

Abstract
This investigation evaluated the impact of potential drug interactions on the incidence of reported toxicities seen with common dosing patterns in children with cancer, with the intent of being able to screen and reduce the incidence of adverse drug reactions (ADRs) in the future. Toxicity reported in pediatric cancer patients treated at the Children’s Hospital of Philadelphia from 2004 to 2010 were abstracted from a cancer tumor registry and merged with drug order profiles from the medical record system. Analysis datasets were created in SAS and permutation algorithms were used to identify pairwise drug combinations associated with specific toxicity occurrence. Relative risk of toxicity based on dosing pattern was assessed via comparison to control patients. A total of 326 of 1,713 patients (19%) had reportable toxicities. Neutrophil count decreases and alanine aminotransferase increases represented the highest occurring, corresponding to 28.8% and 31.9% prevalence among patients reporting toxicity, respectively. Of coadministered drug pairs, acetaminophen–diphenhydramine occurred most frequently; however, methotrexate–vincristine was the highest occurring pair linked to a single toxicity (hepatotoxicity). Toxicity was highly associated with the diagnoses of leukemia (52.1%) or neuroblastoma (28.5%). Comparison of the dosing interval (≤30 versus >30 min) suggested that risk of toxicity can be associated with the timing of coadministration, with ≤30 min increasing the risk of hepatotoxicity with fentanyl–midazolam and methotrexate–midazolam combinations. Knowledge of drug interactions in children with cancer may help reduce the incidence of ADRs by providing pharmacotherapy options that may reduce the likelihood of toxicity.
KEYWORDS: drug interaction, pediatric oncology, pharmacotherapy, toxicity
INTRODUCTION
The complexity of managing toxicities secondary to therapeutics in children, especially chemotherapeutics dosed to children with cancer, is significant. In addition to the adverse events (AEs) and serious adverse events (SAEs) commonly reported in these patients, adverse drug reactions (ADRs) are common.
The World Health Organization describes an ADR as “A response to a drug that is noxious and unintended and which occurs in doses normally used for the treatment, prophylaxis, or diagnosis of disease, or the modification of physiological function” (1). ADRs can be classified according to dose relatedness, time course, and susceptibility. A subset of ADRs includes those mitigated by the interaction of coadministered drugs or the so-called drug-dependent or drug–drug interactions (DDI). Although the potential for DDIs in children with cancer may be great with certain drug combinations, such information may not receive priority when prescribing because of the presumed favorable benefit–risk ratio.
Knowledge of DDI potential to benefit patient care is typically generated in two settings: (1) phase I, well-defined trials conducted by drug sponsors and (2) during the analysis of post-marketing ADR reporting screened against drugs with common mechanisms of drug elimination (primarily common metabolic pathways). However, in many instances, only in vitro (for example, microsome or hepatocyte) data exist and, when studies are performed, they are conducted in healthy adult volunteers (nontoxic anticancer agents only) or in patients that may not reflect the eventual target population (nonresponsive or stage IV patients) using simple pairwise drug combinations that are unlikely to reflect the true polypharmacy when treating children with cancer. While ADR reporting has improved as a source of DDI detection, it has been historically imprecise, and perceived underreporting has cast doubt on the validity of the “signals” identified (2).
In children with cancer, various registries of data, including those that collect AEs and SAEs are available in order to better understand the patterns of toxicities associated with frequently administered medications, including cytotoxic chemotherapeutics. In addition to the known AE profile of these agents, individual hospital pharmacy databases may provide additional information on reported ADRs either (a) not commonly associated with these medications or (b) reported because of suspected DDIs when concomitant noncytotoxic medications are administered.
While ADR registry data have limitations, including granularity on the time of event, time to resolution of the event, presence of preexisting conditions, details of other concomitant treatments, and little or no patient status indicators, there is still value in the toxicity signals and the magnitude and frequency of occurrences that this data can provide (3). The Children’s Hospital of Philadelphia (CHOP) cancer registry is an informational system that captures accurate and complete data on types of cancer diagnoses and treatments and includes patient demographics, treatment details (protocol names/numbers and protocol-mandated cancer therapies), dates and times of AEs and SAEs and their follow-up, and patient mortality. In addition, targeted toxicities unique to the care of pediatric cancer patients are captured in the registry as well. Hence, there is an implicit hierarchy of toxicity events that is qualified by grade and severity. The CHOP electronic medical record (EMR) system contains the complementary demographic, diagnostic, and medical history along with medication dosing information and laboratory data. The merger of these data sources provides an opportunity to evaluate the patient’s drug dosing history in a more comprehensive manner.
Our overall intention is to identify potentially meaningful DDI–toxicity correlations in children with cancer by developing a framework from which real-time patient data collected in toxicity registries and EMRs can be utilized to screen associations. Such relationships can then be challenged for plausibility based on mechanistic rationale and pharmacokinetic/pharmacodynamic (PK/PD) likelihood via in vitro simulation techniques. The objectives for this initial, exploratory investigation were (1) to assess the diversity of toxicities seen in children with cancer, (2) to evaluate the association between these toxicities and the coadministration of drug pairs prior to toxicity occurrence, and (3) to compare the reported incidence of specific toxicities based on differences in dosing patterns between patients reporting toxicity and those that did not to better understand the potential causal role of DDI in these patients. These data and analyses are critical to the challenge of maintaining an informed pediatric formulary in light of drugs coming on and off the market, drug shortages, and the continual stream of new clinical data.
METHODS
Data Collection and Assembly
Research presented herein is based on targeted toxicity data collected over a 7-year period (January 2004–July 2010) from the CHOP cancer registry merged with complementary drug dosing histories from the same patients over the same time period. These data were collected from two operational data sources: the hospital-based tumor registry and the EMR system (EPIC, Verona, WI, USA) for dataset creation and subsequent analyses. The tumor registry system is supported by an Oracle application database and is maintained by a data administrator. Reported AE data for pediatric cancer patients treated at CHOP between 2004 and 2010 were requested; the following fields were provided from the relational database: patient identifier, age in months, gender, diagnosis, diagnosis date, protocol title, protocol number, protocol date, AE description, event onset date, and toxicity grade. Approval for data abstraction for these analyses was given by the Division Chief of Oncology and based on the IRB approval granted to our pediatric knowledgebase initiative (4).
