, and is controlled using equal concentrations of sodium chloride [8, 9]. After 4 hr of treatment, the media was collected and stored at −20° C, and the cells were lysed in ice cold RIPA lysis buffer (pH = 8.0) containing 150 mM sodium chloride, Triton X-100, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulphate, 50 mM. Samples used for Western blot analysis were aliquoted into 100 μL units and stored at −80° C.
Western blot analysis
The Western blot analysis and protein quantification was performed as described previously [8, 9]. The antibody concentrations used for phosphorylated cPLA2, cPLA2, sPLA2 IIA, PLCβ1, PLCγ1, PLCδ1, Cox-1 and Cox- 2 were 1:250, 1:500, 1:500, 1:250, 1:350, 1:350, 1:1000, and 1:1000, respectively. All Western blot data are expressed as the ratio of the optical density of the respective protein normalized to the optical density of α-tubulin. The phosphorylation ration of cPLA2 was calculated by normalizing the optical density of phosphorylated cPLA2 to the optical density of total cPLA2. Image analysis was performed using a UVP EpiChemi3 Imaging system equipped with VisionWorks LP image acquisition and analysis software (Ver. 6.3.1, Upland, CA)
Chromatin immunoprecipitation
After treating the cells for 4 hr, cross linking was performed using 1% paraformaldehyde at room temperature for 10 min, followed by 0.125 M glycine at room temperature for 5 min. The cells were then washed once with phosphate-buffered saline (PBS, pH = 7.4) containing 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4 prior to adding 0.2% trypsin and incubating at 37° C for 5 min (5% carbon dioxide atmosphere). Trypsin was neutralized with fetal bovine serum. Sample used for chromatin immunoprecipitation were suspended in SDS lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris, pH 8.1) containing protease inhibitor cocktail. Cells in SDS lysis buffer were extracted using sonication then centrifuged at 15,000 × g for 30 min at 4° C to remove cellular debris. Immunoprecipitation was performed for 3 hr at 4° C using 7 μg of acetylated H3K9 antibody, normal rabbit IgG, protein A, and protein G magnetic beads. The magnetic beads were washed 4 times with low-salt wash buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl, pH 8.1, 150 mM NaCl), once with high-salt wash buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl, pH 8.1, 500 mM NaCl), and finally with TE buffer (10 mM Tris-HCl, 1 mM EDTA, pH 8.0). The samples were then eluted off the beads at 65° C for 15 min using elution buffer (50 mM Tris-HCl, 10 mM EDTA, 1% SDS, pH 8.0). Samples were incubated at 65° C overnight to reverse the crosslinks, then incubated with RNAse A (0.2 μg/ml) for 2 hr at 37° C, and with proteinase K (0.2 μg/ml) for 2 hr at 55° C. One μg of the Chromatin immunoprecipitation end product, with 1 μg of forward and reverse primers, 10 μl SYBR green, and 8 μl of nuclease-free water were used for quantitative real-time polymerase chain reaction (qrt-PCR).
Prostaglandin quantification
An enzyme linked immunoassay was used to quantify PGE2 release into the media of BV-2 microglia and primary astrocyte cell cultures. The analysis was performed according the manufacturer's instructions and the final absorbance was measured at 405 nm using a Labsystem MultiSkan Plus plate reader (Fisher Scientific).
Statistical analysis
A one-way Analysis of Variance (ANOVA) followed by Tukey's post-hoc test using GraphPad InStat software (Version 3.06 for Windows, San Diego, CA) was used to calculate statistical significance of the data. All results are expressed as means ± SD with statistical significance set at p ≤ 0.05.
Results
Phospholipase levels in BV-2 microglia
To determine the effect acetate treatment had on phospholipase levels, we measured cPLA2 phosphorylation and the total protein levels of cPLA2, sPLA2 IIA and PLC using Western blot analysis form whole cell BV-2 lysates. Protein bands corresponding to cPLA2, phosphorylated cPLA2, sPLA2 IIA, PLCβ1, PLCγ1, PLCδ1, and -tubulin were detected at the molecular weight of 85, 85, 18, 150, 155, 85, and 50 kDa, respectively (Fig 1A). The protein levels of total cPLA2 were not different between groups (Fig 1B), whereas LPS increased phosphorylated cPLA2 by 1.5-fold which was not altered by acetate treatment (Fig 1C). The protein levels of sPLA2 IIA, PLCγ1 and PLCδ1 were not altered by either LPS or acetate treatment (Fig 1D, F and G). By contrast, LPS decreased PLCβ1 by 2-fold which was reversed to control levels with acetate treatment (Fig 1E).
Figure 1. Phospholipases levels in LPS-stimulated BV-2 cell cultures.

