

Abstract
The Ca2+-activated potassium channel of intermediate conductance, KCa3.1, is now emerging as a therapeutic target for a large variety of health disorders. The Ca2+ sensitivity of KCa3.1 is conferred by the Ca2+-binding protein calmodulin (CaM), with the CaM C-lobe constitutively bound to an intracellular domain of the channel C terminus. It was proposed on the basis of the crystal structure obtained for the C-terminal region of the rat KCa2.2 channel (rSK2) with CaM that the binding of Ca2+ to the CaM N-lobe results in CaM interlocking the C-terminal regions of two adjacent KCa3.1 subunits, leading to the formation of a dimeric structure. A study was thus undertaken to identify residues of the CaM N-lobe–KCa3.1 complex that either contribute to the channel activation process or control the channel open probability at saturating Ca2+ (Pomax). A structural homology model of the KCa3.1–CaM complex was first generated using as template the crystal structure of the C-terminal region of the rat KCa2.2 channel with CaM. This model was confirmed by cross-bridging residues R362 of KCa3.1 and K75 of CaM. Patch-clamp experiments were next performed, demonstrating that the solvation energy of the residue at position 367 in KCa3.1 is a key determinant to the channel Pomax and deactivation time toff. Mutations of residues M368 and Q364 predicted to form anchoring points for CaM binding to KCa3.1 had little impact on either toff or Pomax. Finally, our results show that channel activation depends on electrostatic interactions involving the charged residues R362 and E363, added to a nonpolar energy contribution coming from M368. We conclude that electrostatic interactions involving residues R362 and E363 and hydrophobic effects at M368 play a prominent role in KCa3.1 activation, whereas hydrophobic interactions at S367 are determinant to the stability of the CaM–KCa3.1 complex throughout gating.
INTRODUCTION
The calcium-activated channel of intermediate conductance, KCa3.1, plays a prominent role in a large variety of physiological events including immune reactions involving memory B and T cells (Wulff et al., 2004; Lam and Wulff, 2011), control of vascular tone via endothelium-derived hyperpolarizing factor and NO release (Félétou and Vanhoutte, 2007; Sheng et al., 2009; Köhler and Ruth, 2010; Hasenau et al., 2011), modulation of trans epithelial ion transport in Cl−-secreting cells (Singh et al., 2001; Szkotak et al., 2004; Nanda Kumar et al., 2010; Hasenau et al., 2011), and renal fibroblast proliferation and fibrogenesis (Grgic et al., 2009). Of importance is the current view that KCa3.1 is a key cellular effector in several life-threatening diseases such as atherosclerosis, restenosis, and traumatic injury–induced edema (Köhler et al., 2003; Toyama et al., 2008; Tharp and Bowles, 2009). The functional role of KCa3.1 also extends to excitable cells as well. Notably, an inhibition of the KCa3.1 was found to improve tissue protection and locomotor recovery after spinal cord injury, suggesting that blocking the KCa3.1 channel could be a potential therapeutic approach for treating secondary damage after spinal cord injury (Bouhy et al., 2011). Collectively, these data support KCa3.1 as a promising therapeutic target for a large variety of health disorders.
KCa3.1 is a tetrameric membrane protein, with each subunit organized in six transmembrane segments, S1–S6, with a pore motif between segment 5 (S5) and 6 (S6) (see Fig. 1 B). The KCa3.1 trafficking and Ca2+ sensitivity are conferred by calmodulin (CaM), with the CaM C-lobe constitutively bound to a domain in the membrane-proximal region of the intracellular C terminus of the channel (Joiner et al., 2001; Maylie et al., 2004). Several studies have shown that KCa3.1 activity can be stimulated by ATP via phosphorylation by the nucleoside diphosphate kinase NDPK-B of a histidine residue at position 358 located within the channel C-terminal region (Srivastava et al., 2006). The channel regulation by ATP appeared to depend on a multi-basic 15RKR17 motif in the N terminus (Fig. 1 B), suggesting a complex interaction between the channel N-terminal and C-terminal domains (Jones et al., 2007). Work from our laboratory has provided evidence that the C-terminal region of KCa3.1 interacts with the γ1 subunit of the metabolic-sensing kinase AMPK (Klein et al., 2009), suggesting a regulation by AMPK. It follows that KCa3.1 is part of a multi-protein complex involving several protein kinases.
Figure 1.