A report of the requested data fields was provided by the data administrator in a single Excel spreadsheet that contained data from 326 of 1,713 patients treated at this institution over the time period specified, with at least 1 ADR reported, including 2,357 unique records with 11 distinct variables. ADR severity was classified on a scale ranging from 0 to 5 based on the NCI Common Toxicity Criteria Adverse Event grading criteria (CTCAE version 4; http://evs.nci.nih.gov/ftp1/CTCAE/About.html). Patient data included single and/or multiple ADRs of varying grades over the course of their treatment. Toxicity categories for adverse reaction descriptions were also assigned based on NCI CTCAE criteria. Age categories were defined for neonates (≥0 and ≤1 months), infants (>1 and ≤24 months), children (>24 and ≤144 months), adolescents (>144 and ≤216 months), and adults (>216 months) based on US FDA pediatric age category cut-points. The ADR Excel file was subsequently imported into SAS for further data analyses.
Patients diagnosed on the Children’s Oncology Group (COG) or non-COG protocols were identified as the total pediatric cancer patient population (n = 1,713) treated over the targeted observation period. Patients not experiencing an ADR (control patients, n = 1,387) were considered for the comparison group. Comparisons of demographics, diagnosis, and protocol details of pediatric cancer patients across the two sample populations (patients experiencing a targeted toxicity and patients that did not) were made via basic descriptive statistics.
In order to assess drug utilization patterns as well as to associate drug administration time course with toxicity occurrence, drug order profiles (n = 319) of the patients who experienced ADRs were queried and exported from EPIC, the hospital EMR system using a web-based business objects application to abstract records. Output data were stored in multiple Excel files in password-protected locked computer servers without public access. The drug order data contained fields for patient identifiers, drug name, and date in order to provide accurate mapping to toxicity data from the tumor registry. Individual Excel files were imported into SAS and combined into a common dataset. Nutritional products, vitamins, vaccines, mouthwash, mask, and several locally acting or topical drugs (e.g., Neosporin, Chloraseptic, Maxitrol) were excluded from the dataset. Throughout the data abstraction process, incomplete/incorrect records or missing dosing details were observed to prohibit the exact mapping of registry and EMR data. A flow diagram illustrating the various data abstraction steps, dataset dimensions, and resultant analyses output is shown in Fig. 1.
Fig. 1.
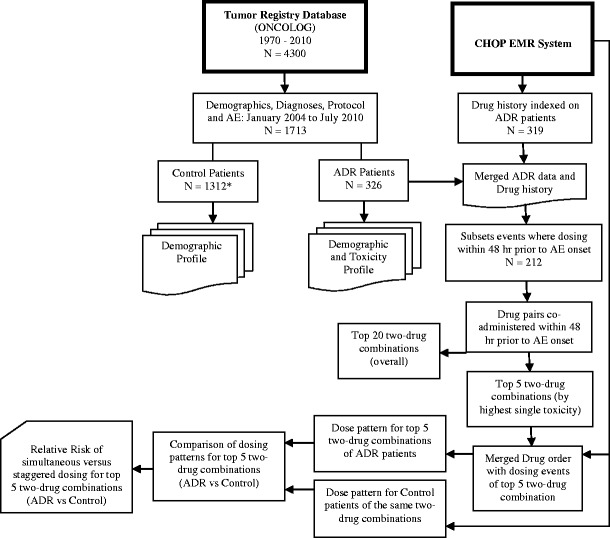
Data abstraction flow diagram illustrating subset sample size and analyses
Analyses
Toxicity Incidence and Patterns
Descriptive statistics and frequency analysis of observed ADRs were calculated by toxicity category in SAS. Categories with ADR frequency counts <5 were excluded from subsequent analysis. Toxicities were ranked by total frequency; the top 20 toxicities within and across age category and by toxicity grade were identified with SAS (data step). Patients on a COG-sponsored protocol were examined for timing of ADR onset based on the designation of protocol initiation as day 1 of a 28-day cycle. Hence, the onset time for ADRs occurring between days 1 and 28 of treatment were projected for the top 10 toxicity categories, and cumulative frequency plots were made to assess common patterns. Incidence rates were calculated based on the total pediatric oncology sampled population (ADR and control patients) and subset by age category.
Drug Utilization and Drug Combinations
Drugs utilized in the ADR and control patient groups were summarized and ranked; the top 20 utilized drugs in each group were assessed. Patients experiencing at least one ADR were further evaluated by identifying drug pairs administered within 48 h prior to reported AE onset in order to assess correlation with toxicity response. Specifically, all paired combinations of drugs administered within a 48-h window were assigned a unique identifier and the frequency of occurrence of these drug-paired dosing events was summarized across all patients in the dataset using SAS data steps and conditional logic. The selection of the 48-h window was based on the presumption that PK-mediated DDIs were observed more acutely within the proximity of actual drug administration. The coding solution is easily modified to more broadly assess associations with toxicity onset and resolution as opposed to peak effects.
The top 5 drug pair combinations within unique toxicity categories were identified and examined further for plausible DDI rationale. Two separate sources were examined for DDI potential: the Lexicomp Online™ drug interaction tool (http://www.lexi.com/institutions/hospitals/online/) and the metabolic pathways for the individual agents from the SuperCYP database (http://bioinformatics.charite.de/supercyp/index.php?site=get_drug_interaction). In addition, we performed a literature search to identify published evidence of metabolic pathways for drug pairs of interest.
The Lexicomp interaction tool provides risk ratings for drugs known or suspected as having a high potential for clinically relevant interactions. It also provides information on adverse reaction. Specifically, Lexicomp’s risk rating “C” indicates that the specified agents may interact with each other in a clinically significant manner and dosage adjustments of one or both agents may be needed in a minority of patients. SuperCYP is a comprehensive database on cytochrome P450 (CYP) enzymes, including a tool for the analysis of CYP–drug interactions. It provides a mechanism-based rationale for drug interaction potential for unique drug pairs.
Variation in Dosing Patterns
In order to assess the variation in the timing of coadministered agents, the top 5 drug pairs with the single highest toxicity occurrence were assigned to a subset and examination of the time interval between each drug administration for each pair was calculated. Dose administration records of patients receiving these five drug pairs were abstracted for both ADR and control patients via the business objects application and converted into Excel files which were subsequently imported into SAS. Each dataset contained drug names (generic or branded), dose amount and route of administration, and date and time the drug was administered. As patients were often given multiple doses of each drug per day, the dosing time between the two drugs was based on the first dosing occasion per day. Patient data were merged with a type variable designating group (ADR or control) assignment. Time windows were used to bin the individual patient concomitant medication dosing events: 0–30 min, 30 min–1 h, 1–2 h, 2–4 h, 4–6 h, and >6 h. Histograms of dosing event frequencies within each window for the five targeted drug pairs were generated to compare dosing patterns between toxicity and control patient groups.