Western blot analysis was performed to show changes in the levels of cPLA2 phosphorylation and total cPLA2, sPLA2 IIA, PLCβ1, PLCγ1 and PLCδ1 protein levels in BV-2 microglial cell cultures stimulated with LPS and/or treated with acetate. Panel A shows representative images of the Western blots. Panels B, D, E, F and G show the optical densities total cPLA2, sPLA2 IIA, PLCβ1, PLCγ1 and PLCδ1 normalized to the loading control α-tubulin. Panel C shows the optical density of phosphorylated cPLA2 normalized to total cPLA2. Bars represent means ± SD where statistical significance was set at p ≤ 0.05. Abbreviations are: a = compared to NaCl-treated group and b = compared to sodium acetate-treated group (n = 6 per group).
Phospholipase levels in primary astrocytes
To determine the effect acetate treatment had on phospholipase levels in LPS-treated astrocytes, we performed Western blot analysis on whole cell lysates as described above. The protein levels of total cPLA2 were not different between groups (Fig 2B), however LPS increased phosphorylated cPLA2 by 2-fold which was returned to control levels by acetate treatment but remained significantly elevated compared to cell treated with acetate alone (Fig 2C). Acetate treatment decreased the protein levels of sPLA2 IIA and PLCβ1 by 20% in the presence of LPS (Fig 2D and E). PLCγ1 and PLCδ1 protein levels were not altered by either LPS or acetate treatment (Fig 2F and G).
Figure 2. Phospholipase levels in LPS-stimulated primary astrocyte cell cultures.

Western blot analysis was performed to show changes in the levels of cPLA2 phosphorylation and total cPLA2, sPLA2 IIA, PLCβ1, PLCγ1 and PLCδ1 protein levels in primary astrocyte cell cultures stimulated with LPS and/or acetate. Panel A shows representative images of the Western blots. Panels B, D, E, F and G show the optical densities total cPLA2, sPLA2 IIA, PLCβ1, PLCγ1 and PLCδ1 normalized to the loading control α-tubulin. Panel C shows the optical density of phosphorylated cPLA2 normalized to total cPLA2. Bars represent means ± SD where statistical significance was set at p ≤ 0.05. Abbreviations are: a = compared to NaCl-treated group, b = compared to sodium acetate-treated group and c = compared to LPS + sodium acetate-treated group (n = 6 per group).
Cox-1 and 2 levels in primary astrocyte and BV-2 microglia cultures
Because cyclooxygenases are the rate-limiting step in the release of eicosanoids, we measured the effect of acetate treatment on Cox-1 and Cox-2 protein levels in BV-2 microglia and primary astrocytes cultures. Using whole cell lysates for Western blot analysis, Cox-1, Cox-2, and -tubulin were detected as protein bands corresponding to molecular weights of 70, 72, and 50 kDa, respectively (Fig 3A). In BV-2 microglia cultures, LPS increased Cox-1 levels by 1.5-fold which was reversed to control levels with acetate treatment (Fig 3B). Cox-2 levels were increased by 4-fold and partially attenuated with acetate treatment (Fig 3C). In astrocyte cultures, LPS increased the protein levels of Cox-1 by 1.5-fold which was reversed to control levels with acetate treatment (Fig 3D). Cox-2 levels were increase in astrocyte cultures treated with LPS by 2.9-fold, but was not altered by acetate treatment (Fig 3E).
Figure 3. Cox-1 and -2 levels in LPS-stimulated BV-2 and primary astrocyte cell cultures.

Panel A shows representative images from a Western blot analysis to show the changes in the levels Cox-1 and Cox-2 in BV-2 microglia and primary astrocyte cell cultures stimulated with LPS and/or treated with acetate. Panels B and C show the optical densities of Cox-1 and Cox 2, respectively normalized to the loading control α-tubulin in BV-2 microglial cell cultures. Panels D and E show the optical densities of Cox-1 and Cox 2, respectively normalized to the loading control α-tubulin in primary astrocyte cell cultures. Bars represent means ± SD where statistical significance was set at p ≤ 0.05. Abbreviations are: a = compared to NaCl-treated group, b = compared to sodium acetate-treated group and c = compared to LPS + sodium acetate-treated group (n = 6 per group).
Prostaglandin E2 levels in BV-2 microglia and primary astrocyte cultures
Having demonstrated the ability of acetate treatment to alter cPLA2 phosphorylation, sPLA2 IIA, Cox-1 and Cox-2 protein levels, we proceeded to determine the effect acetate treatment had PGE2 release using an enzyme immunoassay. In BV-2 microglia, PGE2 levels were not altered by either LPS or acetate treatment (Fig 4A) despite significant changes in Cox-1 and Cox-2 (Fig 3, B and C). On the other hand, LPS increased PGE2 levels by 4-fold in astrocyte cultures which was completely reversed to control levels with acetate treatment (Fig 4B).
Figure 4. Prostaglandin E2 levels released from LPS-stimulated BV-2 microglia and primary astrocyte cultures.
