

The enteric nervous system is the part of the autonomic nervous system that directly controls the gastrointestinal tract. Derived from a multipotent, migratory cell population called the neural crest, a complete enteric nervous system is necessary for proper gut function. Disorders that arise as a consequence of defective neural crest cell development are termed neurocristopathies. One such disorder is Hirschsprung disease, also known as congenital megacolon or intestinal aganglionosis. Hirschsprung disease occurs in 1/5000 live births, and typically presents with the inability to pass meconium, along with abdominal distension and discomfort that usually requires surgical resection of the aganglionic bowel. This disorder is characterized by a congenital absence of neurons in a portion of the intestinal tract, usually the distal colon, due to a disruption of normal neural crest cell migration, proliferation, differentiation, survival and/or apoptosis. The inheritance of Hirschsprung disease is complex, often non-Mendelian, and characterized by variable penetrance. Extensive research has identified a number of key genes that regulate neural crest cell development in the pathogenesis of Hirschsprung disease including RET, GDNF, GFRα1, NRTN, EDNRB, ET3, ZFHX1B, PHOX2b, SOX10, and SHH. However, mutations in these genes account for only ~50% of the known cases of HSCR. Thus, other genetic mutations and combinations of genetic mutations and modifiers likely contribute to the etiology and pathogenesis of HSCR. The aims of this review are to summarize the HSCR phenotype, diagnosis and treatment options; to discuss the major genetic causes and the mechanisms by which they disrupt normal ENCC development; and to explore new pathways that may contribute to HSCR pathogenesis.
Gastrointestinal Tract, Enteric Nervous System and Hirschsprung Disease
The gastrointestinal (GI) tract is an endoderm derived organ system that begins at the mouth and terminates at the anus. The fetal GI tract is divided into three segments based on vascular supply. The foregut, supplied by the celiac artery, consists of the esophagus, stomach, part of duodenum, and biliary apparatus. The midgut, supplied by the superior mesenteric artery, comprises the rest of small and large bowel up to the splenic flexure. Lastly, the hindgut consists of the remainder of the large bowel to the superior part of anal canal, and is supplied by the inferior mesenteric artery. This organ system functions to digest and process foods and liquids taken in through the mouth via two types of digestion: mechanical, such as chewing and gut peristalsis, and chemical, such as enzymatic breakdown. The GI tract also plays a role as a major part of the immune system via the recognition of and response to introduced pathogens.
The ability of the GI tract to respond to the state of the lumen and gut wall by activating peristalsis, controlling blood flow and secretions and thus maintain proper physiological balance depends on the enteric nervous system (ENS) [1]. The ENS is the largest part of the peripheral nervous system and functions almost independently of the central nervous system [2] and is in direct control of the GI system [3]. ENS neurons and glia are organized into ganglia. The enteric ganglia are interconnected to form two plexi that extend along the length of the bowel: an outer myenteric (Auerbach) plexus running the full length of the gut, and an inner submucosal (Meissner) plexus, found only in the small and large intestine. The myenteric plexus develops first and is situated between the longitudinal and circular smooth muscle layers, and is involved in motility, while the submucosal plexus, which forms later in gestation, regulates motility, blood flow, and the transport of ions across the intestinal epithelium.
Gut motility is controlled by interdependent mechanisms including neural, such as the enteric ganglia, and nonneural, such as the interstitial cells of Cajal (ICC)[4, 5]. Interstitial cells of Cajal serve as pacemaker cells creating and propagating slow waves that lead to smooth muscle contraction in the gut [4]. Here, we focus on the necessity of the ENS to form a completely colonized gut that can maintain peristaltic activity of the gut wall and proper gut function. The absence of enteric ganglion cells of the myenteric and submucosal plexi along variable portions of the GI tract results in Hirschsprung disease (HSCR) [6], which is characterized by sustained contraction of the aganglionic bowel segment, leading to intestinal obstruction and distension of proximal segments (megacolon). The ENS is derived from a multipotent, migratory cell population called the neural crest, and the formation of a functional ENS requires coordination of the survival, migration, proliferation, and differentiation of these progenitor cells within the GI tract. Events that disrupt these processes can lead to HSCR. For example, a delay or arrest of NCC migration can result in the failure of ENCCs to reach their correct distal intestinal position [7]. Alternatively, NCCs can fail to survive, proliferate or differentiate after migration has occurred, due to abnormalities in the microenvironment [8–12].
Disorders such as HSCR that arise as a consequence of defective NCC development are termed neurocristopathies. HSCR is a particularly devastating neurocristopathy disorder that usually presents with the inability to pass meconium, together with abdominal distension and discomfort that usually necessitates surgical resection of the aganglionic bowel [13].
Clinical presentation
Histologically, aganglionosis is pathognomonic for HSCR. In 80–85% of HSCR cases, the aganglionotic region is limited to the rectum and sigmoid colon (short-segment disease). Long segment disease occurs in up to 20% of cases, and is characterized by aganglionosis extending proximally to the sigmoid colon. Total colonic agangliononsis is more rare, occurring in 3–8% of patients with HSCR [14]. Another rare variant is ultra-short segment disease, affecting only the distal rectum (because this variant has such a short aganglionic zone, ganglion cells may be present in the biopsy)[15]. The portion of the bowel adjacent to the aganglionotic region with reduced enteric neuron density is termed the transition zone, and it is always cranial to the aganglionic region [16]. Hypoganglionosis and aganglionosis of the terminal gut can be caused by a reduction in enteric progenitor cells [17, 18]. The zone of aganglionosis results in tonic contraction of the affected bowel, leading to obstructive symptoms. Most often, patients are diagnosed in the neonatal period [19], presenting with a distended abdomen, the delayed passage of meconium (>24 hours), and vomiting. When patients are diagnosed later in childhood, they often have short-segment aganglionosis and present with chronic constipation and distension, vomiting and failure to thrive [20]. In 10% of HSCR cases, patients can present with enterocolitis, fever, and even septicemia.
HSCR occurs in 1/5000 live births and has an overall 4:1 male predominance [21]. However, for short segment disease there is a 4.2–4.4 male: female predominance and for long-segment disease there is a 1.2–1.9 male: female predominance [22]. Furthermore, the risk of an HSCR sibling recurrence is 200 times higher than in the general population (4% versus 0.02%) [22, 23]. Up to 30% of patients with HSCR also exhibit other abnormalities [21, 24], such as velocardiofacial defects [25], congenital heart defects [26], gastrointestinal tract malformations, CNS abnormalities, genitourinary problems, craniofacial malformations, and spina bifida [22, 26–29]. In addition, 2–15% of HSCR cases are associated with Down’s syndrome [27, 30].
Diagnosis
The diagnosis of HSCR can be made by a variety of methods. However, the preferred first diagnostic procedure is a contrast enema. This will define the transition zone between normal (dilated) bowel and the narrow aganglionic bowel. This transition zone is seen in 70–90% of cases [31, 32]. The rectosigmoid ratio is used to evaluate the transition zone. A rectosigmoid ratio greater than 1 is normal. A stool-filled proximal bowel will decrease the rectum to sigmoid ratio. Plain radiographs show dilated bowel loops, and anorectal manometry may also help with the diagnosis. When using anorectal manometry, clinicians note an absent rectoanal inhibitory reflex [33]. This absence of internal sphincter relaxation [34] is only a reliable test after neonatal day 12, when the rectoenteric reflex is present [35].
The gold standard for an HSCR diagnosis is a rectal biopsy. It is possible to obtain a submucosal rectal suction biopsy without anesthesia [36]. Analysis of the biopsy specimen is performed, to look for an absence of ganglion cells and hypertrophic nerve trunks. The clinician should be careful to biopsy proximal to the physiologically normal hypoganglionic zone at the pectinate (dentate) line, yet caudal enough to detect very short segment aganglionosis [37].
The biopsy specimen can be stained for an increase in acetylcholinesterase activity, which can contribute to the diagnosis [37, 38]. A full-thickness biopsy should be completed if the suction biopsy is unable to provide an accurate diagnosis. The length of aganglionosis is definitively determined at the time of surgical resection, confirming the absence of ganglionic cells in the myenteric and submucosal plexi.
Treatment
Currently, the only treatment for HSCR is surgery. Failure to surgically treat HSCR can be fatal due to malnutrition or sepsis following bowel perforation. Although surgery is the routine therapy for HSCR patients, surgical outcomes can vary widely, with a range of long term consequences, such as constipation, fecal incontinence and enterocolitis [39, 40]. The surgical treatments aim to remove the aganglionic bowel and anastomose the normal bowel to the anus while preserving sphincter function. The main techniques include the total transanal endorectal pull-through (TERPT) [41–43] and the laparoscopic assisted pull-through [44, 45] procedures.
As compared to traditional transabdominal open surgery, TERPT and laparoscopic assisted pull-through are associated with faster recoveries, shorter hospital stays, improved cosmetic appearance, and fewer perioperative complications [46, 47]. The TERPT is useful for aganglionosis confined to the rectosigmoid area [48], as it minimizes intraabdominal contamination, and adhesion formation, and the risk of damage to pelvic structures. The laparoscopic assisted pull-through has the benefit of collecting seromuscular biopsies for the identification of normal colon, better mobilization and dissection of the aganglionic colon, and minimized dilation of the anal canal [44, 46].
Although the TERPT and laparoscopic assisted pull-through procedures have better outcomes than traditional open surgery, there are risks and complications. Common post-surgical problems include long-term obstructive symptoms and soiling. Milder obstructive symptoms can be treated by dietary changes, laxatives, enemas or botulinum toxin injections every 3–4 months [49, 50]. Patients who are responsive to botulinum toxin injections but are unable to undergo repeated injections can have a myectomy procedure [51]. Although a repeat pull-through procedure can be done if there is residual or acquired aganglionosis, strictures, or dysfunctional, dilated proximal bowel, it is often very difficult due to scarring from the previous procedure [52, 53].
Pathogenesis
Proper neural crest cell migration, proliferation, differentiation, survival, and apoptosis all contribute to a functional ENS. Perturbation in any of these processes can lead to a Hirschsprung disease phenotype. Many genes which play a critical functional role in neural crest cell development have been implicated in HSCR, including the proto-oncogene RET, endothelin signaling genes, and transcription factors [28]. Although over a dozen genes have been identified that contribute to the etiology of HSCR, these pathways only account for about half of the known cases (Table 1).
Table 1.
Hirschsprung disease genes and contribution to neural crest cell migration, proliferation, differentiation and survival
| Gene | Migration | Proliferation | Differentiation | Survival/Cell Death |
|---|---|---|---|---|
| RET | Attracted to GDNF | Promote proliferation | Promote neuronal differentiation | Promote ENCC and enteric neuron survival, Inhibit non-apoptotic cell death |
| GDNF | Gradient to attract RET+ and GFRα1+ ENCCs | Promote proliferation | Promote neuronal differentiation | Promote survival |
| GFRα-1 | Attracted to GDNF, necessary to cross mesentery | Promote enteric neuron survival | ||
| EDNRB | Necessary for normal migration | Maintain proliferative state | ||
| EDN3/ET3 | Necessary for normal migration, may maintain permissive environment | Inhibit neuronal differentiation | ||
| ZFHX1B/SIP1/SMADIP1 | Neural specification, Epithelial-to-mesenchymal transition | |||
| PHOX2b | Expressed in migrating ENCCs | |||
| SHH | Oppose GDNF, reduce migration | Promote proliferation | Inhibit neuronal differentiation by affecting responsiveness to GDNF | |
| SOX10 | Maintain ENS progenitor state | |||
| NRTN/NTN | Promote proliferation | Promote neuronal differentiation | Promote survival |
RET = receptor tyrosine kinase, GDNF = glial cell line-derived neurotrophic factor, GFRα1 =glycosylphosphatidylinositol-linked receptor alpha 1, EDNRB = endothelin receptor type B, ET3/EDN3 = endothelin 3, ZFHX1B/SIP1/SMADIP1 = zinc finger homeobox 1b/SMAL interacting protein, PHOX2b = paired-like homeobox2b, SHH = sonic hedgehog, SOX10 = SRY-related high mobility group-box transcription factor, NRTN/NTN = neurturin
Migration
The ENS is derived from migratory neural crest cells which originate at the vagal (somites 1–7) and sacral (caudal to somite 24) regions of the embryonic axis. These subsets contribute to different gut regions [54–57]. Vagal neural crest cells migrate in a rostral to caudal direction and sequentially contribute to the foregut, midgut, and hindgut [2, 55, 58]. In contrast, sacral NCCs are thought to contribute to the distal hindgut [59, 60]. Migration takes about six days in mice, with vagal NCC migration beginning around embryonic day (E) 8.5 and completing around E14.5 (Figure 1) [56, 61]. In humans, enteric neural crest cell (ENCC) migration takes about three weeks [62], beginning around week 4 and ending by week 7 [30, 63].