Relative Risk
The risk of experiencing a specific toxicity when receiving certain drug pairs simultaneously relative to the risk when these medicines are administered separately, defined as at least 30 min apart, was assessed via conventional case–control analysis; relative risk (RR) ratios and 95% confidence intervals were calculated using SAS version 9.3 (SAS, Cary, NC, USA). The relationship defining the case–control scenarios for each drug pair is defined in the following table for clarity:
| Risk | Toxicity present | Toxicity absent |
|---|---|---|
| Simultaneous administration | a | b |
| Administration ≥30 min apart | c | d |
In this comparison, RR is defined as:
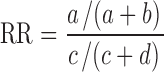 |
Two separate analyses were conducted for the five drug pairs most commonly linked to single agent toxicities. In the first analysis, all cancer diagnoses were considered; in the second analysis, only the two most common diagnoses (see Table I), acute lymphoblastic leukemia (ALL) and neuroblastoma, were considered due to the reduction in sample size of the analysis data with the “by group” analysis.
Table I.
Demographic Characteristics of Pediatric Oncology Patients Evaluated Based on Toxicity Occurrence from January 2004 to July 2010
| Age category | Toxicity population, n = 326, n (%) | Control population, n = 1,312, n (%) |
|---|---|---|
| Neonates, ≥0 and ≤1 months | 2 (0.6) | 26 (1.9) |
| Infants, >1 and ≤24 months | 37 (11.3) | 296 (22.6) |
| Children, >24 and ≤144 months | 210 (64.4) | 568 (43.29) |
| Adolescents, >144 and ≤216 months | 64 (19.6) | 346 (26.4) |
| Adults, >216 months | 13 (4.0) | 76 (5.8) |
| Gender | ||
| Female | 153 (46.9) | 576 (43.9) |
| Male | 173 (53.1) | 735 (56.0) |
| Diagnosis | ||
| Leukemia | 170 (52.1) | 270 (20.6) |
| Neuroblastoma | 93 (28.5) | 164 (12.5) |
| Bone tumors | 16 (4.9) | 71 (5.4) |
| Rhabdomyosarcoma | 14 (4.3) | 53 (4.0) |
| Hodgkin disease | 8 (2.5) | 90 (6.9) |
| Non-Hodgkin lymphoma | 7 (2.1) | 89 (6.8) |
| Kidney tumors | 6 (1.8) | 66 (5.0) |
| Central nervous system tumors | 4 (1.2) | 180 (13.7) |
| Other malignant tumors | 4 (1.2) | 48 (3.7) |
| Other soft tissue sarcomas | 2 (0.6) | 70 (5.3) |
| Retinoblastoma | 2 (0.6) | 116 (8.8) |
| Other | – | 116 (8.8) |
Patients with toxicities are summarized relative to patients not experiencing targeted toxicities. All patients were abstracted from the tumor registry database. Gender was not recorded for one patient in control group
RESULTS
ADR Incidence in Pediatric Cancer Patients
Over the period 2004 to July 2010, 1,713 hospitalized pediatric cancer patients were treated with inpatient cancer chemotherapeutics, with 326 reporting at least 1 ADR during their course of cancer treatment. The annual incidence rate of reported toxicity over this period ranged from 14.4% to 23.5%. There were 12 deaths reported (coincident diagnoses for reported deaths: leukemia (9), neuroblastoma (2), and severe distress syndrome, hypokalemia, and pneumonitis (1)).
Demographic comparisons between patients experiencing at least one ADR and those that did not over the observation period are shown in Table I. The age distribution was similar for both groups though there were more children (>24 and ≤144 months) experiencing ADRs relative to other age strata. This represented a 20% increase in this group compared to control patients (64.4% versus 44%). The gender partition was similar for both groups. There was a difference in the distribution of diagnoses between the two groups. Patients experiencing ADRs were overwhelmingly associated with diagnoses of leukemia (52.1%) and neuroblastoma (28.5%) as opposed to the control group which is more evenly distributed across many diagnoses, though leukemia (20.6%) maintains the largest contribution to the non-ADR group as well.
Toxicity Patterns in Relation to Drug Administration
The distribution of individual toxicities by severity (grades 1 through 4) for the 326 patients experiencing toxicity is shown in Table II. It should be noted that the focus of the tumor registry was to capture grades 3 and 4 toxicities. With respect to grade 3 toxicities, alanine aminotransferase (ALT) increases and febrile neutropenia were the most commonly occurring toxicities. Neutrophil count decrease was the highest occurring grade 4 toxicity. Overall, neutrophil count decrease and ALT increase represented the two highest occurring toxicities regardless of grade and corresponded to 28.8% and 31.9% prevalence rates among those patients reporting toxicity, respectively.
Table II.
Top 20 Toxicities and Age Category Observed in 326 Pediatric Cancer Patients Indexed Across Severity Grade (1 to 4)
| Top 20 toxicities (n = 326) | Grade1, n (%) | Grade 2, n (%) | Grade 3, n (%) | Grade 4, n (%) |
|---|---|---|---|---|
| Neutrophil count decreased | 5 (1.5) | 8 (2.5) | 50 (15.3) | 104 (31.9) |
| Alanine aminotransferase increased | 7 (2.1) | 10 (3.1) | 94 (28.8) | 21 (6.4) |
| Febrile neutropenia | 2 (0.6) | 13 (4.0) | 78 (23.9) | 11 (3.4) |
| Platelet count decreased | 8 (2.5) | 8 (2.5) | 27 (8.3) | 57 (17.5) |
| White blood cell decreased | 2 (0.6) | 9 (2.8) | 34 (10.4) | 54 (16.6) |
| Pain | 11 (3.4) | 15 (4.6) | 30 (9.2) | 3 (0.9) |
| Abdominal infection | 1 (0.3) | – | 50 (15.3) | 3 (0.9) |
| Aspartate aminotransferase increased | 5 (1.5) | 4 (1.2) | 35 (10.7) | 5 (1.5) |
| Hyperglycemia | 8 (2.5) | 2 (0.6) | 23 (7.1) | 16 (4.9) |
| Hypokalemia | 4 (1.2) | 1 (0.3) | 27 (8.3) | 10 (3.1) |
| Anemia | 5 (1.5) | 5 (1.5) | 27 (8.3) | 9 (2.8) |
| Diarrhea | 6 (1.8) | 9 (2.8) | 20 (6.1) | – |
| Hyponatremia | 7 (2.1) | 1 (0.3) | 19 (5.8) | 4 (1.2) |
| Peripheral motor neuropathy | 3 (0.9) | 15 (4.6) | 16 (4.9) | 5 (1.5) |
| Constipation | 3 (0.9) | 19 (5.8) | 7 (2.1) | 1 (0.3) |
| Fever | 14 (4.3) | 9 (2.8) | 5 (1.5) | 1 (0.3) |
| GGT increased | 2 (0.6) | 4 (1.2) | 20 (6.1) | 4 (1.2) |
| Anorexia | 3 (0.9) | 8 (2.5) | 13 (4.0) | – |
| Dehydration | – | 1 (0.3) | 21 (6.4) | 1 (0.3) |
| Mucositis oral | – | 7 (2.1) | 13 (4.0) | 1 (0.3) |
| Age Category | ||||
| Neonates | – | 1 (0.3) | – | 1 (0.3) |
| Infants | 4 (1.2) | 12 (3.7) | 26 (8.0) | 23 (7.1) |
| Children | 47 (14.4) | 92 (28.2) | 179 (54.9) | 124 (38.0) |
| Adolescents | 9 (2.8) | 27 (8.3) | 56 (17.2) | 30 (9.2) |
| Adults | 5 (1.5) | 7 (2.2) | 12 (3.7) | 5 (1.5) |
Toxicity frequency [n, (%)] based on the percentage of patients experiencing individual toxicities
With respect to the timing of ADRs, Fig. 2 shows the cumulative toxicity profiles for the 10 most frequently reported toxicities, based on the percentage of all patients reporting toxicity, over a 28-day window representative of a common single cycle of chemotherapy. This suggests that there are time dependencies that differentiate specific toxicity profiles consistent with mechanism-based progression of certain toxicities. Some ADRs occur early in the cycle and peak (e.g., hyperglycemia), while others show consistent occurrence over the 28 days, albeit not at the same steepness in response (neutrophil count decrease). While some of these differences are clearly associated with pharmacotherapy patterns in certain disease groups (e.g., prednisone use in ALL patients with respect to hyperglycemia), there is also the likelihood that dose intensity or cumulative exposure may be correlated with the event occurrence and the timing of the event. The implication is that algorithms used to associate and/or screen for ADRs must be informed by such clinical knowledge.