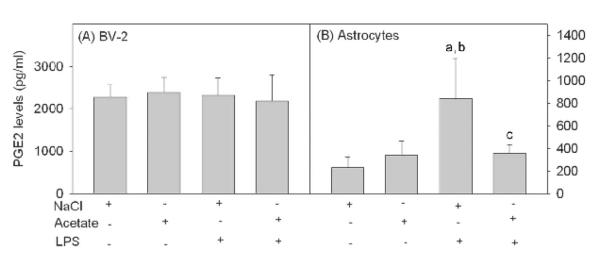
Media levels of secreted PGE2 from BV-2 microglia (panel A) and primary astrocyte cell cultures (panel B) stimulated with LPS and/or acetate as determined using an enzyme-linked immunoassay. Bars represent means ± SD where statistical significance was set at p ≤ 0.05. Abbreviations are: a = compared to NaCl-treated group, b = compared to sodium acetate-treated group and c = compared to LPS + sodium acetate-treated group (n = 6 per group).
Binding of acetylated H3K9 to promoter regions of genes involved in inflammation
Acetate treatment increases H3K9 acetylation in vivo [6, 7] and reverses LPS-induced increase in NF-κB p65 protein but not IL-1β mRNA, and increases IL-4 expression in vitro [8, 9]. Because changes in histone acetylation are associated with alterations in gene expression [42, 43], we measured the enrichment of acetylated H3K9 to promoters of ptgs1, ptgs2 (coding for Cox-1 and Cox-2, respectively), p65, il4 (Il-4) and il1b (IL-1β) genes in BV-2 microglia treated with acetate. Five different primer sets were prepared for each gene with the exception of il1b where 4 primer sets were used. The primers were designed to span different genomic stretches ranging between −750 base pairs and +1000 base pairs in relation to the transcription start site (Table 1). Acetate treatment increased acetylated H3K9 bound to three COX-1 gene sequences: between −393 and −157, −129 and +112, and +668 and +869 (Fig 5A) and one COX-2 gene sequence: between +750 and +971 (Fig 5B). In addition, acetate treatment was found to increase acetylated H3K9 bound to three p65 gene sequences: between −743 and −557, between −518 and −336, and between +146 and +272 (Fig 5C), and three IL-1β gene sequences: between −450 and −229, between −125 and +89, and between +550 and +794 (Fig 5E). Acetate treatment did not alter the binding of acetylated H3K9 to the IL-4 promoter region (Fig 5D). LPS treatment reduced the levels of acetylated H3K9 bound to the promoter region of COX-1 between −393 and −157 as compared to sodium chloride controls. The levels of acetylated H3K9 bound to promoter were also reduced in regions 2 and 3 (COX-1), region 5 (COX-2), regions 1 and 2 (p65), and regions 2, 3, and 4 (IL-1β) compared to cells treated with sodium acetate (Fig 5A. 5B, 5C, and 5E, respectively).
Figure 5. Effect of H3K9 acetylation on binding to transcription start sites of inflammatory genes in BV-2 cultures.

Binding levels of acetylated H3K9 to regions corresponding to ptgs1 (panel A), ptgs2 (panel B), p65 (panel C), il4 (panel D), and il1b (panel E) genes measured using Chromatin immunoprecipitation analysis followed by qrt-PCR. Bars represent means ± SD where statistical significance was set at p ≤ 0.05. Abbreviations are: a = compared to NaCl-treated group, b = compared to sodium acetate-treated group and c = compared to LPS + sodium acetate-treated group (n = 6 per group).
Discussion
In LPS-stimulated BV-2 microglia [8] and primary astrocyte cultures [9], acetate treatment decreases LPS-induced pro-inflammatory cytokines, MAPK signaling and NF-KB p65 protein level and phosphorylation in neuroglial cell type-distinct patterns. Because phospholipase activation and eicosanoids are involved in neuroinflammation that is modulated by inflammatory cytokines and MAPK and NF-κB signaling [22, 23, 44, 45] and is reduced by acetate treatment [8, 9], we tested whether acetate can alter the levels phospholipases, Cox, and PGE2 release in BV-2 microglia and astrocyte cultures. Further, because H3K9 acetylation alters gene expression [42, 43], and acetate treatment induces global histone hyperacetylation [7, 8], we measured the enrichment levels of acetylated H3K9 bound to the promoters of the genes coding for Cox-1, Cox-2, IL-1β, IL-4 and NF-κB p65 in BV-2 microglia. These data suggest that acetate treatment can disrupt eicosanoid signaling in astrocytes, and alters the levels of enzymes involved in eicosanoid release in microglia. The neuroglial cell type-distinct effects of acetate on eicosanoid signaling, and possible chromatin remodeling, may contribute to the distinct anti-inflammatory response found with acetate supplementation both in vivo [3, 4] and in vitro [8, 9].
