

Abstract
The Nanotechnology Risk Assessment Working Group in the Center for Drug Evaluation and Research (CDER) within the United States Food and Drug Administration was established to assess the possible impact of nanotechnology on drug products. The group is in the process of performing risk assessment and management exercises. The task of the working group is to identify areas where CDER may need to optimize its review practices and to develop standards to ensure review consistency for drug applications that may involve the application of nanotechnology. The working group already performed risk management exercises evaluating the potential risks from administering nanomaterial active pharmaceutical ingredients (API) or nanomaterial excipients by various routes of administration. This publication outlines the risk assessment and management process used by the working group, using nanomaterial API by the oral route of administration as an example.
KEY WORDS: CDER, nanomaterial, nanotechnology, risk assessment, risk management
INTRODUCTION
Nanotechnology is currently being applied in the pharmaceutical industry as a means of developing innovative products, including novel dosage forms, complex delivery systems, and targeted therapies (1,2). The use of nanotechnology in pharmaceuticals by industry may result in drugs with improved performance, increased stability, and increased efficacy and consumer compliance (3–5). The United States Food and Drug Administration (USFDA) has a strong interest in contributing to the improvement of product development, including performance, safety, and quality, while ensuring that possible risks to any of the latter are minimized.
In June 2011, the USFDA issued a Draft Guidance to articulate the principles that could be used to consider whether a product contains nanomaterials (6). The considerations listed in the Draft Guidance, while not constituting a formal definition, are related to the likelihood that a product might exhibit properties that might ultimately raise questions about safety, efficacy, quality, or public health impact and in turn require USFDA to revisit its review practices.
In an effort to better understand the ramifications or risks involved with using nanomaterials, USFDA has taken several approaches to better understand how nanomaterials are used in regulated products. As a first step, an internal database of submitted and approved drugs containing nanoscale materials was created, in order to better understand the landscape of products that Center for Drug Evaluation and Research (CDER) was reviewing (7). The next step was to evaluate what might be the possible effects of the nanomaterials on pharmaceutical product development and use in the future. To this end, CDER initiated the risk assessment exercise that is described in this manuscript.
In 2010, CDER issued a Manual of Policy and Procedure (MAPP) 5015.9 called “Reporting Format for Nanotechnology-Related Information in CMC Review (8).” In this MAPP, information is provided to CDER chemistry, manufacturing, and controls (CMC) reviewers on how to consistently capture relevant information from submissions for products that contain materials in the nanoscale for the purposes of database construction. To that end, CMC reviewers in CDER were directed in the MAPP, to collect information on all products with dimensions below 1,000 nm (excluding dissolved molecular entities and biologics). While there is no official FDA definition of nanotechnology, for the purposes of the MAPP, the dimension of 1,000 nm was used by CDER to broadly establish some means to help reviewers identify products for which nanotechnology-relevant data should be collected. The collected information (such as particle size, size distribution, etc.) was and continues to be collated and analyzed in a database, for the purpose of understanding the quality and safety characteristics of drugs that may contain nanomaterials. The goal of the database analysis is to ultimately identify possible trends and generalizations that may help in the review approach for applications containing nanomaterials. A preliminary analysis of particle size information collected in CMC reviews of CDER-regulated drugs was presented at the August 9, 2012 meeting of the advisory committee of the Office of Pharmaceutical Science and Clinical Pharmacology (7).
In order to ensure that CDER could better understand possible effects of using nanomaterials in pharmaceuticals, both from a development and manufacturing standpoint, the Nanotechnology Risk Assessment Working Group was established in 2011. This group is comprised of a multidisciplinary team responsible for conducting assessments of possible risks associated with the application of nanotechnology in the development and manufacturing of drug products. The risk assessment working group is identifying those areas where CDER’s current review processes related to evaluating product safety and quality could be optimized to specifically address considerations associated with nanotechnology product characteristics. The working group has two main goals:
To identify potential risks to safety, quality, and efficacy resulting from the use of nanomaterials in drug products
To identify areas for improvement, based on the results from the risk management exercise
The current regulatory requirements in CDER are robust and have successfully been applied to the evaluation of novel therapies that contain nanomaterials. The intent of the present risk assessment exercise is to comprehensively evaluate how review practices for products containing nanoscale materials could be optimized. The importance of better understanding the potential impact of nanotechnology on drug products was reiterated in the recently passed Food and Drug Administration Safety and Innovation Act (lS.3187, signed July 10, 2012). In the current risk assessment exercise, emphasis is being placed on the review of product quality, although nonclinical safety evaluation has also been addressed. The information that the risk assessment exercise is generating will help CDER continue to ensure the safety, efficacy, and quality of drugs, including those drug products containing nanomaterials, while keeping in mind the benefits of scientific advances in drug development.