Figure 1. Enteric neuron immunostaining in wild-type embryonic guts.

Tuj1 (red) immunostaining of E11.5 to E14.5 whole guts shows wavefront location. At E11.5, the embryonic gut is in a hairpin formation and neurons are present to the cecum. At E12.5 the gut has grown, and the neurons have reached the proximal hindgut. The E12.5 gut pictured is a back-side view to visualize the wavefront. At E14.5 neurons have reached the distal hindgut and grown significantly and the hairpin formation is no longer present. Asterisk (*) marks the mesentery at E12.5 through which tmENCCs migrate. Nuclei are counterstained with 4′,6-diamidino-2-phenylindole (DAPI) (blue).
During migration, chains of interconnected neural crest cells at the leading edge of the population are referred to as the wavefront [63–65]. In these ENCC chains, there are two types of cell: multipolar and monopolar cells [66]. Previously, it had been proposed that the migration of ENCCs through the cecum (circumflex ENCCs: cfENCCs) was important for hindgut colonization [67–69], but recent photo-conversion real-time imaging experiments have shown that ENCCs destined for the hindgut traverse the mesentery (trans-mesenteric ENCSS: tmENCCs) as solitary cells when the midgut and hindgut are opposed in parallel (between E10.5–11.5), and these ENCCs are the major source of the ENS in the hindgut (Figure 2) [66]. Vagal NCC derived neuroblasts had been previously observed in the mesentery along the proximal colon prior to cecal colonization, but the significance of cells along this route were unclear [68]. Moreover cells in the mesentery near the distal hindgut between E11.5–13.5, were thought to be sacral neural crest cells [64].
Figure 2. Trans-mesenteric enteric neural crest cells migrate through the mesentery of E11.75 guts.
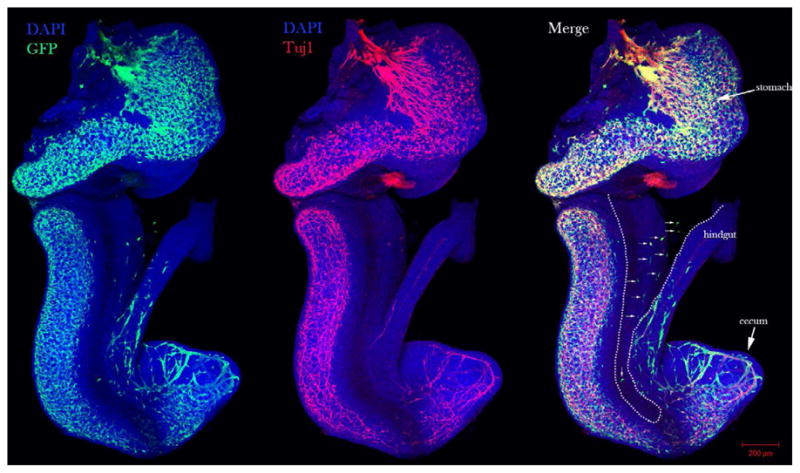
Whole-mount co-immunostain of GFP and Tuj1 staining of E11.75 gut from a RosaeYFP; Wnt1Cre embryo. White arrows depict tmENCCs in the mesentery between the midgut and hindgut. Mesentery is located between dotted lines. Neural crest cells are stained with GFP and mature neurons are stained with Tuj1 (red). Nuclei are counterstained with 4′,6-diamidino-2-phenylindole (DAPI) (blue).
Different populations of ENCCs exhibit different migration behaviors depending on their position along the migratory trail. Wavefront cells display significant caudal expansion, while trailing cells exhibit limited expansion. Therefore, it is the wavefront ENCCs that are responsible for the colonization of the hindgut [66]. This display of regional change in cell behavior is indicative of a positional microenvironment, suggesting the importance of cell-cell interactions during the process of NCC migration [65]. Since the midgut and hindgut are juxtaposed only during E10.5–11.5, and tmENCCs are the primary cell population contributing to the hindgut ENS, it was thought that these cells have only a limited period of time to reach the hindgut. Should these tmENCCs experience delayed migration through the mesentery, the result should be impaired colonization of the hindgut [66]. Furthermore a limited time window critical for complete ENS colonization of the terminal hindgut has also been proposed. In this scenario, a change in the microenvironment occurs at E14.5 when the gut is no longer permissive to migrating ENCCs, and high laminin levels may play a role in this non-permissive environment [70]. In contrast, other data shows that colonization can be completed after E14.5, suggesting that there may not be a strict permissive window of colonization [71].
GDNF/RET
Many genes contribute to normal enteric neural crest cell migration and the formation of a functional enteric nervous system, and mutations in any of these genes may cause an HSCR phenotype (Table 1). Two of the major contributing gene families responsible for HSCR cases are Receptor tyrosine kinase (RET) and Glial cell line-derived neurotrophic factor (GDNF). Mutations in the RET pathway account for 15–35% of patients with sporadic HSCR (HSCR in a single family member) and 50% of familial cases [72]. Patients who are heterozygous for mutations in GDNF have also been diagnosed with HSCR [73]. GDNF is a secreted protein that forms a complex with glycosylphosphatidylinositol-linked receptor (GFRα1), which binds and activates the transmembrane receptor tyrosine kinase RET. RET is then autophosphorylated and activates downstream pathways [74] that influence ENCC proliferation and survival, apoptosis, migration, and differentiation [75].
Prior to the entry of NCCs into the gut, GDNF is expressed in the mesoderm environment [76]. During their entrance into the gut, RET and GFRα1 are expressed in ENCCs [77]. When the NCC wave front reaches the esophagus, GDNF is expressed in the stomach, and is again elevated in the cecum as the NCC approach the distal portion of the small intestine, suggesting GDNF acts to attract RET and GFRα1 expressing ENCCs to the proper location [76, 78]. Following a concentration gradient of GDNF, tmENCCs migrate through the mesentery between the midgut and hindgut. Without GFRα1, tmENCCs are unable to leave the midgut [66]. Thus, without formation of the GDNF-RET-GFRα1 complex, aganglionosis is observed, irrespective of whether the mutation is in RET, GDNF or Gfrα1 [66, 79–84].
Patients with long and short segment disease have been identified as having mutations in RET [85, 86]. However, penetrance is estimated at only between 50–70% [87]. Interestingly, the “gene dosage” necessary for normal ENS development differs between mice and humans. For example, a heterozygous mutation of RET in humans can lead to HSCR. In contrast, Ret+/− mice only exhibit hypoganglionosis [88]. Homozygous mutations in Ret are required for complete intestinal aganglionosis in mice [79]. To mimic the aganglionic phenotype of humans, mice need a loss of 60–70% of RET expression [89]. Due to alternative splicing, 2 RET isoforms can be formed: RET9 and RET51. These isoforms are conserved between human and mice [90]. When Ret51 is absent (Ret9/9), mice have a completely colonized distal colon, whereas when RET9 is absent (Ret51/51), colonization fails in the distal colon [91]. Other targeted RET signaling sites have been used to model aganglionosis such as: Y1062 (RetY1062F), a multidocking site of RET9 (total intestinal aganglionosis)[92]; juxtamembrane serine 697 (RetS697A), a putative protein kinase phosphorylation site (loss of enteric neurons in distal colon) [93]; and a mutation in the extracellular cysteine residue 620 (Ret620R), where Ret620R null mice have total intestinal aganglionsis and heterozygotes have hypoganglionsis [88].
Interestingly, a new Ret9/EGFP hypomorphic mouse mutant (Ret51C681F) has led to the proposition of a new mode of HSCR pathogenesis. These mutant mice display delayed and impaired transmesenteric migration of ENCCs from the midgut to the hindgut. This suggests that the tmENCCs have a limited time window to reach the hindgut, and if there is a delay, the result is intestinal aganglionosis [66]. However, delayed migration and reduced NCC progenitor numbers alone do not necessarily lead to HSCR. Rather, these deficiencies can be overcome through balancing neural crest cell proliferation and differentiation [71].
Noncoding mutations in RET can increase the susceptibility to other HSCR mutations [72, 94, 95]. These noncoding mutations may be involved with regulatory elements and cellular mechanisms, such as transcription, translation, location or level of gene expression, or may be associated with linked susceptible loci. For example, the 9q31 locus segregates with the HSCR phenotype in families with noncoding RET mutations, suggesting that a combination of these specific loci may cause a HSCR phenotype [72]. Additionally, a genome-wide association study of a Mennonite population (with a ten-fold increase in HSCR compared to the general population), identified a HSCR-associated, non-coding RET mutation in the transcriptional enhancer of intron 1, which associates with EDNRB mutations [96]. It was later found that this same RET Mennonite haplotype makes a 20-fold greater contribution to HSCR risk than the rarer protein-coding HSCR mutations [95]. However, not all RET variants increase susceptibility to HSCR. A common variant in the 3′ UTR of the RET gene has been shown to sl mRNA transcript decay [94]. The increasing number of identified non-coding susceptibility RET mutations is a reminder that although they may have low penetrance [95], they are capable of acting synergistically with other mutations to affect a disease phenotype.
Endothelin pathway
Endothelin signaling is also necessary for normal ENCC migration and may help maintain a permissive NCC environment. Endothelin 3 (EDN3 or ET3) is a secreted peptide expressed by gut mesenchyme [97] that binds to the G-protein coupled receptor ENDRB on migrating ENCC. Endothelin converting enzyme 1 (ECE1) post-translationally modifies the immature form of EDN3 into the active form [98, 99]. The EDN3-EDNRB signaling pathway is involved in regulating the normal migration of ENCCs, and maintains enteric progenitors in a proliferative state [69, 100]. Thus ENCC differentiation is inhibited by the presence of EDN3 [100–103].
Mouse models with mutations in Edn3, Ednrb or Ece1 display various aganglionosis and pigmentation defects [98, 104, 105]. Lethal spotted mice have a mutation in the Edn3 ligand, and piebald lethal have an Ednrb mutation. These mice lack enteric neurons in the distal bowel, have ENCC migration defects (delayed NCC arrival to the small intestine), and piebaldism [69, 98, 104, 106, 107].
Studies have shown that the incomplete neuronal colonization of gut [108] can be rescued by transgenic expression of the missing molecule [109, 110]. Additionally, conditional deletion studies of Ednrb show that the critical time window where EDN3-EDNRB signaling is necessary is between E10.5–12.5, as delayed migrating enteric neuroblasts were noted at E11.0 [111]. Additionally, beginning at E10.0, Edn3 mRNA was detected ahead of migrating ENCCs [69]. Thus it has been suggested that because aganlionosis in Ednrb- and Edn3-deficient mice is restricted to the distal colon, that the EDN3-EDNRB signaling pathway is required during later stages of gut colonization [112].