Fig. 2.
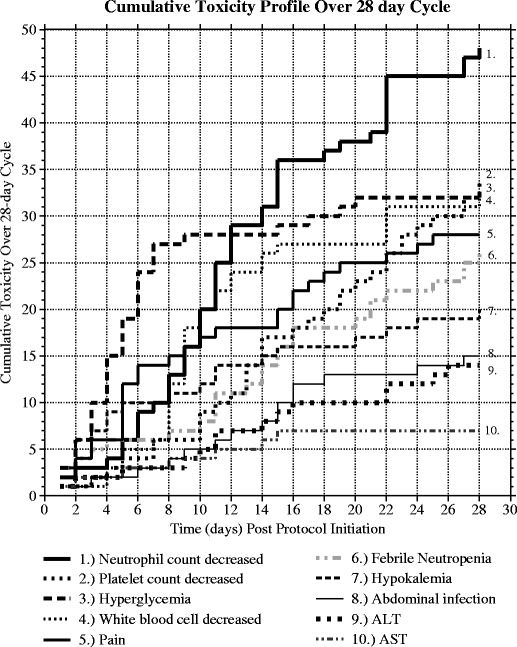
Cumulative frequency time to toxicity profiles over a 28-day observation window based on the first occurrence of study protocol initiation
Table III shows the most commonly utilized single agents among the 326 patients experiencing ADRs. Many of these are prescribed to mitigate the side effects associated with chemotherapy or to manage disease comorbidities (e.g., ondansetron, acetaminophen). The prevalence of chemotherapeutic agents (vincristine, methotrexate) is consistent with the primary diagnoses linked to this population (see Table I). In order to assess the potential for drug combinations to interact and likewise be correlated with toxicity response, the merged drug dosing and toxicity time course data were examined for the occurrence of common events within a 48-h window. Figure 3 illustrates the capture of such data from a representative patient. As this example indicates, during any window of inquiry, any number of drugs may be coadministered. All two-drug permutations were coded to represent targeted drug interaction candidates that have been subsequently summarized. Table IV shows the ranking of these combinations by various criteria. Based purely on occurrence, the ranking favors drug combinations based on utilization. Combinations with acetaminophen are common among the top 20 drug pairs due primarily to the ubiquitous nature of acetaminophen prescription. Controlling for the occurrence of any toxicity with drug pairings slightly modifies the ranking and affords a gross assessment of correlation. When ranking based on the most frequently occurring individual toxicity, a more realistic association of two-drug combinations with specific toxicities is observed. Most importantly, the prevalence of chemotherapeutic agents at higher ranks is far greater, and discriminating single-agent association with toxicity versus toxicities potentially promoted by drug interaction is possible. The top 11 drug combinations associated with a single toxicity are all correlated with hepatotoxicity (ALT increase). Commonly prescribed agents (vincristine, midazolam, and fentanyl) appear numerous times and are all associated with CYP3A4 metabolism, suggesting that some of these ADRs may be the result of PK-mediated DDI. Table V shows the association of these drug pairs with the top 3 single toxicities along with the assessment of common metabolic pathways. It should be noted that the Lexicomp risk rating of “C” (clinically significant interaction with dosage adjustments of one or both agents potentially needed in a minority of patients) is provided only for the fentanyl–midazolam combination; all others in Table V have no risk rating provided in the online formulary guidance at present.
Table III.
Top 20 Utilized Drugs in Oncology Patients Experiencing Toxicity (n = 319)
| Rank | Drug coadministered | Number of patients, n (%) | Frequency |
|---|---|---|---|
| 1 | Ondansetron | 277 (86.8) | 7,941 |
| 2 | Acetaminophen | 275 (86.2) | 6,027 |
| 3 | Diphenhydramine | 242 (75.9) | 5,663 |
| 4 | Cefepime | 166 (52.0) | 4,868 |
| 5 | Ranitidine | 185 (57.9) | 4,644 |
| 6 | Chlorhexidine | 99 (31.03) | 3,755 |
| 7 | Vincristine | 178 (55.80) | 3,621 |
| 8 | Lorazepam | 163 (51.1) | 3,132 |
| 9 | Dexamethasone | 156 (48.9) | 2,727 |
| 10 | Co-trimoxazole | 212 (66.5) | 2,685 |
| 11 | Gentamicin | 195 (61.1) | 2,366 |
| 12 | Vancomycin | 141 (44.2) | 2,248 |
| 13 | Docusate | 137 (42.9) | 1,975 |
| 14 | Morphine | 209 (65.5) | 1,968 |
| 15 | Acyclovir | 71 (22.3) | 1,846 |
| 16 | Fluconazole | 103 (32.3) | 1,843 |
| 17 | Methotrexate | 150 (47) | 1,805 |
| 18 | Midazolam | 228 (71.5) | 1,784 |
| 19 | Oxycodone | 189 (59.3) | 1,783 |
| 20 | Fentanyl | 202 (63.3) | 1,540 |
Fig. 3.