The findings that acetate treatment can modulate eicosanoid signaling are important because of the involvement of eicosanoids in a number of diseases. Prostaglandins are a class of eicosanoids that are formed downstream to the release of arachidonic acid. PGE2 release and cPLA2 and Cox expression are increased after LPS and IL-1β injection, aging and brain trauma [44, 46], Alzheimer's [47], and Parkinson's [48] diseases, among many other neuroinflammatory and degenerative conditions [49]. Pharmacological inhibition of cPLA2, cyclooxygenases and PGE2 release is associated with anti-inflammatory and neuroprotective properties and improved cognitive functions in aging [50], and animal models of Alzheimer's disease [51], traumatic brain injury [52], ischemia [53] and amyotrophic lateral sclerosis [54]. Drugs like corticosteroids and non-steroidal anti-inflammatory drugs (NSAIDs) target primarily the enzymes involved in eicosanoids release. The potential of acetate treatment to disrupt eicosanoid signaling is promising given the possibility of acetate selectively disrupts this pathway only in certain cell types, unlike global cPLA2 and Cox inhibitors that deprive many cell types from the physiological functions of eicosanoid signaling. In this regard, it will be interesting to evaluate whether acetate treatment reduces the side effects associated with NSAIDs like hyperacidity and alteration of blood coagulation, and the side effects associated with glucocorticoids like compromising immunity, blood sugar and blood pressure regulation. Acetate treatment may potentially prove to be more effective compared to current NSAID therapy given its additive effect on other signaling pathways like MAPK, NF-κB and inflammatory cytokines [8, 9].
Acetate supplementation increases the acetylation-state of brain H3K9, H4K8 and H4K16, but not H3K14, H4K5 or H4K16 in normal rats [6] and is similar in a rat model of neuroinflammation [7]. LPS reduces acetylated H3K9 by 2-fold and is reversed with acetate supplementation to a hyperacetylated-state in vivo [7] which is reproducible in BV-2 microglia in vivo [8]. Further, acetate increases H3K9 acetylation in astrocytes, but LPS itself does not reduce acetylated H3K9 in this cell type as found in vivo [9]. Because H3K9 acetylation is implicated in neuroinflammation and neuroglial activation [34, 55, 56], we chose to focus only on H3K9 in this study. For these reasons chromatin immunoprecipitation was performed in BV-2 microglia cultures, where H3K9 acetylation is altered by both LPS and acetate treatment. This study demonstrates the effect of acetate treatment on the protein levels of Cox-1 and 2 which were increased with LPS challenge and reduced upon acetate treatment. Acetate treatment also reverses LPS-induced IL-1β and NF-κB p65 protein levels, and upregulates IL-4 expression in both LPS-stimulated BV-2 microglia [8] and primary astrocyte cultures [9]. In an attempt to determine whether acetate-induced H3K9 hyperacetylation is involved in the regulation of these inflammatory mediators at the gene levels, we measured the binding of acetylated H3K9 to the promoter regions of these genes of interest. Chromatin immunoprecipitation analysis showed that acetate treatment increased the binding of acetylated H3K9 to the promoters of each of these genes with the exception of IL-4, suggesting that acetate-induced H3K9 acetylation may potentially modulate the expression of these specific genes. Acetylated H3K9 is conventionally associated with enhanced gene expression [42, 43]. Of the five genes analyzed using chromatin immunoprecipitation, acetate increases IL-4 transcription, did not alter IL-1β mRNA, and decreases NF-κB p65, Cox-1 and 2 protein levels, while acetylated H3K9 bound to the promoter regions of each of these genes except IL-4 was increased. Because acetate treatment induces histone acetylation changes other than H3K9; including H4K8 or H4K16 [6, 7], warrants more experiments to evaluate the involvement of other histones in mediating changes in pro-inflammatory gene expression. Alternatively, acetate treatment-mediated gene expression changes can possibly be linked to acetylation of non-histone transcription factors disrupting in parallel other inflammatory signaling pathways [8].
Of interest, acetate reduces sPLA2 IIA and LPS-induced cPLA2 phosphorylation in astrocytes and not BV-2 microglia. By contrast, acetate treatment attenuates LPS-induced Cox-2 levels in BV-2 microglia and not in astrocytes. Because cPLA2 is a substrate for phosphorylation by MAPK p38 [22, 23] and ERK [22], both of which are inhibited by acetate treatment in astrocytes and not BV-2 microglia, suggests that acetate treatment reverses cPLA2 phosphorylation primarily by reducing MAPK p38 and ERK activity. This is supported by studies showing that pro-inflammatory cytokines IL-1β and TNF-α increase cPLA2 phosphorylation and sPLA2 IIA expression and activity [45, 57, 58] that is reduced with the administration of TNF-α antibodies and IL-1 receptor antagonists [21]. Similarly, TNF-α enhances Cox-2 promoter activity [59]. It is therefore possible that acetate treatment may reduce cPLA2 phosphorylation, sPLA2 IIA and Cox-2 expression by reducing LPS-induced TNF-α and IL-1β expression. Although generally plausible, this does not explain why acetate treatment did not reduce cPLA2 phosphorylation in BV-2 microglia and Cox-2 levels in astrocytes found in this study.
Distinct neuroglial cell type responses to acetate treatment include the complete reversal of LPS-induced astrocyte activation in vivo, while only partially attenuating microglia activation [3]. In LPS-stimulated BV-2 microglia, acetate treatment transiently reduces LPS-induced MAPK p38 and JNK phosphorylation and increases ERK1/2 phosphorylation [8]. By contrast, in LPS-stimulated primary astrocyte cultures, acetate treatment completely reduces MAPK p38 activation and decreases basal levels of phosphorylated ERK1/2 [9]. These data suggest that the complete inhibition of cPLA2 phosphorylation and PGE2 release in the astrocyte but not in BV-2 microglia may be involved in the attenuation of reactive astrogliosis and partial attenuation of microglia activation found in vivo [3]. Thus it will be important to determine the role that a reduction in astrocytes derived prostaglandin release has in disrupting neuroglia communication both in vivo and in mixed microglia and astrocytes cultures in vitro.