In Ednrbflex3/flex3 mutant mice (null Ednrb allele with NCC labeled with YFP), ENCC advancement is delayed about 24 hours compared with wild-type littermates. By E14.5, wild-type guts are fully occupied by ENCCs, but mutant guts have altered trajectories and reduced speed. Grafting experiments suggested that the age of the recipient tissue, not genotype, restricts donor invasion. Furthermore, the time window when the gut microenvironment changes to become non-permissive to migration is E14.5, suggesting that if ENCCs have not finished migration by E14.5, an HSCR phenotype will result [70]. However, continued colonization has been observed between E14.5 to E18.5 in Tcof1 haploinsufficient mice, even though reduced NCC numbers and delayed migration were observed between E10.5–E14.5 [71]. This evidence argues that delayed migration is not always predictive or sufficient for the pathogenesis of aganglionosis. Rather, a balance between NCC proliferation in concert with differentiation and extrinsic gut microenvironmental influences are required for complete ENS formation [71, 113].
Patients with heterozygous mutations in the EDN3-EDNRB pathway account for 5% of HSCR cases [27], including one case caused by a mutation in ECE1 [114]. EDN3 and EDNRB mutations are associated with both syndromic (Waardenburg Shah) and non-syndromic forms of Hirschsprung disease [27, 72]. Patients with Waardenburg Shah Syndrome have colonic aganglionosis, pigmentation defects and sensorineural deafness. The inheritance of this syndrome can be either recessive (EDNRB and EDN3 cases) or dominant (SOX10 cases) [27, 72]. Patients with EDNRB Hirschsprung disease are associated with a large Mennonite population [115]. Carriers of the Tryp276Cys mutation display incomplete penetrance, with heterozygous mutations leading to HSCR in 21% of the affected patients, and homozygous mutations leading to HSCR in 74% of the affected patients [116]. As with RET mutations, the human ENS is more sensitive to reduced ENDRB signaling than in mice [117].
Additional Genes Regulating Neural Crest Cell Migration
The zinc finger homeobox 1b/SMAL interacting protein (ZFHX1B/SIP1/SMADIP1), is expressed in premigratory and migratory vagal neural crest cells. This transcription factor is involved in neural specification and the epithelial to mesenchymal transition during early NCC development. Zfxh1b−/− mice exhibit complete absence of vagal NCC precursors and die around E9.5 due to cardiovascular and neural defects [118]. A human mutation in ZFHX1B is associated with Mowat-Wilson syndrome of which HSCR is a component [119].
Paired-like homeobox2b (PHOX2b) is a transcription factor expressed in migrating ENCCs, enteric neurons and glial cells [120]. This transcription factor is required for RET expression in ENCCs. Mouse and zebrafish models lacking Phox2b have total intestinal aganglionosis due to a failure of the ENCCs to colonize the gut properly [121, 122]. Consistent with this, mutations in PHOX2b have also recently been described in association with HSCR in humans, particularly in patients with congenital central hypoventilation syndrome (CCHS) and neuroblastoma (NB), referred to as the CCHS-HSCR-NB association [123–125]. Bone morphogenetic proteins (BMPs) also play a role in the regulation of NCC migration and ENS enteric ganglion formation [126, 127]. Targeted inhibition of intestinal BMP activity using noggin leads to delayed ENCC migration by interfering with the migratory effects of GDNF signals, suggesting that appropriate BMP and GDNF interactions are necessary for the proper formation of a complete ENS [126].
Sonic Hedgehog (SHH) is another key regulator of the migration of ENCCs of the gut [128] and plexus formation [129]. SHH and GDNF have opposing effects on ENCC migration. NCCs migrate in the presence of GDNF, but addition of SHH reduces this migration by affecting the responsiveness of NCCs towards GDNF signaling or by regulating proliferation and differentiation of NCCs in the gut [128]. Thus, a balance between GDNF and SHH may be important for the proper positioning of the ENS plexus.
CXCR4 is a cell surface receptor for the CXC chemokine PBSF/SDF-1 (pre-B-cell growth-stimulating factor/stromal-cell derived factor). This factor is expressed in vascular endothelial cells. Cxcr4 null mice display a failure of proper formation of the large vessels that supply the GI tract [130]. These mice also exhibit fewer tmENCCs at E11.5 and impaired hindgut colonization of ENCCs by E14.5 compared to wild type littermates [66]. This implies that tmENCC migration is partially dependent on vascular-derived signals, as CXCR4 indirectly supports trans-mesenteric migration by regulating vasculature development.
Proliferation, Survival and Differentiation
Proliferation rates in the developing GI system are equivalent throughout the ENS, with active ENCC proliferation at the wavefront necessary to colonize the gut, and behind the wavefront to fully populate the expanding intestine [131] and generate the millions of enteric neurons and glia present in the adult intestine [17]. The balance between proliferation and differentiation is necessary to maintain a sufficient progenitor pool of cells necessary to ensure complete ENS colonization [1, 18, 71, 113, 132].
Many of the genes discussed above also contribute to maintain ENCC and postmitotic enteric neuron survival in the gastrointestinal tract. Ret, Gdnf, and Gfrα1 are necessary for early ENCC survival [133]. Gfrα1 and Ret are also essential for survival of colonic enteric neurons. Furthermore, enteric neuron survival is sensitive to Ret dosage [89, 134]. Low Ret dosage does not result in increased apoptosis in ENCCs, but can lead to increased nonapoptotic cell death [89]. Ret hypomorphic mice have delayed migration and elevated nonapoptotic ENCC death. Consistent with this, when cell death is inhibited, a normal ENS is formed. This result led to the suggestion that cell death, not migratory delay of ENCCs, is a principle cause of isolated HSCR [135].
The EDN3-EDNRB signaling pathway is not only involved with regulating ENCC migration, but also plays a role in maintaining the enteric progenitors in a proliferative state [69, 100]. By keeping progenitors in a self-renewing state, the presence of Edn3 inhibits ENCC differentiation [100–103]. Gdnf and Ret promote proliferation of ENS progenitors and differentiation into neurons and glia [136, 137]. Gdnf also has a synergistic effect with Edn3 to enhance this proliferative effect, but inhibits neuronal differentiation to maintain the precursor cell population [101, 103].
Neurturin (NRTN or NTN) is a ligand in the GDNF family, which binds to GFRα2 to activate RET and promote the projection and branching of some enteric neuron axons. This trophic factor is important for the maintenance of neuronal projections, as neuturin-deficient mice have reduced nerve fiber density in the ENS, abnormal neurotransmitter release, and abnormal GI motility [17, 138]. Homozygous mice with null mutations in Ntn and Gfrα2 have reduced excitatory fibers present in the ENS and decreased gut motility along the entire GI tract [17, 138, 139]. Myenteric ganglion cells are smaller in Ntn−/− adult mice compared to wild-type mice as well, suggesting that Ntn plays a supportive role for neurons and cell size maintenance [138], and promotes the proliferation of ENS precursors and neuronal differentiation to increase the number of neurons and glia [136]. In addition, Ret+ cells from embryonic rat guts were cultured and failed to survive in the absence of GDNF or NTN [137]. Thus, it has been shown both in vivo and in vitro that Ntn promotes survival of enteric NCCs as well as the maintenance of enteric neurons and ganglia. Rare human cases of HSCR have been documented with NTN mutations, and of those cases, occasional mutations have also been found in RET or another HSCR gene, suggesting a modifier role for NTN in the etiology and pathogenesis of HSCR [28, 73, 140–144].
SOX10
Sox10 (SRY-related high mobility group-box transcription factor) is expressed in vagal NCCs as they emigrate from the neural tube. Sox10 is used as a marker of ENS progenitors as it is expressed during NCC migration, and maintains the ENS progenitor state [100, 145]. This transcription factor regulates key genes required for ENS, melanocyte, and glial development [146], such as Ednrb and Ret in ENCCs [147–149]
Mouse and zebrafish Sox10 homozygous mutants display abnormal ENS and melanocyte phenotypes. Sox10Dom mice have a single base pair insertion leading to a spontaneous dominant negative mutation. This truncates Sox10 downstream of its DNA binding domain [150]. These mice have a smaller pool of ENS progenitors as compared to wild-type mice [151, 152], and lack enteric neurons through the entire GI tract [150, 153]. The vagal NCC die prior to their entry into the gut, and these homozygous mutant mice die at birth [153]. Furthermore, up to 20% of the heterozygous mice have colonic aganglionosis and coat-color abnormalities, but the incidence is background-dependent [18, 150, 154, 155].
SOX10 mutations account for less than 5% of both syndromic and nonsyndromic forms of HSCR. In humans, SOX10 mutations are associated with the dominant inheritance of HSCR in Waardenburg Syndrome [156], and some patients have additional neurologic phenotypes [157–159], due to dysmyelination of their CNS and PNS [157].
A model for CCHS-HSCR-NB, created by introducing a non polyalanine repeat expansion mutation (NPARM) PHOX2B in the Phox2b mouse locus, resulted in persistent Sox10 expression in mutant embryos in enteric ganglion precursors, leading to reduced proliferation and biased glial differentiation [123]. This demonstrates that reciprocal inhibition between Sox10 and Phox2b is necessary to maintain appropriate differentiation of neurons and glia in the enteric ganglia.
Vitamin A Metabolism
Of all the human genes and mouse models identified, the RET pathway and its interacting components are the most commonly disrupted genes contributing to the HSCR phenotype. However, the mutations discussed above account for only about 50% of the documented cases of HSCR. Thus, there must be other pathways involved in the pathogenesis of Hirschsprung disease. Recently, a role for retinoid signaling in ENS development and the pathogenesis of colonic aganglionosis has been suggested [160, 161].
Vitamin A, an essential nutrient, plays a role in embryonic development in its active form, retinoic acid (RA). Insufficient or excess dietary vitamin A can result in congenital anomalies and even fetal death. Retinoic acid acts as a ligand for nuclear RA receptors (RARs) and retinoid X receptors (RXRs) to regulate transcriptional activity of target genes [162]. Vitamin A (retinol) is taken up by retinol binding protein 4 (RBP4), mediated by cell surface RBP receptor Stra6 (stimulated by retinoic acid gene 6). Once inside the cell, retinol is bound by cellular retinol binding proteins (CRBP) and undergoes two consecutive oxidation reactions to become retinoic acid: retinol is oxidized to all-trans-retinal by microsomal retinol dehydrogenase 10 (RDH10) or cytosolic alcohol dehydrogenases (ADH). Although studies initially indicated that members of the ADH family catalyzed this reaction ubiquitously throughout the embryo [163] a homozygous mouse mutation in Rdh10 [164] which displays a classic RA-deficiency phonotype, indicates RDH10 is the critical enzyme necessary for the regulation of this first oxidation step during embryogenesis [165].
All-trans retinal is then oxidized by retinaldehyde dehydrogenases RALDH1, RALDH2 and RALDH3 to retinoic acid. Retinoic acid can also be produced via an alternative route through beta carotene. Beta-carotene is cleaved by beta-carotene 15/15′-monooxygenase 1 (BCMO1/BCOX) to retinaldehyde which is then oxidized by RALDH1-3. RALDH expression patterns closely correlate with RA signaling, as shown through RA response element (RARE) reporter mice [166, 167]. Raldh2, the first retinaldehyde dehydrogenase to be expressed, is induced in the primitive streak and mesoderm during gastrulation, and is later restricted to the posterior embryonic region. This enzyme is responsible for almost all RA production during early embryogenesis. When necessary, to avoid the accumulation of excess RA, RA can be transformed to 4-hydoxy-RA for metabolism and elimination by cytochrome P450 26A1, B1, and C1 (Cyp26) enzymes. Collectively, the balance between retinoic acid synthesis and catabolism is critical to regulate the precise spatiotemporal domains of retinoid signaling during embryogenesis.