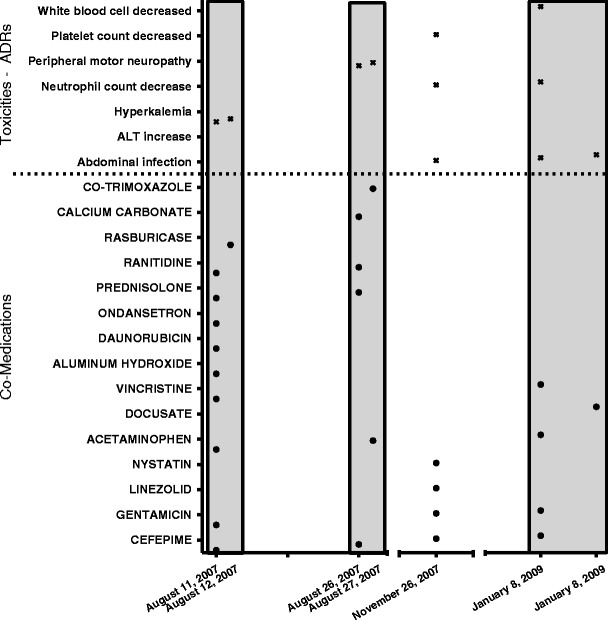
Representative patient drug coadministration and toxicity profile
Table IV.
Ranking of Two-Drug Combinations Among Pediatric Oncology Patients Experiencing Toxicities Based on Occurrence of Drug Administration (Within 48 h) and Toxicity Occurrence (Total Toxicities and Highest Single Toxicity)
| Rank | Occurrence of drug pair within 48 h | All toxicity events | Highest single toxicity | ||||
|---|---|---|---|---|---|---|---|
| Drug pair | Count | Drug pair | Count | Drug pair | Toxicity | Count | |
| 1 | Acetaminophen, diphenhydramine | 193 | Acetaminophen, diphenhydramine | 256 | Methotrexate, vincristine | ALT | 59 |
| 2 | Acetaminophen, cefepime | 161 | Acetaminophen, cefepime | 229 | Lidocaine, midazolam | ALT | 46 |
| 3 | Acetaminophen, ondansetron | 154 | Acetaminophen, ondansetron | 210 | Fentanyl, midazolam | ALT | 45 |
| 4 | Acetaminophen, gentamicin | 125 | Acetaminophen, gentamicin | 179 | Methotrexate, midazolam | ALT | 45 |
| 5 | Diphenhydramine, ondansetron | 122 | Diphenhydramine, ondansetron | 167 | Midazolam, vincristine | ALT | 45 |
| 6 | Cefepime, gentamicin | 110 | Cefepime, gentamicin | 160 | Lidocaine, vincristine | ALT | 42 |
| 7 | Ondansetron, vincristine | 108 | Fentanyl, midazolam | 153 | Fentanyl, methotrexate | ALT | 41 |
| 8 | Fentanyl, midazolam | 107 | Cefepime, diphenhydramine | 145 | Lidocaine, methotrexate | ALT | 41 |
| 9 | Cefepime, diphenhydramine | 102 | Methotrexate, vincristine | 141 | Fentanyl, lidocaine | ALT | 40 |
| 10 | Methotrexate, vincristine | 102 | Cefepime, ondansetron | 135 | Fentanyl, vincristine | ALT | 39 |
| 11 | Cefepime, ondansetron | 98 | Ondansetron, vincristine | 128 | Ondansetron, vincristine | ALT | 38 |
| 12 | Ondansetron, ranitidine | 94 | Acetaminophen, ranitidine | 123 | Acetaminophen, cefepime | Neutrophil count decreased | 37 |
| 13 | Acetaminophen, ranitidine | 89 | Ondansetron, ranitidine | 120 | Methotrexate, ondansetron | ALT | 33 |
| 14 | Acetaminophen, vancomycin | 81 | Lidocaine, midazolam | 118 | Acetaminophen, cefepime | Febrile neutropenia | 32 |
| 15 | Lidocaine, midazolam | 80 | Fentanyl, lidocaine | 108 | Acetaminophen, gentamicin | Febrile neutropenia | 31 |
| 16 | Methotrexate, ondansetron | 77 | Diphenhydramine, gentamicin | 107 | Cefepime, gentamicin | Febrile neutropenia | 30 |
| 17 | Cefepime, ranitidine | 76 | Acetaminophen, co-trimoxazole | 106 | Pentamidine, vincristine | ALT | 30 |
| 18 | Diphenhydramine, gentamicin | 76 | Methotrexate, ondansetron | 106 | Acetaminophen, diphenhydramine | Febrile neutropenia | 27 |
| 19 | Diphenhydramine, ranitidine | 76 | Acetaminophen, vancomycin | 103 | Acetaminophen, diphenhydramine | ALT | 25 |
| 20 | Midazolam, vincristine | 76 | Midazolam, vincristine | 103 | Cefepime, gentamicin | Neutrophil count decreased | 23 |
ALT alanine aminotransferase
Table V.
Association of Metabolic Pathways and Specific Toxicities for Top 5 Drug Pairs Correlated with Unique Targeted Toxicities
| Drug combination | Type of interaction (CYPs involved for each drug) | Count | Top 3 toxicities (frequency) |
|---|---|---|---|
| Fentanyl, midazolam | S (3A4, 3A5) | 107 | ALT increased (45); neutrophil count decreased (18); peripheral motor neuropathy (11) |
| S (3A4, 3A5, 3A7), I (3A4) | |||
| Methotrexate, vincristine | None | 102 | ALT increased (59); neutrophil count decreased (13); anorexia (9) |
| S (3A4, 3A5), I (3A4) | |||
| Lidocaine, midazolam | 3A4 and others | 80 | ALT increased (46); neutrophil count decreased (13); peripheral motor neuropathy (10) |
| S (3A4, 3A5, 3A7), I (3A4) | |||
| Vincristine, midazolam | S (3A4, 3A5), I (3A4) | 76 | ALT increased (45); neutrophil count decreased (13); peripheral motor neuropathy (6) |
| S (3A4, 3A5, 3A7), I (3A4) | |||
| Methotrexate, midazolam | None | 69 | ALT increased (45); neutrophil count decreased (10); peripheral motor neuropathy (10) |
| S (3A4, 3A5, 3A7), I (3A4) |
ALT alanine aminotransferase, S substrate, I inhibitor
Relative Risk—Association of Dosing Practices with Toxicity
Drug combinations linked to single toxicities identified in toxicity patients were also explored in the control patient group (n = 612). A comparison of the time interval between dosing was made and Fig. 4 shows a histogram of dosing time between the most common two combination drugs in both toxicity (top) and control (bottom) patients. While the general shape of these distributions is similar within a drug pair, there are some differences. Figure 5 shows the RR and 95% confidence intervals comparing toxicity and control patients based on whether they received drug pairs simultaneously or in a staggered manner (>30 min gap). RR for methotrexate–vincristine and midazolam–vincristine were close to 1 with confidence intervals that bracket 1 in both directions, indicating no evidence of dosing variation impacting risk of toxicity. Fentanyl–midazolam and methotrexate–midazolam had RRs of 3.72 and 1.64, respectively, suggesting an increased risk of developing hepatotoxicity with simultaneous administration of the two agents. Interestingly, lidocaine–midazolam had an RR of 0.24, suggesting that the risk of toxicity was actually greater with staggered administration as opposed to simultaneous administration. As Table V suggests, some of these relationships would be predicted based on known, common (CYP3A predominantly) metabolic pathways, while others lack a PK rationale (methotrexate combinations). Controlling for diagnoses yielded evaluable comparison for leukemia patients (predominantly ALL patients based on the CHOP population) only. Figure 5 shows the RR and 95% confidence intervals comparing toxicity and control patients based on whether they received drug pairs simultaneously or in a staggered manner, as in Fig. 5, but for leukemia patients only. While subtle shifts in the RR point estimate are observed, all relationships are maintained.