RISK ASSESSMENT STRATEGY OVERVIEW
The following section describes the strategy used by the nanotechnology risk assessment working group to look at the factors that may result in potential risks to quality, efficacy, and safety, when using nanomaterials in the development and manufacturing of pharmaceutical products. According to ICH Q9, risk management is comprised of risk assessment (identification, analysis, and evaluation), risk control (reduction and acceptance), and risk review (continuous evaluation and adjustment) (9). Some applications which contain elements of quality by design have included the risk management tools delineated in the ICH Q9 (10). In addition, risk-based strategies are increasingly being applied to the development of products containing nanotechnology (11,12).
A flowchart for the risk assessment exercise undertaken by CDER is presented in Fig. 1. The first step in the risk assessment process was to form a multidisciplinary team of experts in CDER from regulatory, policy, and research components within the organization. The nanotechnology risk assessment working group used traditional risk assessment tools and developed a strategy that is a combination of Ishikawa diagrams (for identification and categorization of potential risk) and risk management tables for the description and analysis of current risk management approaches used in CDER. In the context of this risk assessment exercise, a hazard or potential risk factor was defined as something that may potentially impact quality, safety, and/or efficacy of the product, as a result of particle size change of the active pharmaceutical ingredients (API). Additional CDER experts were brought into discussions by the working group as needed.
Fig. 1.
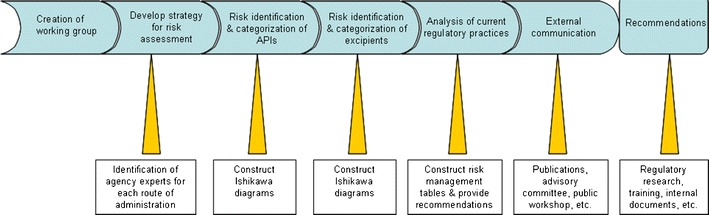
Flowchart of project plan for CDER’s Nanotechnology Risk Assessment Working Group
Common routes of administration were used to organize the risk assessment exercise. The following routes of administration were selected: oral, dermal (topical and transdermal), inhalation, and parenteral. Both API and excipients were evaluated. Once potential risks were identified, risk management tables were completed. The results were analyzed to develop and prioritize recommendations.
RISK IDENTIFICATION AND CHARACTERIZATION
The working group’s approach involved the use of Ishikawa (fishbone) diagrams as the tool to help identify factors, causes, or sources of variation that might lead to a specific result in a product or process. More specifically, as noted in ICH Q8(R2), Ishikawa diagrams identify “potential variables that can have an impact on the desired quality attribute” of a drug product. Ishikawa diagrams are a useful tool for providing an inventory of significant factors in a process and for identifying relationships among these factors (13).
For the risk assessment strategy involving pharmaceuticals containing nanomaterials, the working group has developed Ishikawa diagrams that identify potential risks to quality, safety, and efficacy that should be addressed in the regulatory review process. For example, for the oral route of administration, five phases of the drug product were selected for specific analysis: (1) product manufacture, (2) ingestion and dissolution, (3) absorption and distribution for particles not intended for absorption or (4) absorption and distribution of particles intended for absorption, and (5) elimination. Figure 2 shows the five phases for the oral route of administration used to organize the Ishikawa diagrams.
Fig. 2.
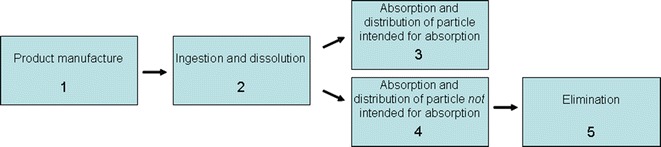
Summary of the five Ishikawa diagrams for the oral route of administration
Figure 3 provides an example of a representative Ishikawa diagram for orally administered drugs, in order to highlight CDER’s approach to the risk assessment exercise. The diagram represents risks to quality, safety, and efficacy at the ingestion and dissolution phase for orally administered drugs that have a change in API particle size.
Fig. 3.