Retinoid Deficient mouse models
Currently, there are a two mouse models of depleted retinoid signaling that may phenotypically display some evidence for colonic aganglionosis, Rbp4 −/− and Raldh2−/− [160, 161]. Rbp4−/− mice are unable to store retinol in their livers, and when subjected to an RA deficient diet during embryogenesis via maternal restriction from E7.5, these mice become fully depleted of Vitamin A and its active form, RA. Subsequently, these Rbp4−/− mice exhibit mild retinoid deficiency, with distal bowel aganglionosis and this phenotype is associated with increased phosphatase and tensin homolog (Pten) accumulation in migrating cells. Consistent with this, Pten overexpression leads to slowed ENS precursor migration. Thus RA has been proposed to maintain signals for migration and may do so through maintaining lamellipodia formation in migrating cells. This led to the suggestion that Vitamin A deficiency may be a non-genetic risk factor that increases HSCR penetrance and expressivity [161]. Consistent with this, the Raldh2−/− mouse model, which is also deficient in RA signaling, suggests that RA may play a role in NCC colonization of the gut. These mice typically die by E9.5, prior to development of the ENS. However, with RA maternal supplementation from E7.5, the animals live long enough to observe agenesis of the enteric ganglia as a result vagal NCC deficiency [160].
Although these studies suggest a role for retinoid signaling in the pathogenesis of colonic aganglionosis, the HSCR phenotype in the Rbp4−/− mice is only observed in response to a vitamin A deficient diet, creating a somewhat artificial situation. Similarly, the Raldh2−/− mutants die too early to examine a HSCR phenotype without providing RA rescue. These partially-rescued mice develop with a range of physical anomalies, so the precise role of RA remains to be characterized. Additional experiments are necessary to fully characterize the contribution of retinoic acid during gastrointestinal tract development, as the effects of RA on NCC formation, migration, proliferation and differentiation are not completely known. A mouse model without the need for dietary control or partial RA rescue would be ideal to properly investigate the role of RA in ENS development. This could be accomplished using conditional knockout mice. By genetically disrupting retinoic acid production, the precise temporal and tissue-specific requirement for RA during ENS development could be elucidated. Human studies are also needed to complement these animal models, and it would be ideal to conduct whole exome or whole genome sequencing to potentially identify new HSCR candidate genes and modifiers in the retinoid metabolism and/or signaling pathways. This would validate the clinical importance of the disrupted genes in many mouse models currently available to study HSCR, while also providing new avenues for clinical research into treatments or prevention of individuals at-risk.
Discussion
Although more than half of the cases associated with Hirschsprung disease have associated genetic mutations, there is still much more to be learned about this disease. A continued effort is needed to identify responsible genes in experimental models, but also to identify relevant mutations in humans. Whole exome and whole genome sequencing of patient samples are an ideal way to identify these genes and mutations. With new genetic models of HSCR and new tools to examine them, new modes of pathogenesis may be proposed. For example, recent time lapse imaging using a photo-convertible mouse model showed that ENCCs reach the hindgut by migrating through the mesentery between the midgut and hindgut, as opposed to a sequential migration from midgut to cecum to hindgut [66]. Even this single deviation from the previous model of ENCC migration necessitates that mutants with migratory dysfunction be revisited to examine the possible changes in tmENCC and cfENCC behaviors [66], and the effects those changes may have on the progression of colonic aganglionosis. This new finding may also affect the way in which in vitro culture experiments are conducted to study the dymanics of NCC colonization and formation of the ENS. Instead of draping the midgut and hindgut across a well with a separated mesentery [168], experiments should now be conducted with the mesentery and blood vessels left in-tact, to allow for transmesenteric migration.
As discussed above, Hirschsprung disease is a complex, and often non-Mendelian disease, with high phenotypic variability and incomplete penetrance. Additionally, the same phenotype (HSCR) is associated with mutations of multiple distinct genes. These characteristics suggest the involvement of modifier genes or environmental influences [28, 87, 140, 143]. For example, rare human cases have been documented with NTN mutations, and many of these cases had a concomitant mutation in RET or another known HSCR gene, suggesting a modifier effect [144]. Although retinoid signaling has been implicated in the pathogenesis of HSCR, conditional knockout models are still required to fully elucidate the precise spatiotemporal role for RA during ENS development.
Great strides are being made to better understand HSCR, but much still remains unknown. The generation of new animal models, together with the continued identification of genes associated with HSCR in humans, and the development of new techniques to study the effects of these genetic disruptions are necessary. This may ultimately lead to better screening programs and prevention for at-risk individuals.
Acknowledgments
All experiments were approved by the Institutional Animal Care and Use Committee of the Stowers Institute for Medical Research. We are grateful to Melissa Childers for excellent technical assistance and maintenance of our mouse lines. Research in the Trainor laboratory is supported by the Stowers Institute for Medical Research, the National Institute for Dental and Craniofacial Research (DE 016082) and the March of Dimes FY08-265.
Abbreviations
- ADH
alcohol dehydrogenase
- BCMO1/BCOX
beta-carotene 15/15′-monooxygenase 1
- BMP
bone morphogenetic protein
- cfENCC
circumflex enteric neural crest cell
- CCHS
congenital central hypoventilation syndrome
- CRABP
cellular retinoic acid binding protein
- CRBP
cellular retinol binding proteins
- CXCR4
cell surface receptor for the CXC chemokine PBSF/SDF-1
- Cyp26
cytochrome P450 26
- DAPI
4′,6-diamidino-2-phenylindole
- E
embryonic day
- ECE1
endothelin converting enzyme 1
- EDNRB
endothelin receptor type B
- ENCC
enteric neural crest cell
- ENS
enteric nervous system
- ET3/EDN3
endothelin 3
- GDNF
glial cell line-derived neurotrophic factor
- GFP
green flourescent protein
- GFRα1
glycosylphosphatidylinositol-linked receptor alpha 1
- GFRα2
glycosylphosphatidylinositol-linked receptor alpha 2
- GI
gastrointestinal
- HSCR
Hirschsprung Disease
- ICC
Interstitial cells of Cajal
- NB
neuroblastoma
- NCC
neural crest cell
- NPARM
non polyalanine repeat expansion mutation
- NRTN/NTN
neurturin
- PBSF/SDF-1
pre-B-cell growth-stimulating factor/stromal-cell derived factor
- PHOX2B
paired-like homeobox2b
- Pten
phosphatase and tensin homolog
- RA
retinoic acid
- RALDH
retinaldehyde dehydrogenase
- RAR
retinoic acid receptor
- RARE
response element
- RBP4
retinol binding protein
- RDH10
retinol dehydrogenase 10
- RET
receptor tyrosine kinase
- RXR
retinoid X receptor
- SHH
sonic hedgehog
- SOX10
SRY-related high mobility group-box transcription factor
- Stra6
stimulated by retinoic acid gene 6
- TCOF1
Treacher Collins-Franceschetti Syndrome 1
- TERPT
transanal endorectal pull-through
- tmENCC
trans-mesenteric enteric neural crest cell
- ZFHX1B/SIP1/SMADIP1
zinc finger homeobox 1b/SMAL interacting protein
Footnotes
The authors declare they have no conflicts of interest to disclose and have read the journal’s policy on disclosure of potential conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Heanue TA, Pachnis V. Enteric nervous system development and Hirschsprung’s disease: advances in genetic and stem cell studies. Nat Rev Neurosci. 2007;8:466–79. doi: 10.1038/nrn2137. [DOI] [PubMed] [Google Scholar]
- 2.Gershon MD. Genes and lineages in the formation of the enteric nervous system. Current Opinion in Neurobiology. 1997;7:101–9. doi: 10.1016/s0959-4388(97)80127-4. [DOI] [PubMed] [Google Scholar]
- 3.Karaosmanoglu T. Regional differences in the number of neurons in the myenteric plexus of the guinea pig small intestine and colon: an evaluation of markers used to count neurons. The Anatomical Record. 1996;244:470–80. doi: 10.1002/(SICI)1097-0185(199604)244:4<470::AID-AR5>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 4.Sanders KM, Koh SD, Ward SM. INTERSTITIAL CELLS OF CAJAL AS PACEMAKERS IN THE GASTROINTESTINAL TRACT. Annual Review of Physiology. 2006;68:307–43. doi: 10.1146/annurev.physiol.68.040504.094718. [DOI] [PubMed] [Google Scholar]
- 5.Huizinga JD, Lammers WJ. Gut peristalsis is governed by a multitude of cooperating mechanisms. American journal of physiology Gastrointestinal and liver physiology. 2009;296:G1–8. doi: 10.1152/ajpgi.90380.2008. [DOI] [PubMed] [Google Scholar]
- 6.Whitehouse FrKJ. Myenteric plexus in congenital megacolon: Study of eleven cases. Archives of Internal Medicine. 1948;82:75–111. doi: 10.1001/archinte.1948.00220250085005. [DOI] [PubMed] [Google Scholar]
- 7.Webster W. Embryogenesis of the enteric ganglia in normal mice and in mice that develop congenital aganglionic megacolon. Journal of Embryology and Experimental Morphology. 1973;30:573–85. [PubMed] [Google Scholar]
- 8.Hoehner JC, Wester T, Påhlman S, Olsen L. Alterations in neurotrophin and neurotrophin-receptor localization in Hirschsprung’s disease. Journal of Pediatric Surgery. 1996;31:1524–9. doi: 10.1016/s0022-3468(96)90170-0. [DOI] [PubMed] [Google Scholar]
- 9.Gaillard D. Colonic nerve network demonstrated by quinacrine. Bulletin de l’Association des anatomistes. 1982;66:63–70. [PubMed] [Google Scholar]