Fig. 4.
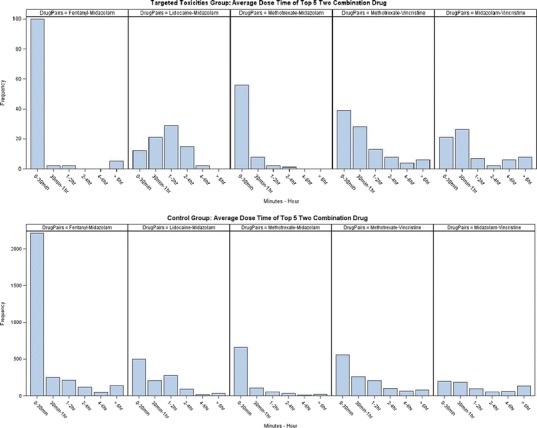
Dose time between drug pairs for ADR and control patients
Fig. 5.
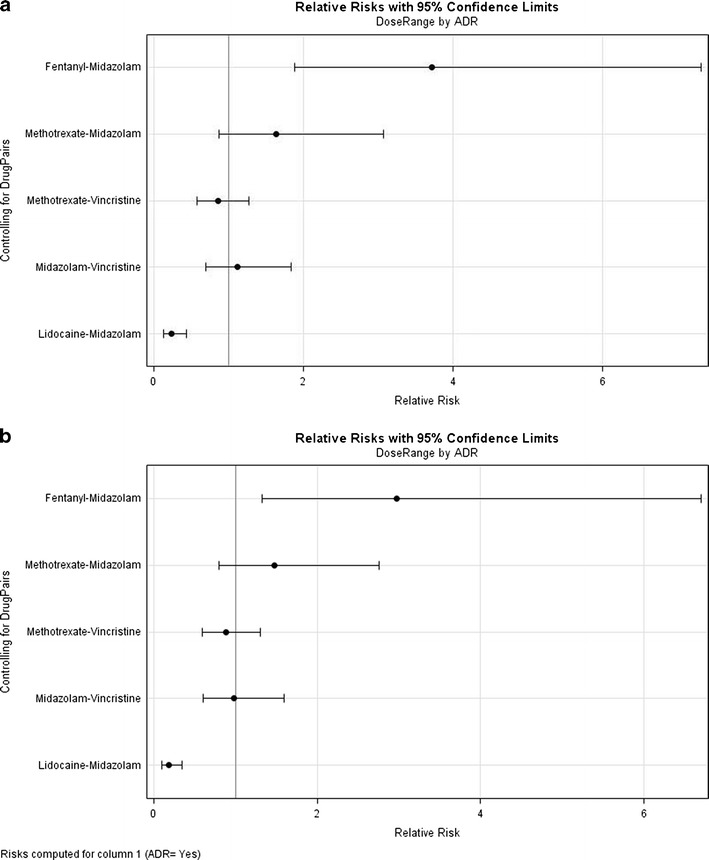
a RR rate comparing ADR and control patients based on whether they received drug pairs simultaneously or in a staggered manner (>30 min gap). b RR rate comparing ADR and control patients controlling for cancer diagnosis of leukemia (predominantly ALL patients based on CHOP population). 95% confidence intervals provided
DISCUSSION
The management of drug therapy in children can be a daunting task especially when precise dosing information and detailed reports on drug interaction potential is absent. Even when there is some information regarding DDI potential available, it often lacks quantitative description, making assessment of expected timing of events and, more importantly, the magnitude of the potential severity difficult to extrapolate to the individual patient. ADRs affect about 5–10% of adult medical inpatients (5,6); in children, it has been estimated at around 10% (7). Coupling this with the reported incidence of AEs in children with cancer treated with cytotoxic agents makes understanding the nature and impact of ADRs in children difficult.
Healthcare costs associated with AEs have been appreciated for some time (8,9) and have been the focus of numerous pharmacoeconomic investigations (10,11). Costs are substantial and correlated with the severity of the toxicity. Cancer patients are especially susceptible to such reactions because of the number and nature of medications administered. Detecting ADRs in pediatric patients is especially challenging (12); limited examples indicate diverse response and detection imprecision (12–14). Hence, the burden of DDIs to the patient, the caregiver, and the healthcare system is substantial and in need of concerted efforts for real gains to be made in each setting. Johnson and Bootman (15) have previously reported that drug-related morbidity and mortality was estimated to cost $76.6 billion in the ambulatory setting in the USA, with the largest component of total cost associated with drug-related hospitalizations. As the authors point out, there is considerable evidence that much of this morbidity is preventable. One potential solution for pediatric patients is the creation of surveillance networks to identify predictive genomic markers for ADRs (16,17). With this approach, children at risk would be identified before therapy is initiated, enabling personalized adjustments to therapy based on genetics.