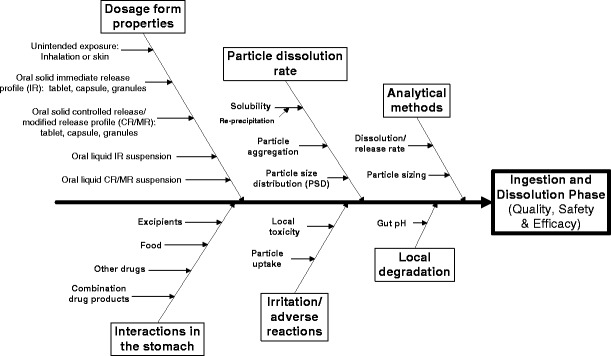
Representative Ishikawa diagram for orally administered drugs. This diagram represents potential risks to safety, quality, and efficacy at the ingestion and dissolution phase for orally administered drugs that have a change in API particle size. Please note that this example diagram is not meant to be a comprehensive depiction of the risk analysis process
For the absorption and distribution Ishikawa diagrams (phases 3 and 4), there were two main scenarios considered: one in which the API is intended for absorption (phase 3 or systemic delivery) and one when it is not intended for absorption (phase 4 or locally acting). Because these two scenarios were considered to have different risk profiles, they were treated independently. The risk assessment approach for particles meant for absorption (i.e., systemic delivery) focused on potential risk factors related to pharmacokinetic changes, such as differences in the rate and extent of absorption, caused by a change in particle size. On the other hand, for changes to nano-sized particles not intended for absorption (i.e., locally acting), potential risk factors resulting from a locally acting API were considered, such as a change in absorption.
The example selected in this paper is not meant to be a comprehensive depiction of the risk assessment process. Rather, it is intended to provide a framework for how CDER approached this risk assessment exercise. To illustrate the approach, the following section describes how to read the Ishikawa diagram shown in Fig. 3. The head of the Ishikawa diagram typically represents the desired attribute, which, in the case of the selected example, corresponds to quality, safety, and efficacy, when considering ingestion and dissolution of an orally administered drug that is not locally acting. The caption boxes at the top and bottom of the diagrams represent related categories or factors that impact the quality, safety, and efficacy of the product. The arrows pointing directly to these categories are primary causes or factors, and the branching arrows depict secondary causes or factors. For example, in Fig. 3, the box “Analytical methods,” corresponds to a potential risk category. The possibility of inadequate “dissolution/release rate” and “particle sizing” methods are identified as factors that may impact the evaluation of quality, safety, and efficacy. Similarly, within the box “Particle dissolution rate,” particle aggregation is a factor that could ultimately impact the quality, safety, and efficacy of the drug product containing nano-sized APIs (during ingestion and dissolution).
Ishikawa diagrams serve as visual shorthand for representing potential harms to quality, safety, and efficacy and the relationship of these possible harms to each other. It is important to note that the identification of potential risks in an Ishikawa diagram is meant to be qualitative, and that no quantification of severity or probability has been given to these potential risk factors. Such an analysis would be product specific. Note that some of the potential risk factors identified on the diagrams appear not to be unique to products containing nanomaterials, as some potential risks identified may be generally applicable to any change in physical properties of a drug substance. However, these potential risks were identified and analyzed because they were considered to be relevant to materials that undergo a change in particle size. Also, the graphical representation does not always capture the complex relationships between the factors and the implications on their review practices. Therefore, a second tool was applied to analyze the potential risk factors and to capture the adequacy of current review practices as applied to drugs that involve the application of nanotechnology
DEVELOPMENT OF RISK MANAGEMENT TABLES
Following the development of each Ishikawa diagram, a risk management table was created, in order to describe and analyze the review practices currently used to address the potential risks identified. An example of a risk management table is presented in Table I for illustration purposes. For each risk category, the following questions were addressed by the working group and reported in separate columns of the risk management table:
What information do reviewers currently evaluate to address each potential risk? In this column of the risk management table, the group listed any guidances, policies, submitted data, or research that currently addresses the potential risks.
Are the potential risks that were identified by CDER’s nanotechnology risk assessment working group being addressed by current review approaches?
What are preliminary recommendations for review approaches for drugs involving the application of nanotechnology, such that the potential risks are managed appropriately?
Table I.