- 10.Tosney KW, Watanabe M, Landmesser L, Rutishauser U. The distribution of NCAM in the chick hindlimb during axon outgrowth and synaptogenesis. Developmental Biology. 1986;114:437–52. doi: 10.1016/0012-1606(86)90208-3. [DOI] [PubMed] [Google Scholar]
- 11.Clavel C. Distribution of fibronectin and laminin during development of the human myenteric plexus and Hirschsprung’s disease. Gastroentérologie clinique et biologique. 1988;12:193–7. [PubMed] [Google Scholar]
- 12.Langer JC. Smooth muscle from aganglionic bowel in Hirschsprung’s disease impairs neuronal development in vitro. Cell and Tissue Research. 1994;276:181–6. doi: 10.1007/BF00354798. [DOI] [PubMed] [Google Scholar]
- 13.Parisi MA. Hirschsprung Disease Overview. GeneReviews. 2006 [Google Scholar]
- 14.N-Fékété C. Total colonic aganglionosis (with or without ileal involvement): a review of 27 cases. Journal of Pediatric Surgery. 1986;21:251–4. doi: 10.1016/s0022-3468(86)80847-8. [DOI] [PubMed] [Google Scholar]
- 15.Neilson IR, Yazbeck S. Ultrashort Hirschsprung’s disease: Myth or reality. Journal of Pediatric Surgery. 1990;25:1135–8. doi: 10.1016/0022-3468(90)90748-x. [DOI] [PubMed] [Google Scholar]
- 16.Kapur RP. Practical pathology and genetics of Hirschsprung’s disease. Seminars in Pediatric Surgery. 2009;18:212–23. doi: 10.1053/j.sempedsurg.2009.07.003. [DOI] [PubMed] [Google Scholar]
- 17.Gianino S, Grider JR, Cresswell J, Enomoto H, Heuckeroth RO. GDNF availability determines enteric neuron number by controlling precursor proliferation. Development. 2003;130:2187–98. doi: 10.1242/dev.00433. [DOI] [PubMed] [Google Scholar]
- 18.Stanchina L, Baral V, Robert F, Pingault V, Lemort N, Pachnis V, et al. Interactions between Sox10, Edn3 and Ednrb during enteric nervous system and melanocyte development. Developmental Biology. 2006;295:232–49. doi: 10.1016/j.ydbio.2006.03.031. [DOI] [PubMed] [Google Scholar]
- 19.Singh SJ, Croaker GDH, Manglick P, Wong CL, Athanasakos H, Elliott E, et al. Hirschsprung’s disease: the Australian Paediatric Surveillance Unit’s experience. Pediatric Surgery International. 2003;19:247–50. doi: 10.1007/s00383-002-0842-z. [DOI] [PubMed] [Google Scholar]
- 20.Parc R. Megacolon in adults. Apropos of 76 cases. Annales de gastroentérologie et d’hépatologie. 1984;20:133–41. [PubMed] [Google Scholar]
- 21.Spouge D. Hirschsprung disease in a large birth cohort. Teratology (Philadelphia) 1985;32:171–7. doi: 10.1002/tera.1420320204. [DOI] [PubMed] [Google Scholar]
- 22.Badner JA, WKS, Garver KL, Chakravarti A. A genetic study of Hirschsprung disease. Am J Hum Genet. 1990;46:568–80. [PMC free article] [PubMed] [Google Scholar]
- 23.Bodian M, Carter OO. A family study of Hirschsprung’s disease. Annals of Human Genetics. 1963;26:261–77. [Google Scholar]
- 24.Passarge E. The Genetics of Hirschsprung’s Disease. New England Journal of Medicine. 1967;276:138–43. doi: 10.1056/NEJM196701192760303. [DOI] [PubMed] [Google Scholar]
- 25.Kerstjens-Frederikse WS, Hofstra RMW, van Essen AJ, Meijers JHC, Buys CHCM. A Hirschsprung disease locus at 22q11? Journal of Medical Genetics. 1999;36:221–4. [PMC free article] [PubMed] [Google Scholar]
- 26.Ryan ET, Ecker JL, Christakis NA, Folkman J. Hirschsprung’s disease: Associated abnormalities and demography. Journal of Pediatric Surgery. 1992;27:76–81. doi: 10.1016/0022-3468(92)90111-j. [DOI] [PubMed] [Google Scholar]
- 27.Amiel J, Lyonnet S. Hirschsprung disease, associated syndromes, and genetics: a review. Journal of Medical Genetics. 2001;38:729–39. doi: 10.1136/jmg.38.11.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Parisi MA, Kapur RP. Genetics of Hirschsprung disease. Current Opinion in Pediatrics. 2000;12:610–7. doi: 10.1097/00008480-200012000-00017. [DOI] [PubMed] [Google Scholar]
- 29.Sarioglu A. Hirschsprung-associated congenital anomalies. European journal of pediatric surgery. 1997;7:331–7. doi: 10.1055/s-2008-1071186. [DOI] [PubMed] [Google Scholar]
- 30.Fu M, Chi Hang Lui V, Har Sham M, Nga Yin Cheung A, Kwong Hang Tam P. HOXB5 expression is spatially and temporarily regulated in human embryonic gut during neural crest cell colonization and differentiation of enteric neuroblasts. Developmental Dynamics. 2003;228:1–10. doi: 10.1002/dvdy.10350. [DOI] [PubMed] [Google Scholar]
- 31.Rosenfield NS. Hirschsprung disease: accuracy of the barium enema examination. Radiology. 1984;150:393–400. doi: 10.1148/radiology.150.2.6691093. [DOI] [PubMed] [Google Scholar]
- 32.Smith GHH, Cass D. Infantile Hirschsprung’s disease — is a barium enema useful? Pediatric Surgery International. 1991;6:318–21. [Google Scholar]
- 33.Tobon F, Reid NCRW, Talbert JL, Schuster MM. Nonsurgical Test for the Diagnosis of Hirschsprung’s Disease. New England Journal of Medicine. 1968;278:188–94. doi: 10.1056/NEJM196801252780404. [DOI] [PubMed] [Google Scholar]
- 34.Emir H, Akman M, Sarimurat N, Kilic N, Erdogan E, Soylet Y. Anorectal manometry during the neonatal period: its specificity in the diagnosis of Hirschsprung’s disease. Eur J Pediatr Surg. 1999;9:101–3. doi: 10.1055/s-2008-1072221. [DOI] [PubMed] [Google Scholar]
- 35.Lopez Alonso M. Manometric study in the newborn. Cir Pediatr. 1992;5:66–71. [PubMed] [Google Scholar]
- 36.Andrassy RJ, Weitzman HIJJ. Rectal suction biopsy for the diagnosis of Hirschsprung’s disease. Annals of Surgery. 1981;193:419–24. doi: 10.1097/00000658-198104000-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kapur RP. Can We Stop Looking?: Immunohistochemistry and the Diagnosis of Hirschsprung Disease. American Journal of Clinical Pathology. 2006;126:9–12. doi: 10.1309/T7RE-Y1N4-3FML-7AA8. [DOI] [PubMed] [Google Scholar]
- 38.Kurer MH, Lawson JO, Pambakian H. Suction biopsy in Hirschsprung’s disease. Archives of Disease in Childhood. 1986;61:83–4. doi: 10.1136/adc.61.1.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baillie CT, Kenny SE, Rintala RJ, Booth JM, Lloyd DA. Long-term outcome and colonic motility after the Duhamel procedure for Hirschsprung’s disease. Journal of Pediatric Surgery. 1999;34:325–9. doi: 10.1016/s0022-3468(99)90201-4. [DOI] [PubMed] [Google Scholar]
- 40.Menezes M, Corbally M, Puri P. Long-term results of bowel function after treatment for Hirschsprung’s disease: a 29-year review. Pediatric Surgery International. 2006;22:987–90. doi: 10.1007/s00383-006-1783-8. [DOI] [PubMed] [Google Scholar]
- 41.De la Torre-Mondragón L, Ortega-Salgado JA. Transanal endorectal pull-through for Hirschsprung’s disease. Journal of Pediatric Surgery. 1998;33:1283–6. doi: 10.1016/s0022-3468(98)90169-5. [DOI] [PubMed] [Google Scholar]
- 42.Albanese CT, Jennings RW, Smith B, Bratton B, Harrison MR. Perineal one-stage pull-through for Hirschsprung’s disease. Journal of Pediatric Surgery. 1999;34:377–80. doi: 10.1016/s0022-3468(99)90480-3. [DOI] [PubMed] [Google Scholar]
- 43.Langer JC, Minkes RK, Mazziotti MV, Skinner MA, Winthrop AL. Transanal one-stage soave procedure for infants with Hirschsprung’s disease. Journal of Pediatric Surgery. 1999;34:148–52. doi: 10.1016/s0022-3468(99)90246-4. [DOI] [PubMed] [Google Scholar]
- 44.Georgeson KE, Fuenfer MM, Hardin WD. Primary laparoscopic pull-through for Hirschsprung’s disease in infants and children. Journal of Pediatric Surgery. 1995;30:1017–22. doi: 10.1016/0022-3468(95)90333-x. [DOI] [PubMed] [Google Scholar]
- 45.Jona JZ. Laparoscopic pull-through procedure for Hirschsprung’s disease. Seminars in Pediatric Surgery. 1998;7:228–31. doi: 10.1016/s1055-8586(98)70036-8. [DOI] [PubMed] [Google Scholar]
- 46.Georgeson KE. Primary laparoscopic-assisted endorectal colon pull-through for Hirschsprung’s disease: a new gold standard. Annals of surgery. 1999;229:678–82. doi: 10.1097/00000658-199905000-00010. discussion 82–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fujiwara N, Kaneyama K, Okazaki T, Lane GJ, Kato Y, Kobayashi H, et al. A comparative study of laparoscopy-assisted pull-through and open pull-through for Hirschsprung’s disease with special reference to postoperative fecal continence. Journal of Pediatric Surgery. 2007;42:2071–4. doi: 10.1016/j.jpedsurg.2007.08.033. [DOI] [PubMed] [Google Scholar]
- 48.De la Torre L, Ortega A. Transanal versus open endorectal pull-through for Hirschsprung’s disease. Journal of Pediatric Surgery. 2000;35:1630–2. doi: 10.1053/jpsu.2000.18338. [DOI] [PubMed] [Google Scholar]
- 49.Minkes RK, Langer JC. A prospective study of botulinum toxin for internal anal sphincter hypertonicity in children with Hirschsprung’s disease. Journal of Pediatric Surgery. 2000;35:1733–6. doi: 10.1053/jpsu.2000.19234. [DOI] [PubMed] [Google Scholar]
- 50.Patrus B, Nasr A, Langer JC, Gerstle JT. Intrasphincteric botulinum toxin decreases the rate of hospitalization for postoperative obstructive symptoms in children with Hirschsprung disease. Journal of Pediatric Surgery. 2011;46:184–7. doi: 10.1016/j.jpedsurg.2010.09.089. [DOI] [PubMed] [Google Scholar]
- 51.Wildhaber BE, Pakarinen M, Rintala RJ, Coran AG, Teitelbaum DH. Posterior myotomy/myectomy for persistent stooling problems in Hirschsprung’s disease. Journal of Pediatric Surgery. 2004;39:920–6. doi: 10.1016/j.jpedsurg.2004.02.016. [DOI] [PubMed] [Google Scholar]
- 52.Langer JC. Repeat pull-through surgery for complicated Hirschsprung’s disease: Indications, techniques, and results. Journal of Pediatric Surgery. 1999;34:1136–41. doi: 10.1016/s0022-3468(99)90585-7. [DOI] [PubMed] [Google Scholar]
- 53.Teitelbaum DH, Coran AG. Reoperative Surgery for Hirschsprung’s Disease. Seminars in Pediatric Surgery. 2003;12:124–31. doi: 10.1016/s1055-8586(02)00023-9. [DOI] [PubMed] [Google Scholar]
- 54.Burns AJ, Champeval D, Le Douarin NM. Sacral Neural Crest Cells Colonise Aganglionic Hindgut in Vivo but Fail to Compensate for Lack of Enteric Ganglia. Developmental Biology. 2000;219:30–43. doi: 10.1006/dbio.1999.9592. [DOI] [PubMed] [Google Scholar]
- 55.Durbec PL, Larsson-Blomberg LB, Schuchardt A, Costantini F, Pachnis V. Common origin and developmental dependence on c-ret of subsets of enteric and sympathetic neuroblasts. Development. 1996;122:349–58. doi: 10.1242/dev.122.1.349. [DOI] [PubMed] [Google Scholar]