In conclusion, acetate treatment can disrupt eicosanoid signaling in LPS-stimulated primary astrocyte, and alters the levels of enzymes involved in eicosanoids release in microglia. Acetate treatment also induces increases in the binding of acetylated H3K9 to the promoters of certain inflammatory genes which may potentially be involved in chromatin remodeling and changes in inflammatory gene expression.
Acknowledgements
This work was funded by grants from the National Institutes of Health (2P20RR17699 and P30GM103329). We would like to thank Kendra L. Puig and Dr. Colin K. Combs (University of North Dakota School of Medicine and Health Sciences) for providing primary astrocytes and BV-2 microglia.
Abbreviations
- PLA2
phospholipases A2
- cPLA2
cytosolic phospholipases A2
- sPLA2
secretory phospholipases A2
- Cox-1
cyclooxygenase-1
- Cox-2
cyclooxygenase-2
- IL-1β
interleukin-1beta
- IL-6
interleukin-6
- IL-4
interleukin-4
- TNF-α
tumor necrosis factor-alpha
- TGF-β1
transforming growth factor-beta 1
- MAPK
mitogen-activated protein kinases
- JNK
c-Jun N-terminal kinase
- ERK
extracellular signal-regulated kinase
- NF-κB
nuclear factor-kappa B
- H3K9
histone H3 at lysine 9
Footnotes
Conflict of interest: The authors declare that they have no conflict of interest.
References
- 1.Arun P, Madhavarao CN, Moffett JR, Hamilton K, Grunberg NE, Ariyannur PS, Gahl WA, Anikster Y, Mog S, Hallows WC, Denu JM, Namboodiri AM. Metabolic acetate therapy improves phenotype in the tremor rat model of Canavan disease. J Inherit Metab Dis. 2010;33:195–210. doi: 10.1007/s10545-010-9100-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arun P, Ariyannur PS, Moffett JR, Xing G, Hamilton K, Grunberg NE, Ives JA, Namboodiri AM. Metabolic acetate therapy for the treatment of traumatic brain injury. J Neurotrauma. 2010;27:293–298. doi: 10.1089/neu.2009.0994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reisenauer CJ, Bhatt DP, Mitteness DJ, Slanczka ER, Gienger HM, Watt JA, Rosenberger TA. Acetate supplementation attenuates lipopolysaccharide-induced neuroinflammation. J Neurochem. 2011;117:264–274. doi: 10.1111/j.1471-4159.2011.07198.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brissette CA, Houdek HM, Floden AM, Rosenberger TA. Acetate supplementation reduces microglia activation and brain interleukin-1beta levels in a rat model of Lyme neuroborreliosis. J Neuroinflammation. 2012;9:249. doi: 10.1186/1742-2094-9-249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bhatt DP, Houdek HM, Watt JA, Rosenberger TA. Acetate supplementation increases brain phosphocreatine and reduces AMP levels with no effect on mitochondrial biogenesis. Neurochem Int. 2013;62:296–305. doi: 10.1016/j.neuint.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Soliman ML, Rosenberger TA. Acetate supplementation increases brain histone acetylation and inhibits histone deacetylase activity and expression. Mol Cell Biochem. 2011;352:173–180. doi: 10.1007/s11010-011-0751-3. [DOI] [PubMed] [Google Scholar]
- 7.Soliman ML, Smith MD, Houdek HM, Rosenberger TA. Acetate supplementation modulates brain histone acetylation and decreases interleukin-1beta expression in a rat model of neuroinflammation. J Neuroinflammation. 2012;9:51. doi: 10.1186/1742-2094-9-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Soliman ML, Puig KL, Combs CK, Rosenberger TA. Acetate reduces microglia inflammatory signaling in vitro. J Neurochem. 2012;123:555–567. doi: 10.1111/j.1471-4159.2012.07955.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Soliman ML, Combs CK, Rosenberger TA. Modulation of Inflammatory Cytokines and Mitogen-activated Protein Kinases by Acetate in Primary Astrocytes. J Neuroimmune Pharmacol. 2013;8:287–300. doi: 10.1007/s11481-012-9426-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lima IV, Bastos LF, Limborco-Filho M, Fiebich BL, de Oliveira AC. Role of prostaglandins in neuroinflammatory and neurodegenerative diseases. Mediators Inflamm. 2012;2012:946813. doi: 10.1155/2012/946813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Phillis JW, O'Regan MH. A potentially critical role of phospholipases in central nervous system ischemic, traumatic, and neurodegenerative disorders. Brain Res Brain Res Rev. 2004;44:13–47. doi: 10.1016/j.brainresrev.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 12.Farooqui AA, Yang HC, Rosenberger TA, Horrocks LA. Phospholipase A2 and its role in brain tissue. J Neurochem. 1997;69:889–901. doi: 10.1046/j.1471-4159.1997.69030889.x. [DOI] [PubMed] [Google Scholar]