Representative Excerpt of a Risk Management Table for Analysis of “Analytical Method” Risk Factors, in the Ingestion and Dissolution Phase, for Orally Administered Drugs that Have a Change in API Particle Size
| What do we do or require currently to address this risk? | Is this sufficient to address nanomaterial API effects and/or causes? | Potential approach to gap, e.g., proposed solution, references to future, or proposed work, if any | ||
|---|---|---|---|---|
| Risk identified: risk factor category | Sub-risk factor, primary, and/or secondary cause | Guidelines, policies, submitted data, or research that currently address this risk | Identified area for improvement | Area of focus |
| Analytical methods | Dissolution/release rate method | Evaluate dissolution/release rate method development report for discrimination and justification of parameters | For IR, BE studies may need to take into consideration API PSD impact on dissolution for BCS class II and BCS class IV | Reminder that for nanomaterials to focus on understanding the effect of particle size distribution on bioavailability and dissolution for immediate release, particularly for BCS II and IV, where API PSD may have impact on dissolution |
| Evaluate method against changes in formulation or IV/IVR | Monograph methods may or may not be suitable for reformulated materials if change to nano API has occurred. Any in vitro methods that use filtration and are being used for comparative evaluation of quality may need to be evaluated further | Request studies to show API PSD impact on dissolution, a dissolution specification is requested that covers ranges in dissolution may need to show in vivo data (“clinically relevant specs”) | ||
| Methods are reviewed following the same requirements for discrimination, development information, etc., regardless of case A, B, C, or D | The review of any unconventional methodology | Conventional methodology involving filtration of materials in in vitro analytical methods (e.g., dissolution, assay) may need to be revaluated when applied to nanomaterials | ||
| For OTC, methods are compendial and evaluation is done against compendial methods | ||||
| BE data would also catch differences in modified release formulations and could trigger more work on method development information |
Please note that this example diagram is not meant to be a comprehensive depiction of the risk analysis process
OTC over-the-counter
Since current review practices may differ for the various types of drug submissions, when addressing a potential risk factor in the risk management table, the working group considered different submission types, such as 501(b)(1), 505(b)(2), and 505(j) (14–16). In addition, the group also considered the scenario where a change might occur after the major clinical safety and efficacy studies are completed. In this case, the group considered the situation where changes could occur to the size of an API, both after phase 3 clinical testing but before approval as well as post-approval for an already approved application.
RISK ASSESSMENT AND RECOMMENDATIONS
The described CDER risk assessment exercise indicates that, in most cases, current review practices are adequate for evaluating nano-sized APIs, if the sponsor undertakes the appropriate studies early in the development process. However, if the change in particle size occurs after phase 3 testing, or involves an abbreviated new drug application, the working group identified areas where CDER can develop appropriate review processes and clarify the applicable policies. The working group also identified areas for educational opportunities for reviewers, with the goal of ensuring consistency throughout the review process.
While final recommendations are pending the completion of the risk management exercise, several key points have emerged. In general, the group has identified four areas where improvements could be made. These include: (1) improvements in analytical methods to characterize nanomaterials and reviewer training on these methods; (2) better understanding of how particle size change can affect product performance, including product quality; (3) additional need for clarification of policy in some situations where safety testing is typically not required or where there may be unintended exposure; and (4) development of nanotechnology-related educational opportunities for review staff. The four areas highlighted above are currently being addressed by both review and research staff within CDER. However, within the agency, in all the various centers, similar activities are ongoing, focusing on those areas that are of particular need to individual centers.
SUMMARY
This paper describes the approach undertaken by CDER’s Nanotechnology Risk Assessment Working Group, in developing a framework for identifying potential risks associated with the use of nanomaterials in drug products. The tools used by CDER in performing the risk assessment exercise are described, using as an example the oral route of administration. The working group is carrying out similar exercises for a number of routes of administration, excipients, and drugs that fall under the over-the-counter monograph system. It is expected that these exercises will lead to future discussions between stakeholders and CDER staff to develop a common understanding of nanotechnology-related product development and product review that will ensure product quality, safety, and efficacy.
ACKNOWLEDGMENTS
The authors would like to thank the following for their assistance in this exercise: Edward Bashaw, Scott Furness, Narayan Nair, Marie Angeline O’Shea, Arlene Solbeck, Reynold Tan, Douglas Throckmorton, and Helen Winkle.
The findings and conclusions in this article have not been formally disseminated by the USFDA and should not be construed to represent any USFDA determination or policy. The mention of commercial products, their sources, or their use in connection with material reported herein is not to be construed as either an actual or implied endorsement of such products by the Department of Health and Human Services.
Conflict of Interest
The authors are employed by the US Food and Drug Administration. The authors do not declare any other conflict of interest.