- 56.Young HM, Newgreen D. Enteric neural crest-derived cells: Origin, identification, migration, and differentiation. The Anatomical Record. 2001;262:1–15. doi: 10.1002/1097-0185(20010101)262:1<1::AID-AR1006>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 57.Wallace AS, Burns AJ. Development of the enteric nervous system, smooth muscle and interstitial cells of Cajal in the human gastrointestinal tract. Cell and Tissue Research. 2005;319:367–82. doi: 10.1007/s00441-004-1023-2. [DOI] [PubMed] [Google Scholar]
- 58.Young HM, Hearn CJ, Ciampoli D, Southwell BR, Brunet JF, Newgreen DF. A Single Rostrocaudal Colonization of the Rodent Intestine by Enteric Neuron Precursors Is Revealed by the Expression of Phox2b, Ret, and p75 and by Explants Grown under the Kidney Capsule or in Organ Culture. Developmental Biology. 1998;202:67–84. doi: 10.1006/dbio.1998.8987. [DOI] [PubMed] [Google Scholar]
- 59.Kapur RP. Colonization of the Murine Hindgut by Sacral Crest-Derived Neural Precursors: Experimental Support for an Evolutionarily Conserved Model. Developmental Biology. 2000;227:146–55. doi: 10.1006/dbio.2000.9886. [DOI] [PubMed] [Google Scholar]
- 60.Burns AJ, Le Douarin NM. The sacral neural crest contributes neurons and glia to the post-umbilical gut: spatiotemporal analysis of the development of the enteric nervous system. Development. 1998;125:4335–47. doi: 10.1242/dev.125.21.4335. [DOI] [PubMed] [Google Scholar]
- 61.Newgreen D, Young HM. Enteric Nervous System: Development and Developmental Disturbances—Part 2. Pediatric and Developmental Pathology. 2002;5:329–49. doi: 10.1007/s10024-002-0002-4. [DOI] [PubMed] [Google Scholar]
- 62.Anderson R, Stewart A, Young H. Phenotypes of neural-crest-derived cells in vagal and sacral pathways. Cell and Tissue Research. 2006;323:11–25. doi: 10.1007/s00441-005-0047-6. [DOI] [PubMed] [Google Scholar]
- 63.Druckenbrod NR, Epstein ML. The pattern of neural crest advance in the cecum and colon. Developmental Biology. 2005;287:125–33. doi: 10.1016/j.ydbio.2005.08.040. [DOI] [PubMed] [Google Scholar]
- 64.Young HM, Bergner AJ, Anderson RB, Enomoto H, Milbrandt J, Newgreen DF, et al. Dynamics of neural crest-derived cell migration in the embryonic mouse gut. Developmental Biology. 2004;270:455–73. doi: 10.1016/j.ydbio.2004.03.015. [DOI] [PubMed] [Google Scholar]
- 65.Druckenbrod NR, Epstein ML. Behavior of enteric neural crest-derived cells varies with respect to the migratory wavefront. Developmental Dynamics. 2007;236:84–92. doi: 10.1002/dvdy.20974. [DOI] [PubMed] [Google Scholar]
- 66.Nishiyama C, Uesaka T, Manabe T, Yonekura Y, Nagasawa T, Newgreen DF, et al. Trans-mesenteric neural crest cells are the principal source of the colonic enteric nervous system. Nat Neurosci. 2012;15:1211–8. doi: 10.1038/nn.3184. [DOI] [PubMed] [Google Scholar]
- 67.Kruger GM, Mosher JT, Tsai Y-H, Yeager KJ, Iwashita T, Gariepy CE, et al. Temporally Distinct Requirements for Endothelin Receptor B in the Generation and Migration of Gut Neural Crest Stem Cells. Neuron. 2003;40:917–29. doi: 10.1016/s0896-6273(03)00727-x. [DOI] [PubMed] [Google Scholar]
- 68.Coventry S, Yost C, Palmiter RD, Kapur RP. Migration of ganglion cell precursors in the ileoceca of normal and lethal spotted embryos, a murine model for Hirschsprung disease. Lab Invest. 1994;71:82–93. [PubMed] [Google Scholar]
- 69.Barlow A, de Graaff E, Pachnis V. Enteric Nervous System Progenitors Are Coordinately Controlled by the G Protein-Coupled Receptor EDNRB and the Receptor Tyrosine Kinase RET. Neuron. 2003;40:905–16. doi: 10.1016/s0896-6273(03)00730-x. [DOI] [PubMed] [Google Scholar]
- 70.Druckenbrod NR, Epstein ML. Age-dependent changes in the gut environment restrict the invasion of the hindgut by enteric neural progenitors. Development. 2009;136:3195–203. doi: 10.1242/dev.031302. [DOI] [PubMed] [Google Scholar]
- 71.Barlow AJ, Dixon J, Dixon MJ, Trainor PA. Balancing neural crest cell intrinsic processes with those of the microenvironment in Tcof1 haploinsufficient mice enables complete enteric nervous system formation. Human Molecular Genetics. 2012;21:1782–93. doi: 10.1093/hmg/ddr611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Brooks AS, Oostra BA, Hofstra RMW. Studying the genetics of Hirschsprung’s disease: unraveling an oligogenic disorder. Clinical Genetics. 2005;67:6–14. doi: 10.1111/j.1399-0004.2004.00319.x. [DOI] [PubMed] [Google Scholar]
- 73.Eketjäll S, Ibáñez CF. Functional characterization of mutations in the GDNF gene of patients with Hirschsprung disease. Human Molecular Genetics. 2002;11:325–9. doi: 10.1093/hmg/11.3.325. [DOI] [PubMed] [Google Scholar]
- 74.Tansey MG, Baloh RH, Milbrandt J, Johnson EM., Jr GFRα-Mediated Localization of RET to Lipid Rafts Is Required for Effective Downstream Signaling, Differentiation, and Neuronal Survival. Neuron. 2000;25:611–23. doi: 10.1016/s0896-6273(00)81064-8. [DOI] [PubMed] [Google Scholar]
- 75.Eng C. RET Proto-Oncogene in the Development of Human Cancer. Journal of Clinical Oncology. 1999;17:380. doi: 10.1200/JCO.1999.17.1.380. [DOI] [PubMed] [Google Scholar]
- 76.Natarajan D, Marcos-Gutierrez C, Pachnis V, de Graaff E. Requirement of signalling by receptor tyrosine kinase RET for the directed migration of enteric nervous system progenitor cells during mammalian embryogenesis. Development. 2002;129:5151–60. doi: 10.1242/dev.129.22.5151. [DOI] [PubMed] [Google Scholar]
- 77.Tomac AC, Grinberg A, Huang SP, Nosrat C, Wang Y, Borlongan C, et al. Glial cell line-derived neurotrophic factor receptor α1 availability regulates glial cell line-derived neurotrophic factor signaling: evidence from mice carrying one or two mutated alleles. Neuroscience. 1999;95:1011–23. doi: 10.1016/s0306-4522(99)00503-5. [DOI] [PubMed] [Google Scholar]
- 78.Young HM, Hearn CJ, Farlie PG, Canty AJ, Thomas PQ, Newgreen DF. GDNF Is a Chemoattractant for Enteric Neural Cells. Developmental Biology. 2001;229:503–16. doi: 10.1006/dbio.2000.0100. [DOI] [PubMed] [Google Scholar]
- 79.Schuchardt A, D’Agati V, Larsson-Blomberg L, Costantini F, Pachnis V. Defects in the kidney and enteric nervous system of mice lacking the tyrosine kinase receptor Ret. Nature. 1994;367:380–3. doi: 10.1038/367380a0. [DOI] [PubMed] [Google Scholar]
- 80.Shepherd IT, Pietsch J, Elworthy S, Kelsh RN, Raible DW. Roles for GFRα1 receptors in zebrafish enteric nervous system development. Development. 2004;131:241–9. doi: 10.1242/dev.00912. [DOI] [PubMed] [Google Scholar]
- 81.Sanchez MP, Silos-Santiago I, Frisen J, He B, Lira SA, Barbacid M. Renal agenesis and the absence of enteric neurons in mice lacking GDNF. Nature. 1996;382:70–3. doi: 10.1038/382070a0. [DOI] [PubMed] [Google Scholar]
- 82.Pichel JG, Shen L, Sheng HZ, Granholm A-C, Drago J, Grinberg A, et al. Defects in enteric innervation and kidney development in mice lacking GDNF. Nature. 1996;382:73–6. doi: 10.1038/382073a0. [DOI] [PubMed] [Google Scholar]
- 83.Moore MW, Klein RD, Farinas I, Sauer H, Armanini M, Phillips H, et al. Renal and neuronal abnormalities in mice lacking GDNF. Nature. 1996;382:76–9. doi: 10.1038/382076a0. [DOI] [PubMed] [Google Scholar]
- 84.Cacalano G, Fariñas I, Wang L-C, Hagler K, Forgie A, Moore M, et al. GFRα1 Is an Essential Receptor Component for GDNF in the Developing Nervous System and Kidney. Neuron. 1998;21:53–62. doi: 10.1016/s0896-6273(00)80514-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Edery P, Lyonnet S, Mulligan LM, Pelet A, Dow E, Abel L, et al. Mutations of the RET proto-oncogene in Hirschsprung’s disease. Nature. 1994;367:378–80. doi: 10.1038/367378a0. [DOI] [PubMed] [Google Scholar]
- 86.Romeo G, Ronchetto P, Luo Y, Barone V, Seri M, Ceccherini I, et al. Point mutations affecting the tyrosine kinase domain of the RET proto-oncogene in Hirschsprung’s disease. Nature. 1994;367:377–8. doi: 10.1038/367377a0. [DOI] [PubMed] [Google Scholar]
- 87.Bolk S, Pelet A, Hofstra RMW, Angrist M, Salomon R, Croaker D, et al. A human model for multigenic inheritance: Phenotypic expression in Hirschsprung disease requires both the RET gene and a new 9q31 locus. Proceedings of the National Academy of Sciences. 2000;97:268–73. doi: 10.1073/pnas.97.1.268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Carniti C, Belluco S, Riccardi E, Cranston AN, Mondellini P, Ponder BAJ, et al. The RetC620R Mutation Affects Renal and Enteric Development in a Mouse Model of Hirschsprung’s Disease. The American Journal of Pathology. 2006;168:1262–75. doi: 10.2353/ajpath.2006.050607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Uesaka T, Nagashimada M, Yonemura S, Enomoto H. Diminished Ret expression compromises neuronal survival in the colon and causes intestinal aganglionosis in mice. The Journal of Clinical Investigation. 2008;118:1890–8. doi: 10.1172/JCI34425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Carter MT. Conservation of RET proto-oncogene splicing variants and implications for RET isoform function. Cytogenetics and cell genetics. 2001;95:169–76. doi: 10.1159/000059341. [DOI] [PubMed] [Google Scholar]
- 91.de Graaff E, Srinivas S, Kilkenny C, D’Agati V, Mankoo BS, Costantini F, et al. Differential activities of the RET tyrosine kinase receptor isoforms during mammalian embryogenesis. Genes & Development. 2001;15:2433–44. doi: 10.1101/gad.205001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Jain S, Knoten A, Hoshi M, Wang H, Vohra B, Heuckeroth RO, et al. Organotypic specificity of key RET adaptor-docking sites in the pathogenesis of neurocristopathies and renal malformations in mice. The Journal of Clinical Investigation. 2010;120:778–90. doi: 10.1172/JCI41619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Asai N, Fukuda T, Wu Z, Enomoto A, Pachnis V, Takahashi M, et al. Targeted mutation of serine 697 in the Ret tyrosine kinase causes migration defect of enteric neural crest cells. Development. 2006;133:4507–16. doi: 10.1242/dev.02616. [DOI] [PubMed] [Google Scholar]
- 94.Griseri P, Lantieri F, Puppo F, Bachetti T, Di Duca M, Ravazzolo R, et al. A common variant located in the 3′UTR of the RET gene is associated with protection from Hirschsprung disease. Human Mutation. 2007;28:168–76. doi: 10.1002/humu.20397. [DOI] [PubMed] [Google Scholar]