- 13.Dennis EA. Diversity of group types, regulation, and function of phospholipase A2. J Biol Chem. 1994;269:13057–13060. [PubMed] [Google Scholar]
- 14.Balboa MA, Varela-Nieto I, Killermann Lucas K, Dennis EA. Expression and function of phospholipase A2 in brain. FEBS Lett. 2002;531:12–17. doi: 10.1016/s0014-5793(02)03481-6. [DOI] [PubMed] [Google Scholar]
- 15.Smith WL, DeWitt DL, Garavito RM. Cyclooxygenases: structural, cellular, and molecular biology. Annu Rev Biochem. 2000;69:145–182. doi: 10.1146/annurev.biochem.69.1.145. [DOI] [PubMed] [Google Scholar]
- 16.Sun GY, Shelat PB, Jensen MB, He Y, Sun AY, Simonyi A. Phospholipases A2 and inflammatory responses in the central nervous system. Neuromolecular Med. 2010;12:133–148. doi: 10.1007/s12017-009-8092-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fujimori Y, Murakami M, Kim DK, Hara S, Takayama K, Kudo I, Inoue K. Immunochemical detection of arachidonoyl-preferential phospholipase A2. J Biochem. 1992;111:54–60. doi: 10.1093/oxfordjournals.jbchem.a123718. [DOI] [PubMed] [Google Scholar]
- 18.Lauritzen I, Heurteaux C, Lazdunski M. Expression of group II phospholipase A2 in rat brain after severe forebrain ischemia and in endotoxic shock. Brain Res. 1994;651:353–356. doi: 10.1016/0006-8993(94)90719-6. [DOI] [PubMed] [Google Scholar]
- 19.Farooqui AA, Horrocks LA. Signaling and interplay mediated by phospholipases A2, C, and D in LA-N-1 cell nuclei. Reprod Nutr Dev. 2005;45:613–631. doi: 10.1051/rnd:2005049. [DOI] [PubMed] [Google Scholar]
- 20.Grinberg S, Hasko G, Wu D, Leibovich SJ. Suppression of PLCbeta2 by endotoxin plays a role in the adenosine A(2A) receptor-mediated switch of macrophages from an inflammatory to an angiogenic phenotype. Am J Pathol. 2009;175:2439–2453. doi: 10.2353/ajpath.2009.090290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Adibhatla RM, Hatcher JF. Secretory phospholipase A2 IIA is up-regulated by TNF-alpha and IL-1alpha/beta after transient focal cerebral ischemia in rat. Brain Res. 2007;1134:199–205. doi: 10.1016/j.brainres.2006.11.080. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 22.Hiller G, Sundler R. Activation of arachidonate release and cytosolic phospholipase A2 via extracellular signal-regulated kinase and p38 mitogen-activated protein kinase in macrophages stimulated by bacteria or zymosan. Cell Signal. 1999;11:863–869. doi: 10.1016/s0898-6568(99)00058-3. [DOI] [PubMed] [Google Scholar]
- 23.Kramer RM, Roberts EF, Um SL, Borsch-Haubold AG, Watson SP, Fisher MJ, Jakubowski JA. p38 mitogen-activated protein kinase phosphorylates cytosolic phospholipase A2 (cPLA2) in thrombin-stimulated platelets. Evidence that proline-directed phosphorylation is not required for mobilization of arachidonic acid by cPLA2. J Biol Chem. 1996;271:27723–27729. doi: 10.1074/jbc.271.44.27723. [DOI] [PubMed] [Google Scholar]
- 24.Gorisch SM, Wachsmuth M, Toth KF, Lichter P, Rippe K. Histone acetylation increases chromatin accessibility. J Cell Sci. 2005;118:5825–5834. doi: 10.1242/jcs.02689. [DOI] [PubMed] [Google Scholar]
- 25.Anderson JD, Lowary PT, Widom J. Effects of histone acetylation on the equilibrium accessibility of nucleosomal DNA target sites. J Mol Biol. 2001;307:977–985. doi: 10.1006/jmbi.2001.4528. [DOI] [PubMed] [Google Scholar]
- 26.Polach KJ, Lowary PT, Widom J. Effects of core histone tail domains on the equilibrium constants for dynamic DNA site accessibility in nucleosomes. J Mol Biol. 2000;298:211–223. doi: 10.1006/jmbi.2000.3644. [DOI] [PubMed] [Google Scholar]
- 27.Krajewski WA, Becker PB. Reconstitution of hyperacetylated, DNase I-sensitive chromatin characterized by high conformational flexibility of nucleosomal DNA. Proc Natl Acad Sci U S A. 1998;95:1540–1545. doi: 10.1073/pnas.95.4.1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mu C, Liu H, Zheng GC. The modification and variants of histone. Mol Biol (Mosk) 2007;41:395–407. [PubMed] [Google Scholar]