We have attempted to create a preliminary framework from which ADRs can be assessed as well as decision logic that links toxicity occurrence with drug dosing practices. This initial “proof-of-concept” effort will be expanded to encompass associations based on dose intensity as well as toxicity patterns which occur at varying time courses, reflecting mechanistic understanding of cascade effects where such time courses are well appreciated. We also intend to explore the contributions of potential pharmacogenomic (PGx) interactions, PGx contributions to variability in PK, and sequence effects relating to the order of the administered medicines. While there are limitations of currently available data sources and tools to study such association, future efforts will also focus on refinement and clinical validation of these associations in order to provide clinical evidence and practical guidance to the ordering clinician. An important area of consideration is the influence of test frequency on the associations (e.g., toxicity “signal” influenced by the frequency of sampling to explain associations with liver enzyme elevations). Likewise, the exact observance of toxicity time course (onset, peak, and resolution) is not guaranteed with the routine care of these patients; future attempts to model toxicity will have to incorporate uncertainty regarding temporal effects, hopefully benefiting from well-studied time course evaluations. The first step and biggest challenge is to establish compelling evidence of causality as well as identify favorable pharmacotherapy practices which correlate with improved clinical outcomes (18). Reducing the reported incidence of toxicity attributed to DDIs is certainly a goal of future efforts.
Given that CYP3A4 is the most abundant CYP enzyme in the liver and gut, metabolizing approximately 50% of currently available drugs, it is not surprising that this enzyme is prominent in our analysis (Table V). Many important drugs have been identified as substrates, inducers, and/or inhibitors of CYP3A4. Substrates of CYP3A4 overlap considerably with those of P-glycoprotein, and both CYP3A4 and P-glycoprotein are subject to inhibition and induction. While such information is known about the majority of drugs on pediatric formularies, it is static and not tailored to the individual patient (19). A dynamic, real-time assessment of the potential risk of a drug-mediated ADR that was unique to an individual patient would provide a more compelling tool for prescribers, especially if alternative dosing strategies were recommended.
The timing of DDIs is not well appreciated. Competitive interactions typically occur soon after drug administration and are acutely observed. Likewise, the cascade of effects and the overall ADR time course resultant from a PK-mediated DDI would include exposure differences in parent or metabolite (beyond what was expected) and then some form of toxic reaction. However, these relationships are better appreciated in the care of cancer patients. Recent investigations confirm improved efficacy based on synergistic interactions from combination therapies and schedule dependence (20). Similar results should be expected when projecting toxicity outcomes. Our cumulative frequency plots reveal gross timing expectations for individual toxicities over a chemotherapy cycle, but they lack the granularity to provide guidance on the patient level.
The identification of liver enzyme increases as an indicator of hepatotoxicity should also be somewhat expected as this is an ADR commonly associated with polypharmacy. Recent studies in HIV-infected children also point to hepatotoxicity as a frequently occurring side effect during antiretroviral therapy (21). The key question is: Can hepatotoxicity be better managed via modified dosing paradigms for problem combinations? The first step is to differentiate toxicity attributed to the chemotherapeutic cytotoxic agents from those related to DDIs. Our RR analysis suggests that there may be a rationale for such strategies, though future analyses will also assess the impact with the frequency of sampling on these associations. When looking at the five drug pairs most commonly linked to single-agent toxicities across all cancer patients, the results suggest that simultaneous administration of some drug pairs (e.g., fentanyl–midazolam and methotrexate–midazolam) may indeed increase the risk of hepatotoxicity. Of course, there are many confounders with this analysis, and assignment of case and control groups to registry data is difficult given the variation in diagnosis, age, disease status, etc. When controlling for diagnosis (looking only at leukemia patients), this relationship is maintained but will require verification as well as extension to other cancer diagnoses. Our intention is also to examine the generalizability of these findings based on observational data collected by the Children’s Hospital Corporation of America (PHIS database). Obviously, these initial findings are relevant to pediatric cancer patients only. It is likely that associations defined herein may not be generalizable to other pediatric populations where cancer is not prevalent and when drug combinations and/or the rationale for coadministration differ.
The efforts summarized herein do not represent a definitive path forward given the plethora of unknowns regarding the mechanisms of ADRs, causality of DDIs, time dependencies, and developmental considerations which may promote age-specific considerations. Much research is still needed. Nonetheless, the merging of dosing history with ADR time course in children with cancer offers a previously underappreciated connection between drug utilization and toxicity. The selection of pairwise versus more complex drug combination permutations was simply for convenience at this initial stage. In silico strategies to consider multidrug interactions are in development and more sophisticated detection algorithms (22,23) could further improve the ability to discriminate problem combinations in a polypharmacy setting. The selection of the 48-h window to construct associations and the choice of dosing bins for comparing ADR and control patient groups were arbitrary as well. More rigorous justification for these cut-points and an appropriate sensitivity analysis on time windows for association will be critical next steps. In all likelihood, the concept of dynamic windows for unique toxicities will be required as previously alluded. The trajectories of these toxicities themselves will certainly depend on numerous factors. Given the works of Carleton and others (18,24–28), the benefit of linking genetic information to the data mart would seem obvious as well as the potential to improve association at the individual patient level.
CONCLUSIONS
Reducing the frequency and severity of pediatric toxicities would seem possible once more quantitative relationships between dosing practices, patient susceptibility, and toxicity time course can be constructed. Given the expense in healthcare costs related to ADRs in adults and children, it would seem that research in this area would have an immediate return on investment. A more coordinated effort with multi-institutional collaboration will be essential. Mechanistic understanding of ADRs may indeed be possible, though this will require more granular toxicity data, the addition of genetic information, drug-specific PK and PD knowledge, and an integrated medical informatics approach with the ability for machine learning. As always, this will necessitate a multidisciplinary collaboration of pediatric clinical pharmacology, drug metabolism, and clinical and medical informatics professionals.
ACKNOWLEDGMENTS
This work was partially supported by NICHD/NLM, Grant No. 1RC1LM010367-01, Decision Support System to Guide Pediatric Pharmacotherapy and an internal grant from the Pediatrics Chair’s Initiative of the Children’s Hospital of Philadelphia.
Competing Interests
Financial competing interests:
In the past 5 years, we have not received reimbursements, fees, funding, or salary from an organization that may in any way gain or lose financially from the publication of this manuscript, either now or in the future.
We do not hold any stocks or shares in an organization that may in any way gain or lose financially from the publication of this manuscript, either now or in the future.
We do not hold or have applied for any patents relating to the content of the manuscript. We have not received reimbursements, fees, funding, or salary from an organization that holds or has applied for patents relating to the content of the manuscript.
We do not have any other financial competing interests.
Nonfinancial competing interests:
There are no nonfinancial competing interests (political, personal, religious, ideological, academic, intellectual, commercial, or any other) to declare in relation to this manuscript.