- 95.Emison ES, McCallion AS, Kashuk CS, Bush RT, Grice E, Lin S, et al. A common sex-dependent mutation in a RET enhancer underlies Hirschsprung disease risk. Nature. 2005;434:857–63. doi: 10.1038/nature03467. [DOI] [PubMed] [Google Scholar]
- 96.Carrasquillo MM, McCallion AS, Puffenberger EG, Kashuk CS, Nouri N, Chakravarti A. Genome-wide association study and mouse model identify interaction between RET and EDNRB pathways in Hirschsprung disease. Nat Genet. 2002;32:237–44. doi: 10.1038/ng998. [DOI] [PubMed] [Google Scholar]
- 97.Nataf V, Amemiya A, Yanagisawa M, Le Douarin NM. The expression pattern of endothelin 3 in the avian embryo. Mechanisms of Development. 1998;73:217–20. doi: 10.1016/s0925-4773(98)00048-3. [DOI] [PubMed] [Google Scholar]
- 98.Baynash AG, Hosoda K, Giaid A, Richardson JA, Emoto N, Hammer RE, et al. Interaction of endothelin-3 with endothelin-B receptor is essential for development of epidermal melanocytes and enteric neurons. Cell. 1994;79:1277–85. doi: 10.1016/0092-8674(94)90018-3. [DOI] [PubMed] [Google Scholar]
- 99.Xu D, Emoto N, Giaid A, Slaughter C, Kaw S, deWit D, et al. ECE-1: A membrane-bound metalloprotease that catalyzes the proteolytic activation of big endothelin-1. Cell. 1994;78:473–85. doi: 10.1016/0092-8674(94)90425-1. [DOI] [PubMed] [Google Scholar]
- 100.Bondurand N, Natarajan D, Barlow A, Thapar N, Pachnis V. Maintenance of mammalian enteric nervous system progenitors by SOX10 and endothelin 3 signalling. Development. 2006;133:2075–86. doi: 10.1242/dev.02375. [DOI] [PubMed] [Google Scholar]
- 101.Nagy N, Goldstein AM. Endothelin-3 regulates neural crest cell proliferation and differentiation in the hindgut enteric nervous system. Developmental Biology. 2006;293:203–17. doi: 10.1016/j.ydbio.2006.01.032. [DOI] [PubMed] [Google Scholar]
- 102.Wu JJ, Chen JX, Rothman TP, Gershon MD. Inhibition of in vitro enteric neuronal development by endothelin-3: mediation by endothelin B receptors. Development. 1999;126:1161–73. doi: 10.1242/dev.126.6.1161. [DOI] [PubMed] [Google Scholar]
- 103.Hearn CJ, Murphy M, Newgreen D. GDNF and ET-3 Differentially Modulate the Numbers of Avian Enteric Neural Crest Cells and Enteric Neurons in Vitro. Developmental Biology. 1998;197:93–105. doi: 10.1006/dbio.1998.8876. [DOI] [PubMed] [Google Scholar]
- 104.Hosoda K, Hammer RE, Richardson JA, Baynash AG, Cheung JC, Giaid A, et al. Targeted and natural (piebald-lethal) mutations of endothelin-B receptor gene produce megacolon associated with spotted coat color in mice. Cell. 1994;79:1267–76. doi: 10.1016/0092-8674(94)90017-5. [DOI] [PubMed] [Google Scholar]
- 105.Yanagisawa H, Yanagisawa M, Kapur RP, Richardson JA, Williams SC, Clouthier DE, et al. Dual genetic pathways of endothelin-mediated intercellular signaling revealed by targeted disruption of endothelin converting enzyme-1 gene. Development. 1998;125:825–36. doi: 10.1242/dev.125.5.825. [DOI] [PubMed] [Google Scholar]
- 106.Lee H-O, Levorse JM, Shin MK. The endothelin receptor-B is required for the migration of neural crest-derived melanocyte and enteric neuron precursors. Developmental Biology. 2003;259:162–75. doi: 10.1016/s0012-1606(03)00160-x. [DOI] [PubMed] [Google Scholar]
- 107.Ro S, Hwang SJ, Muto M, Jewett WK, Spencer NJ. Anatomic modifications in the enteric nervous system of piebald mice and physiological consequences to colonic motor activity. American Journal of Physiology - Gastrointestinal and Liver Physiology. 2006;290:G710–G8. doi: 10.1152/ajpgi.00420.2005. [DOI] [PubMed] [Google Scholar]
- 108.Kapur RP, Yost C, Palmiter RD. A transgenic model for studying development of the enteric nervous system in normal and aganglionic mice. Development. 1992;116:167–75. doi: 10.1242/dev.116.Supplement.167. [DOI] [PubMed] [Google Scholar]
- 109.Svensson P-J, Von Tell D, Molander M-L, Anvret M, Nordenskjold A. A Heterozygous Frameshift Mutation in the Endothelin-3 (EDN-3) Gene in Isolated Hirschsprung’s Disease. Pediatr Res. 1999;45:714–7. doi: 10.1203/00006450-199905010-00018. [DOI] [PubMed] [Google Scholar]
- 110.Gariepy CE, Williams SC, Richardson JA, Hammer RE, Yanagisawa M. Transgenic expression of the endothelin-B receptor prevents congenital intestinal aganglionosis in a rat model of Hirschsprung disease. The Journal of Clinical Investigation. 1998;102:1092–101. doi: 10.1172/JCI3702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Shin MK, Levorse JM, Ingram RS, Tilghman SM. The temporal requirement for endothelin receptor-B signalling during neural crest development. Nature. 1999;402:496–501. doi: 10.1038/990040. [DOI] [PubMed] [Google Scholar]
- 112.Leibl MA, Ota T, Woodward MN, Kenny SE, Lloyd DA, Vaillant CR, et al. Expression of endothelin 3 by mesenchymal cells of embryonic mouse caecum. Gut. 1999;44:246–52. doi: 10.1136/gut.44.2.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Barlow AJ, Dixon J, Dixon M, Trainor PA. Tcof1 acts as a modifier of Pax3 during enteric nervous system development and in the pathogenesis of colonic aganglionosis. Human Molecular Genetics. 2013 doi: 10.1093/hmg/dds528. [in press] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hofstra RMW, Valdenaire O, Arch E, Osinga J, Kroes H, Löffler B-M, et al. A Loss-of-Function Mutation in the Endothelin-Converting Enzyme 1 (ECE-1) Associated with Hirschsprung Disease, Cardiac Defects, and Autonomic Dysfunction. The American Journal of Human Genetics. 1999;64:304–7. doi: 10.1086/302184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Mulligan LM, Kwok JBJ, Healey CS, Elsdon MJ, Eng C, Gardner E, et al. Germ-line mutations of the RET proto-oncogene in multiple endocrine neoplasia type 2A. Nature. 1993;363:458–60. doi: 10.1038/363458a0. [DOI] [PubMed] [Google Scholar]
- 116.Puffenberger EG, Hosoda K, Washington SS, Nakao K, deWit D, Yanagisawa M, et al. A missense mutation of the endothelin-B receptor gene in multigenic hirschsprung’s disease. Cell. 1994;79:1257–66. doi: 10.1016/0092-8674(94)90016-7. [DOI] [PubMed] [Google Scholar]
- 117.Wallace AS, Anderson RB. Genetic interactions and modifier genes in Hirschsprung’s disease. World J Gastroenterol. 2011 Dec 7;17(45):4937–44. doi: 10.3748/wjg.v17.i45.4937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Van de Putte T, Maruhashi M, Francis A, Nelles L, Kondoh H, Huylebroeck D, et al. Mice Lacking Zfhx1b, the Gene That Codes for Smad-Interacting Protein-1, Reveal a Role for Multiple Neural Crest Cell Defects in the Etiology of Hirschsprung Disease–Mental Retardation Syndrome. The American Journal of Human Genetics. 2003;72:465–70. doi: 10.1086/346092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Yamada K, Yamada Y, Nomura N, Miura K, Wakako R, Hayakawa C, et al. Nonsense and Frameshift Mutations in ZFHX1B, Encoding Smad-Interacting Protein 1, Cause a Complex Developmental Disorder with a Great Variety of Clinical Features. The American Journal of Human Genetics. 2001;69:1178–85. doi: 10.1086/324343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Corpening JC, Cantrell VA, Deal KK, Southard-Smith EM. A Histone2BCerulean BAC transgene identifies differential expression of Phox2b in migrating enteric neural crest derivatives and enteric glia. Developmental Dynamics. 2008;237:1119–32. doi: 10.1002/dvdy.21498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Pattyn A, Morin X, Cremer H, Goridis C, Brunet J-F. The homeobox gene Phox2b is essential for the development of autonomic neural crest derivatives. Nature. 1999;399:366–70. doi: 10.1038/20700. [DOI] [PubMed] [Google Scholar]
- 122.Elworthy S, Pinto JP, Pettifer A, Cancela ML, Kelsh RN. Phox2b function in the enteric nervous system is conserved in zebrafish and is sox10-dependent. Mechanisms of Development. 2005;122:659–69. doi: 10.1016/j.mod.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 123.Nagashimada M, Ohta H, Li C, Nakao K, Uesaka T, Brunet J-F, et al. Autonomic neurocristopathy-associated mutations in PHOX2B dysregulate Sox10 expression. The Journal of Clinical Investigation. 2012;122:3145–58. doi: 10.1172/JCI63401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Amiel J, Laudier B, Attie-Bitach T, Trang H, de Pontual L, Gener B, et al. Polyalanine expansion and frameshift mutations of the paired-like homeobox gene PHOX2B in congenital central hypoventilation syndrome. Nat Genet. 2003;33:459–61. doi: 10.1038/ng1130. [DOI] [PubMed] [Google Scholar]
- 125.Trochet D, O’Brien LM, Gozal D, Trang H, Nordenskjöld A, Laudier B, et al. PHOX2B Genotype Allows for Prediction of Tumor Risk in Congenital Central Hypoventilation Syndrome. The American Journal of Human Genetics. 2005;76:421–6. doi: 10.1086/428366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Goldstein AM, Brewer KC, Doyle AM, Nagy N, Roberts DJ. BMP signaling is necessary for neural crest cell migration and ganglion formation in the enteric nervous system. Mechanisms of Development. 2005;122:821–33. doi: 10.1016/j.mod.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 127.Faure C, Chalazonitis A, Rhéaume C, Bouchard G, Sampathkumar SG, Yarema KJ, et al. Gangliogenesis in the enteric nervous system: Roles of the polysialylation of the neural cell adhesion molecule and its regulation by bone morphogenetic protein-4. Developmental Dynamics. 2007;236:44–59. doi: 10.1002/dvdy.20943. [DOI] [PubMed] [Google Scholar]
- 128.Fu M, Lui VCH, Sham MH, Pachnis V, Tam PKH. Sonic hedgehog regulates the proliferation, differentiation, and migration of enteric neural crest cells in gut. The Journal of Cell Biology. 2004;166:673–84. doi: 10.1083/jcb.200401077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ramalho-Santos M, Melton DA, McMahon AP. Hedgehog signals regulate multiple aspects of gastrointestinal development. Development. 2000;127:2763–72. doi: 10.1242/dev.127.12.2763. [DOI] [PubMed] [Google Scholar]
- 130.Tachibana K, Hirota S, Iizasa H, Yoshida H, Kawabata K, Kataoka Y, et al. The chemokine receptor CXCR4 is essential for vascularization of the gastrointestinal tract. Nature. 1998;393:591–4. doi: 10.1038/31261. [DOI] [PubMed] [Google Scholar]