- 29.Eberharter A, Becker PB. Histone acetylation: a switch between repressive and permissive chromatin. Second in review series on chromatin dynamics. EMBO Rep. 2002;3:224–229. doi: 10.1093/embo-reports/kvf053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Glozak MA, Sengupta N, Zhang X, Seto E. Acetylation and deacetylation of non-histone proteins. Gene. 2005;363:15–23. doi: 10.1016/j.gene.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 31.Polevoda B, Sherman F. The diversity of acetylated proteins. Genome Biol. 2002;3:6.1–6.6. doi: 10.1186/gb-2002-3-5-reviews0006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim HJ, Rowe M, Ren M, Hong JS, Chen PS, Chuang DM. Histone deacetylase inhibitors exhibit anti-inflammatory and neuroprotective effects in a rat permanent ischemic model of stroke: multiple mechanisms of action. J Pharmacol Exp Ther. 2007;321:892–901. doi: 10.1124/jpet.107.120188. [DOI] [PubMed] [Google Scholar]
- 33.Rouaux C, Panteleeva I, Rene F, Gonzalez de Aguilar JL, Echaniz-Laguna A, Dupuis L, Menger Y, Boutillier AL, Loeffler JP. Sodium valproate exerts neuroprotective effects in vivo through CREB-binding protein-dependent mechanisms but does not improve survival in an amyotrophic lateral sclerosis mouse model. J Neurosci. 2007;27:5535–5545. doi: 10.1523/JNEUROSCI.1139-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang B, West EJ, Van KC, Gurkoff GG, Zhou J, Zhang XM, Kozikowski AP, Lyeth BG. HDAC inhibitor increases histone H3 acetylation and reduces microglia inflammatory response following traumatic brain injury in rats. Brain Res. 2008;1226:181–191. doi: 10.1016/j.brainres.2008.05.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ryu H, Lee J, Olofsson BA, Mwidau A, Dedeoglu A, Escudero M, Flemington E, Azizkhan-Clifford J, Ferrante RJ, Ratan RR. Histone deacetylase inhibitors prevent oxidative neuronal death independent of expanded polyglutamine repeats via an Sp1-dependent pathway. Proc Natl Acad Sci U S A. 2003;100:4281–4286. doi: 10.1073/pnas.0737363100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McCampbell A, Taye AA, Whitty L, Penney E, Steffan JS, Fischbeck KH. Histone deacetylase inhibitors reduce polyglutamine toxicity. Proc Natl Acad Sci U S A. 2001;98:15179–15184. doi: 10.1073/pnas.261400698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Steffan JS, Bodai L, Pallos J, Poelman M, McCampbell A, Apostol BL, Kazantsev A, Schmidt E, Zhu YZ, Greenwald M, Kurokawa R, Housman DE, Jackson GR, Marsh JL, Thompson LM. Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature. 2001;413:739–743. doi: 10.1038/35099568. [DOI] [PubMed] [Google Scholar]
- 38.Langley B, Gensert JM, Beal MF, Ratan RR. Remodeling chromatin and stress resistance in the central nervous system: histone deacetylase inhibitors as novel and broadly effective neuroprotective agents. Curr Drug Targets CNS Neurol Disord. 2005;4:41–50. doi: 10.2174/1568007053005091. [DOI] [PubMed] [Google Scholar]
- 39.Blanchard F, Chipoy C. Histone deacetylase inhibitors: new drugs for the treatment of inflammatory diseases? Drug Discov Today. 2005;10:197–204. doi: 10.1016/S1359-6446(04)03309-4. [DOI] [PubMed] [Google Scholar]
- 40.Adcock IM. HDAC inhibitors as anti-inflammatory agents. Br J Pharmacol. 2007;150:829–831. doi: 10.1038/sj.bjp.0707166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dhawan G, Floden AM, Combs CK. Amyloid-beta oligomers stimulate microglia through a tyrosine kinase dependent mechanism. Neurobiol Aging. 2012;33:2247–2261. doi: 10.1016/j.neurobiolaging.2011.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 43.Rice JC, Allis CD. Histone methylation versus histone acetylation: new insights into epigenetic regulation. Curr Opin Cell Biol. 2001;13:263–273. doi: 10.1016/s0955-0674(00)00208-8. [DOI] [PubMed] [Google Scholar]
- 44.Hein AM, Stutzman DL, Bland ST, Barrientos RM, Watkins LR, Rudy JW, Maier SF. Prostaglandins are necessary and sufficient to induce contextual fear learning impairments after interleukin-1 beta injections into the dorsal hippocampus. Neuroscience. 2007;150:754–763. doi: 10.1016/j.neuroscience.2007.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sun GY, Xu J, Jensen MD, Yu S, Wood WG, Gonzalez FA, Simonyi A, Sun AY, Weisman GA. Phospholipase A2 in astrocytes: responses to oxidative stress, inflammation, and G protein-coupled receptor agonists. Mol Neurobiol. 2005;31:27–41. doi: 10.1385/MN:31:1-3:027. [DOI] [PubMed] [Google Scholar]