REFERENCES
- 1.Cobert BL. Manual of drug safety and pharmacovigilance. Massachusetts: Jones and Bartlett; 2007. [Google Scholar]
- 2.van Puijenbroek EP, et al. Determinants of signal selection in a spontaneous reporting system for adverse drug reactions. Br J Clin Pharmacol. 2001;52(5):579–86. doi: 10.1046/j.0306-5251.2001.01501.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Willis CD, et al. Monitoring drug safety with registries: useful components of postmarketing pharmacovigilance systems. J Clin Epidemiol. 2012;65(2):121–5. doi: 10.1016/j.jclinepi.2011.06.017. [DOI] [PubMed] [Google Scholar]
- 4.Barrett JS, et al. Integration of modeling and simulation into hospital-based decision support systems guiding pediatric pharmacotherapy. BMC Med Inform Decis Mak. 2008;8:6. doi: 10.1186/1472-6947-8-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hallas J, et al. Drug related admissions to medical wards: a population based survey. Br J Clin Pharmacol. 1992;33(1):61–8. doi: 10.1111/j.1365-2125.1992.tb04001.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van der Hooft CS, et al. Adverse drug reaction-related hospitalisations: a population-based cohort study. Pharmacoepidemiol Drug Saf. 2008;17(4):365–71. doi: 10.1002/pds.1565. [DOI] [PubMed] [Google Scholar]
- 7.Clavenna A, Bonati M. Adverse drug reactions in childhood: a review of prospective studies and safety alerts. Arch Dis Child. 2009;94(9):724–8. doi: 10.1136/adc.2008.154377. [DOI] [PubMed] [Google Scholar]
- 8.Ioannides-Demos LL, Eckert GM, McLean AJ. Pharmacoeconomic consequences of measurement and modification of hospital drug use. PharmacoEconomics. 1992;2(1):15–33. doi: 10.2165/00019053-199202010-00004. [DOI] [PubMed] [Google Scholar]
- 9.Venulet J. Increasing threat to man as a result of frequently uncontrolled and widespread use of various drugs. Int J Clin Pharmacol Biopharm. 1975;12(4):387–94. [PubMed] [Google Scholar]
- 10.Lundkvist J, Jonsson B. Pharmacoeconomics of adverse drug reactions. Fundam Clin Pharmacol. 2004;18(3):275–80. doi: 10.1111/j.1472-8206.2004.00239.x. [DOI] [PubMed] [Google Scholar]
- 11.Paessens BJ, et al. Health resource consumption and costs attributable to chemotherapy-induced toxicity in German routine hospital care in lymphoproliferative disorder and NSCLC patients. Ann Oncol. 2011;22(10):2310–9. doi: 10.1093/annonc/mdq759. [DOI] [PubMed] [Google Scholar]
- 12.Weiss J, et al. Survey of adverse drug reactions on a pediatric ward: a strategy for early and detailed detection. Pediatrics. 2002;110(2 Pt 1):254–7. doi: 10.1542/peds.110.2.254. [DOI] [PubMed] [Google Scholar]
- 13.Aagaard L, Christensen A, Hansen EH. Information about adverse drug reactions reported in children: a qualitative review of empirical studies. Br J Clin Pharmacol. 2010;70(4):481–91. doi: 10.1111/j.1365-2125.2010.03682.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Clarkson A, et al. A novel scheme for the reporting of adverse drug reactions. Arch Dis Child. 2001;84(4):337–9. doi: 10.1136/adc.84.4.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Johnson JA, Bootman JL. Drug-related morbidity and mortality. A cost-of-illness model. Arch Intern Med. 1995;155(18):1949–56. doi: 10.1001/archinte.1995.00430180043006. [DOI] [PubMed] [Google Scholar]
- 16.Carleton B, et al. Adverse drug reaction active surveillance: developing a national network in Canada’s children’s hospitals. Pharmacoepidemiol Drug Saf. 2009;18(8):713–21. doi: 10.1002/pds.1772. [DOI] [PubMed] [Google Scholar]
- 17.Wong E, et al. Genotypic Approaches to Therapy in Children (GATC): using information technology to improve drug safety. Stud Health Technol Inform. 2009;143:209–14. [PubMed] [Google Scholar]
- 18.Carleton BC, et al. Paediatric adverse drug reaction reporting: understanding and future directions. Can J Clin Pharmacol. 2007;14(1):e45–57. [PubMed] [Google Scholar]
- 19.Enders SJ, Enders JM, Holstad SG. Drug-information software for Palm operating system personal digital assistants: breadth, clinical dependability, and ease of use. Pharmacotherapy. 2002;22(8):1036–40. doi: 10.1592/phco.22.12.1036.33601. [DOI] [PubMed] [Google Scholar]
- 20.Makiyama A, et al. Schedule-dependent synergistic interaction between gemcitabine and oxaliplatin in human gallbladder adenocarcinoma cell lines. Anticancer Drugs. 2009;20(2):123–30. doi: 10.1097/CAD.0b013e3283218080. [DOI] [PubMed] [Google Scholar]
- 21.Shah I. Adverse effects of antiretroviral therapy in HIV-1 infected children. J Trop Pediatr. 2006;52(4):244–8. doi: 10.1093/tropej/fmi086. [DOI] [PubMed] [Google Scholar]
- 22.Bate A. Bayesian confidence propagation neural network. Drug Saf. 2007;30(7):623–5. doi: 10.2165/00002018-200730070-00011. [DOI] [PubMed] [Google Scholar]
- 23.Bate A, et al. A Bayesian neural network method for adverse drug reaction signal generation. Eur J Clin Pharmacol. 1998;54(4):315–21. doi: 10.1007/s002280050466. [DOI] [PubMed] [Google Scholar]
- 24.Carleton B, Primmett D. Recognizing idiosyncratic adverse drug reactions in children: a practice imperative. Paediatr Child Health. 2001;6(4):187–9. doi: 10.1093/pch/6.4.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Castro-Pastrana LI, Carleton BC. Improving pediatric drug safety: need for more efficient clinical translation of pharmacovigilance knowledge. J Popul Ther Clin Pharmacol. 2011;18:e76–88. [PubMed] [Google Scholar]
- 26.Castro-Pastrana LI, et al. Cutaneous adverse drug reactions in children: an analysis of reports from the Canadian Pharmacogenomics Network for Drug Safety (CPNDS) J Popul Ther Clin Pharmacol. 2011;18:e106–20. [PubMed] [Google Scholar]
- 27.Lagnaoui R, et al. Adverse drug reactions in a department of systemic diseases-oriented internal medicine: prevalence, incidence, direct costs and avoidability. Eur J Clin Pharmacol. 2000;56(2):181–6. doi: 10.1007/s002280050738. [DOI] [PubMed] [Google Scholar]
- 28.Loo TT, et al. Pharmacogenomics and active surveillance for serious adverse drug reactions in children. Pharmacogenomics. 2010;11(9):1269–85. doi: 10.2217/pgs.10.111. [DOI] [PubMed] [Google Scholar]