- 131.Young HM, Turner KN, Bergner AJ. The location and phenotype of proliferating neural-crest-derived cells in the developing mouse gut. Cell and Tissue Research. 2005;320:1–9. doi: 10.1007/s00441-004-1057-5. [DOI] [PubMed] [Google Scholar]
- 132.Laranjeira C, Pachnis V. Enteric nervous system development: Recent progress and future challenges. Autonomic Neuroscience. 2009;151:61–9. doi: 10.1016/j.autneu.2009.09.001. [DOI] [PubMed] [Google Scholar]
- 133.Taraviras S, Pachnis V. Development of the mammalian enteric nervous system. Current Opinion in Genetics & Development. 1999;9:321–7. doi: 10.1016/s0959-437x(99)80048-3. [DOI] [PubMed] [Google Scholar]
- 134.Uesaka T, Jain S, Yonemura S, Uchiyama Y, Milbrandt J, Enomoto H. Conditional ablation of GFRα1 in postmigratory enteric neurons triggers unconventional neuronal death in the colon and causes a Hirschsprung’s disease phenotype. Development. 2007;134:2171–81. doi: 10.1242/dev.001388. [DOI] [PubMed] [Google Scholar]
- 135.Uesaka T, Enomoto H. Neural Precursor Death Is Central to the Pathogenesis of Intestinal Aganglionosis in Ret Hypomorphic Mice. The Journal of Neuroscience. 2010;30:5211–8. doi: 10.1523/JNEUROSCI.6244-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Heuckeroth RO, Lampe PA, Johnson EM, Jr, Milbrandt J. Neurturin and GDNF Promote Proliferation and Survival of Enteric Neuron and Glial Progenitorsin Vitro. Developmental Biology. 1998;200:116–29. doi: 10.1006/dbio.1998.8955. [DOI] [PubMed] [Google Scholar]
- 137.Taraviras S, Marcos-Gutierrez CV, Durbec P, Jani H, Grigoriou M, Sukumaran M, et al. Signalling by the RET receptor tyrosine kinase and its role in the development of the mammalian enteric nervous system. Development. 1999;126:2785–97. doi: 10.1242/dev.126.12.2785. [DOI] [PubMed] [Google Scholar]
- 138.Heuckeroth RO, Enomoto H, Grider JR, Golden JP, Hanke JA, Jackman A, et al. Gene Targeting Reveals a Critical Role for Neurturin in the Development and Maintenance of Enteric, Sensory, and Parasympathetic Neurons. Neuron. 1999;22:253–63. doi: 10.1016/s0896-6273(00)81087-9. [DOI] [PubMed] [Google Scholar]
- 139.Rossi J, Herzig K-H, Võikar V, Hiltunen PH, Segerstråle M, Airaksinen MS. Alimentary tract innervation deficits and dysfunction in mice lacking GDNF family receptor α2. The Journal of Clinical Investigation. 2003;112:707–16. doi: 10.1172/JCI17995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Angrist M, Bolk S, Halushka M, Lapchak PA, Chakravarti A. Germline mutations in glial cell line-derived neurotrophic factor (GDNF) and RET in a Hirschsprung disease patient. Nat Genet. 1996;14:341–4. doi: 10.1038/ng1196-341. [DOI] [PubMed] [Google Scholar]
- 141.Hofstra RMW, Osinga J, Tan-Sindhunata G, Wu Y, Kamsteeg E-J, Stulp RP, et al. A homozygous mutation in the endothelin-3 gene associated with a combined Waardenburg type 2 and Hirschsprung phenotype (Shah-Waardenburg syndrome) Nat Genet. 1996;12:445–7. doi: 10.1038/ng0496-445. [DOI] [PubMed] [Google Scholar]
- 142.Ivanchuk SM, Myers SM, Eng C, Mulligan LM. De Novo Mutation of GDNF, Ligand for the RET/GDNFR-α Receptor Complex, in Hirschsprung Disease. Human Molecular Genetics. 1996;5:2023–6. doi: 10.1093/hmg/5.12.2023. [DOI] [PubMed] [Google Scholar]
- 143.Salomon R, Attie T, Pelet A, Bidaud C, Eng C, Amiel J, et al. Germline mutations of the RET ligand GDNF are not sufficient to cause Hirschsprung disease. Nat Genet. 1996;14:345–7. doi: 10.1038/ng1196-345. [DOI] [PubMed] [Google Scholar]
- 144.Doray B. Mutation of the RET ligand, neurturin, supports multigenic inheritance in Hirschsprung disease. Human Molecular Genetics. 1998;7:1449–52. doi: 10.1093/hmg/7.9.1449. [DOI] [PubMed] [Google Scholar]
- 145.Bondurand N, Natarajan D, Thapar N, Atkins C, Pachnis V. Neuron and glia generating progenitors of the mammalian enteric nervous system isolated from foetal and postnatal gut cultures. Development. 2003;130:6387–400. doi: 10.1242/dev.00857. [DOI] [PubMed] [Google Scholar]
- 146.Kelsh RN. Sorting out Sox10 functions in neural crest development. BioEssays. 2006;28:788–98. doi: 10.1002/bies.20445. [DOI] [PubMed] [Google Scholar]
- 147.Lang D, Chen F, Milewski R, Li J, Lu MM, Epstein JA. Pax3 is required for enteric ganglia formation and functions with Sox10 to modulate expression of c-ret. The Journal of Clinical Investigation. 2000;106:963–71. doi: 10.1172/JCI10828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Lang D, Epstein JA. Sox10 and Pax3 physically interact to mediate activation of a conserved c-RET enhancer. Human Molecular Genetics. 2003;12:937–45. doi: 10.1093/hmg/ddg107. [DOI] [PubMed] [Google Scholar]
- 149.Zhu L, Lee H-O, Jordan CS, Cantrell VA, Southard-Smith EM, Shin MK. Spatiotemporal regulation of endothelin receptor-B by SOX10 in neural crest-derived enteric neuron precursors. Nat Genet. 2004;36:732–7. doi: 10.1038/ng1371. [DOI] [PubMed] [Google Scholar]
- 150.Southard-Smith EM, Kos L, Pavan W. Sox10 mutation disrupts neural crest development in Dom Hirschsprung mouse model. Nat Genetics. 1998;18:60–4. doi: 10.1038/ng0198-60. [DOI] [PubMed] [Google Scholar]
- 151.Britsch S, Goerich DE, Riethmacher D, Peirano RI, Rossner M, Nave K-A, et al. The transcription factor Sox10 is a key regulator of peripheral glial development. Genes & Development. 2001;15:66–78. doi: 10.1101/gad.186601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Paratore C, Eichenberger C, Suter U, Sommer L. Sox10 haploinsufficiency affects maintenance of progenitor cells in a mouse model of Hirschsprung disease. Human Molecular Genetics. 2002;11:3075–85. doi: 10.1093/hmg/11.24.3075. [DOI] [PubMed] [Google Scholar]
- 153.Kapur RP. Early death of neural crest cells is responsible for total enteric aganglionosis in Sox10(Dom)/Sox10(Dom) mouse embryos. Pediatric and Developmental Pathology. 1999;2:559–69. doi: 10.1007/s100249900162. [DOI] [PubMed] [Google Scholar]
- 154.Cantrell VA, Owens SE, Chandler RL, Airey DC, Bradley KM, Smith JR, et al. Interactions between Sox10 and EdnrB modulate penetrance and severity of aganglionosis in the Sox10Dom mouse model of Hirschsprung disease. Human Molecular Genetics. 2004;13:2289–301. doi: 10.1093/hmg/ddh243. [DOI] [PubMed] [Google Scholar]
- 155.Maka M, Claus Stolt C, Wegner M. Identification of Sox8 as a modifier gene in a mouse model of Hirschsprung disease reveals underlying molecular defect. Developmental Biology. 2005;277:155–69. doi: 10.1016/j.ydbio.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 156.Pingault V, Bondurand N, Kuhlbrodt K, Goerich DE, Prehu M-O, Puliti A, et al. SOX10 mutations in patients with Waardenburg-Hirschsprung disease. Nat Genet. 1998;18:171–3. doi: 10.1038/ng0298-171. [DOI] [PubMed] [Google Scholar]
- 157.Touraine RL, Attié-Bitach T, Manceau E, Korsch E, Sarda P, Pingault V, et al. Neurological Phenotype in Waardenburg Syndrome Type 4 Correlates with Novel SOX10 Truncating Mutations and Expression in Developing Brain. The American Journal of Human Genetics. 2000;66:1496–503. doi: 10.1086/302895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Southard-Smith EM, Angrist M, Ellison JS, Agarwala R, Baxevanis AD, Chakravarti A, et al. The Sox10Dom Mouse: Modeling the Genetic Variation of Waardenburg-Shah (WS4) Syndrome. Genome Research. 1999;9:215–25. [PubMed] [Google Scholar]
- 159.Inoue K, Tanabe Y, Lupski JR. Myelin deficiencies in both the central and the peripheral nervous systems associated with a SOX10 mutation. Annals of Neurology. 1999;46:313–8. doi: 10.1002/1531-8249(199909)46:3<313::aid-ana6>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 160.Niederreither K, Vermot J, Roux IL, Schuhbaur B, Chambon P, Dollé P. The regional pattern of retinoic acid synthesis by RALDH2 is essential for the development of posterior pharyngeal arches and the enteric nervous system. Development. 2003;130:2525–34. doi: 10.1242/dev.00463. [DOI] [PubMed] [Google Scholar]
- 161.Fu M, Sato Y, Lyons-Warren A, Zhang B, Kane MA, Napoli JL, et al. Vitamin A facilitates enteric nervous system precursor migration by reducing Pten accumulation. Development. 2010;137:631–40. doi: 10.1242/dev.040550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Niederreither K, Dolle P. Retinoic acid in development: towards an integrated view. Nat Rev Genet. 2008;9:541–53. doi: 10.1038/nrg2340. [DOI] [PubMed] [Google Scholar]
- 163.Molotkov A, Fan X, Deltour L, Foglio MH, Martras S, Farrés J, et al. Stimulation of Retinoic Acid Production and Growth by Ubiquitously Expressed Alcohol Dehydrogenase Adh3. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:5337–42. doi: 10.1073/pnas.082093299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Sandell LL, Sanderson BW, Moiseyev G, Johnson T, Mushegian A, Young K, et al. RDH10 is essential for synthesis of embryonic retinoic acid and is required for limb, craniofacial, and organ development. Genes & Development. 2007;21:1113–24. doi: 10.1101/gad.1533407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Cammas L, Romand R, Fraulob V, Mura C, Dollé P. Expression of the murine retinol dehydrogenase 10 (Rdh10) gene correlates with many sites of retinoid signalling during embryogenesis and organ differentiation. Developmental Dynamics. 2007;236:2899–908. doi: 10.1002/dvdy.21312. [DOI] [PubMed] [Google Scholar]
- 166.Rossant J, Zirngibl R, Cado D, Shago M, Giguère V. Expression of a retinoic acid response elemen-thsplacZ transgene defines specific domains of transcriptional activity during mouse embryogenesis. Genes & Development. 1991;5:1333–44. doi: 10.1101/gad.5.8.1333. [DOI] [PubMed] [Google Scholar]
- 167.Haskell GT, LaMantia A-S. Retinoic Acid Signaling Identifies a Distinct Precursor Population in the Developing and Adult Forebrain. The Journal of Neuroscience. 2005;25:7636–47. doi: 10.1523/JNEUROSCI.0485-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Hearn CJ, Young HM, Ciampoli D, Lomax AEG, Newgreen D. Catenary cultures of embryonic gastrointestinal tract support organ morphogenesis, motility, neural crest cell migration, and cell differentiation. Developmental Dynamics. 1999;214:239–47. doi: 10.1002/(SICI)1097-0177(199903)214:3<239::AID-AJA7>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]