Footnotes
The findings and conclusions in this article have not been formally disseminated by the USFDA and should not be construed to represent any USFDA determination or policy. The mention of commercial products, their sources, or their use in connection with material reported herein is not to be construed as either an actual or implied endorsement of such products by the Department of Health and Human Services.
REFERENCES
- 1.Kim BYS, Kutka JT, Chan WCW. Nanomedicine. N Engl J Med. 2010;363:434–43. doi: 10.1056/NEJMra0912273. [DOI] [PubMed] [Google Scholar]
- 2.Bosselmann S, Williams RO. Has nanotechnology led to improved therapeutic outcomes? Drug Dev Ind Pharm. 2012;38(2):158–70. doi: 10.3109/03639045.2011.597764. [DOI] [PubMed] [Google Scholar]
- 3.Hu J, Johnston KP, Williams RO. Nanoparticle engineering processes for enhancing the dissolution rates of poorly water soluble drugs. Drug Dev Ind Pharm. 2004;30(3):233–45. doi: 10.1081/DDC-120030422. [DOI] [PubMed] [Google Scholar]
- 4.Lu WL, Qi XR, Zhang Q, Li RY, Wang GL, et al. A pegylated liposomal platform: pharmacokinetics, pharmacodynamics, and toxicity in mice using doxorubicin as a model drug. J Pharmacol Sci. 2004;95:381–9. doi: 10.1254/jphs.FPJ04001X. [DOI] [PubMed] [Google Scholar]
- 5.Schlossman D, Shao Y. Inorganic ultraviolet filters. In: Shaath NA, editor. Sunscreens: regulation and commercial development. Florida: Taylor & Francis; 2005. pp. 239–80. [Google Scholar]
- 6.FDA.gov. Draft guidance: considering whether an FDA-regulated product involves the application of nanotechnology. 2011. http://www.fda.gov/ScienceResearch/SpecialTopics/Nanotechnology/ucm257926.htm. Accessed Jun 2012.
- 7.Sadrieh N. Overview of CDER experience with nanotechnology-related drugs. In: Slides for the August 9, 2012 Meeting of the Advisory Committee for Pharmaceutical Science and Clinical Pharmacology. 2012. http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/AdvisoryCommitteeforPharmaceuticalScienceandClinicalPharmacology/UCM315773.pdf. Accessed 18 January 2013.
- 8.Office of Pharmaceutical Science. Reporting format for nanotechnology-related information in CMC review. 2010. http://inside.fda.gov:9003/downloads/ProgramsInitiatives/Drugs/ScienceResearch/UCM285306.pdf. Accessed May 2012.
- 9.FDA.gov. Guidance for industry. Q9 quality risk management. 2006. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM073511.pdf. Accessed May 2012.
- 10.Martin-Moe S, Lim FJ, Wong L, Sreedhara A, Sundaram J, Sane SU. A new roadmap for biopharmaceutical drug product development: integrating development, validation, and quality by design. J Pharm Sci. 2011;100(8):3031–43. doi: 10.1002/jps.22545. [DOI] [PubMed] [Google Scholar]
- 11.Scientific Committee on Emerging and Newly Identified Health Risks (SCENIHR). In: The appropriateness of existing methodologies to assess the potential risks associated with engineered and adventitious products of nanotechnologies. 2006. http://ec.europa.eu/health/ph_risk/documents/synth_report.pdf. Accessed May 2012.
- 12.Tsuji JS, Maynard AD, Howard PC, James JT, Lam CW, Warheit DB, et al. Research strategies for safety evaluation of nanomaterials, part IV: risk assessment of nanoparticles. Toxicol Sci. 2006;89(1):42–50. doi: 10.1093/toxsci/kfi339. [DOI] [PubMed] [Google Scholar]
- 13.FDA.gov. Guidance for industry. In: Q8(R2) pharmaceutical development. 2012. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM073507.pdf. Accessed Jun 2012
- 14.FDA.gov. Guidance for industry. Content and format of investigational new drug applications (INDs) for phase 1 studies of drugs, including well-characterized, therapeutic biotechnology-derived products. 1995. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM071597.pdf. Accessed Jun 2012.
- 15.FDA.gov. Guidance for industry. Applications covered by section 505(b)(2). 1999. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM079345.pdf?utm_campaign=Google2&utm_source=fdaSearch&utm_medium=website&utm_term=Applications covered by section 505(b)(2).&utm_content=1. Accessed Jun 2012.
- 16.FDA.Gov. Guidance for industry. Submission of summary bioequivalence data for ANDAs. 2011. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM134846.pdf. Accessed Jun 2012.