- 46.Quan N, Whiteside M, Herkenham M. Cyclooxygenase 2 mRNA expression in rat brain after peripheral injection of lipopolysaccharide. Brain Res. 1998;802:189–197. doi: 10.1016/s0006-8993(98)00402-8. [DOI] [PubMed] [Google Scholar]
- 47.Yermakova A, O'Banion MK. Cyclooxygenases in the central nervous system: implications for treatment of neurological disorders. Curr Pharm Des. 2000;6:1755–1776. doi: 10.2174/1381612003398672. [DOI] [PubMed] [Google Scholar]
- 48.Teismann P, Tieu K, Choi DK, Wu DC, Naini A, Hunot S, Vila M, Jackson-Lewis V, Przedborski S. Cyclooxygenase-2 is instrumental in Parkinson's disease neurodegeneration. Proc Natl Acad Sci U S A. 2003;100:5473–5478. doi: 10.1073/pnas.0837397100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hein AM, O'Banion MK. Neuroinflammation and memory: the role of prostaglandins. Mol Neurobiol. 2009;40:15–32. doi: 10.1007/s12035-009-8066-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Casolini P, Catalani A, Zuena AR, Angelucci L. Inhibition of COX-2 reduces the age-dependent increase of hippocampal inflammatory markers, corticosterone secretion, and behavioral impairments in the rat. J Neurosci Res. 2002;68:337–343. doi: 10.1002/jnr.10192. [DOI] [PubMed] [Google Scholar]
- 51.Kotilinek LA, Westerman MA, Wang Q, Panizzon K, Lim GP, Simonyi A, Lesne S, Falinska A, Younkin LH, Younkin SG, Rowan M, Cleary J, Wallis RA, Sun GY, Cole G, Frautschy S, Anwyl R, Ashe KH. Cyclooxygenase-2 inhibition improves amyloid-beta-mediated suppression of memory and synaptic plasticity. Brain. 2008;131:651–664. doi: 10.1093/brain/awn008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gopez JJ, Yue H, Vasudevan R, Malik AS, Fogelsanger LN, Lewis S, Panikashvili D, Shohami E, Jansen SA, Narayan RK, Strauss KI. Cyclooxygenase-2-specific inhibitor improves functional outcomes, provides neuroprotection, and reduces inflammation in a rat model of traumatic brain injury. Neurosurgery. 2005;56:590–604. doi: 10.1227/01.NEU.0000154060.14900.8F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Iadecola C, Niwa K, Nogawa S, Zhao X, Nagayama M, Araki E, Morham S, Ross ME. Reduced susceptibility to ischemic brain injury and N-methyl-D-aspartate-mediated neurotoxicity in cyclooxygenase-2-deficient mice. Proc Natl Acad Sci U S A. 2001;98:1294–1299. doi: 10.1073/pnas.98.3.1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Klivenyi P, Kiaei M, Gardian G, Calingasan NY, Beal MF. Additive neuroprotective effects of creatine and cyclooxygenase 2 inhibitors in a transgenic mouse model of amyotrophic lateral sclerosis. J Neurochem. 2004;88:576–582. doi: 10.1046/j.1471-4159.2003.02160.x. [DOI] [PubMed] [Google Scholar]
- 55.Silva PF, Garcia VA, Dornelles Ada S, Silva VK, Maurmann N, Portal BC, Ferreira RD, Piazza FC, Roesler R, Schroder N. Memory impairment induced by brain iron overload is accompanied by reduced H3K9 acetylation and ameliorated by sodium butyrate. Neuroscience. 2012;200:42–49. doi: 10.1016/j.neuroscience.2011.10.038. [DOI] [PubMed] [Google Scholar]
- 56.Govindarajan N, Agis-Balboa RC, Walter J, Sananbenesi F, Fischer A. Sodium butyrate improves memory function in an Alzheimer's disease mouse model when administered at an advanced stage of disease progression. J Alzheimers Dis. 2011;26:187–197. doi: 10.3233/JAD-2011-110080. [DOI] [PubMed] [Google Scholar]
- 57.Anthonsen MW, Solhaug A, Johansen B. Functional coupling between secretory and cytosolic phospholipase A2 modulates tumor necrosis factor-alpha- and interleukin-1beta-induced NF-kappa B activation. J Biol Chem. 2001;276:30527–30536. doi: 10.1074/jbc.M008481200. [DOI] [PubMed] [Google Scholar]
- 58.Jupp OJ, Vandenabeele P, MacEwan DJ. Distinct regulation of cytosolic phospholipase A2 phosphorylation, translocation, proteolysis and activation by tumour necrosis factor-receptor subtypes. Biochem J. 2003;374:453–461. doi: 10.1042/BJ20030705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Deng WG, Zhu Y, Wu KK. Up-regulation of p300 binding and p50 acetylation in tumor necrosis factor-alpha-induced cyclooxygenase-2 promoter activation. J Biol Chem. 2003;278:4770–4777. doi: 10.1074/jbc.M209286200. [DOI] [PubMed] [Google Scholar]