Structural model obtained for the Ca2+-CaM N-lobe–KCa3.1 complex. (A) Sequence alignment between the KCa3.1 segment extending from D304 to K373 and the corresponding sequence used as template for homology modeling based on the 1G4Y (Protein Data Bank; SK2) structure. Alignment was generated by MUSTER. Identical residues are identified in open boxes for an overall sequence identity of 45%. (B) Membrane topology of KCa3.1. KCa3.1 is a tetrameric protein, with each monomer organized in six transmembrane segments plus a pore region between segments 5 and 6. The channel Ca2+ sensitivity is conferred by CaM, with the CaM C-lobe constitutively bound to the 312–329 segment of the channel C-terminal region (CaMBD1). The Ca2+-dependent binding of the CaM to the channel involves the segment 344 to 353 (CaMBD2A) and a stretch of 13 amino acids from 360 to 373 (CaMBD2B). In the N terminus, a 15RKR17 motif has been identified as being potentially involved in N terminus/C terminus coupling. Also illustrated are two leucine zipper (LZ) domains important for channel expression at the membrane. (C) Ribbon representation of the Ca2+-CaM N-lobe–KCa3.1 complex. The initial model was generated with MODELLER using the 1G4Y (Protein Data Bank) crystal structure as template. Each KCa3.1 subunit comprises the amino acids D304 to K373, with the CaM represented in light yellow. The CaMBD2B (K360-K373) is colored cyan, and the CaMBD2A is represented in purple. (D) Close-up of the complex, illustrating residue R362 of KCa3.1 in close proximity of K75 of CaM, and R352 facing both E84 of CaM and E363 on the adjacent KCa3.1 subunit. (E) Illustration of M368 projecting inside the CaM N-lobe together with S367 and Q364. This figure also includes a space-filling representation of two calcium ions as pink-colored spheres. (F) Detailed representation of the interaction domain of S367, with S367 contributing to the intersubunit interface, which includes residues L345 and N349 of the adjacent subunit, and to the CaM N-lobe–CaMBD2B interface with residues M36, L39 (not depicted), and Q41 of CaM. Visualization was generated with Discovery Studio (Accelrys, Inc.).
There is not yet a high resolution 3-D structure of KCa3.1. Work from our laboratory has led to a molecular description of the open/closed KCa3.1 pore region using an approach combining substituted cysteine accessibility method (SCAM), computer-based homology modeling, and single-channel recordings (Banderali et al., 2004; Klein et al., 2007). It was proposed that the KCa3.1 Ca2+-activated gate was located at the level of the selectivity filter or close to it (Garneau et al., 2009), a conclusion confirmed in experiments demonstrating that the accessibility of the thiol-modifying agent, Ag+, to cysteines engineered in the channel cavity was independent of the channel-conducting state (Garneau et al., 2009). Such proposal is in agreement with experiments on the Ca2+-activated KCa2.2 channel that suggested a channel gate located deep in the channel central cavity, perhaps in the selectivity filter itself (Bruening-Wright et al., 2007).
The exact molecular mechanism underlying KCa3.1 opening in response to Ca2+ binding to the CaM–KCa3.1 complex in the C terminus remains to be elucidated. However, structural information pertinent to channel gating was obtained through the crystallization of CaM bound to the rat KCa2.2–CaM-binding domain (CaMBD) in the presence of Ca2+ (Schumacher et al., 2001). On the basis of this structure, it was proposed that a large-scale conformational rearrangement is taking place in the presence of Ca2+, where the N-lobe of CaM binds to a C-terminal segment of an adjacent channel monomer, resulting in a dimerization of contiguous subunits. This rearrangement would in turn lead to a rotation/translation of the associated S6 transmembrane domains and to the opening of the ion-conducting pore (Schumacher et al., 2001, 2004; Wissmann et al., 2002; Maylie et al., 2004). Kinetic measurements of KCa2.2, where terbium was used as CaM ligand instead of Ca2+, have indicated that ligand binding at the N-lobe of CaM is indeed responsible for channel opening (Li and Aldrich, 2009; Li et al., 2009). However, additional results have indicated that the CaM N-lobe also contributes to Ca2+-independent stable interactions between CaM and the KCa2.2 subunits (Li et al., 2009).
One of the distinguishing features of the KCa3.1 Ca2+ dependence is that Pomax, the channel open probability at saturating Ca2+ concentrations (>20 µM), remains low, typically 0.1–0.2 for the WT channel, compared with a Pomax of 0.8 reported for the KCa2.2 channel in the high activity mode (Hirschberg et al., 1998). A higher Pomax value would essentially lead to an increased channel activity without a modification of the channel Ca2+ sensitivity. Such behavior was observed using hydrolyzable ATP analogues (Gerlach et al., 2000) and is at variance with the shift in Ca2+ sensitivity observed for KCa2.2 because of the action of the CK2 kinase 1. In addition, the observation of a low Pomax at saturating Ca2+ conditions clearly argues for the binding of Ca2+ to the CaM–KCa3.1 complex, favoring the formation of a closed-state configuration from which an open configuration can be reached. Such behavior has been documented in numerous ligand-gated channels where the agonist does not alter the open state but brings the channel to a preopen configuration. In this regard, results obtained with constitutively active KCa3.1 channel mutants, or after modifications of cysteine residues engineered along the S6 segment by selective reagents, support a prominent role of the S6 segment conformational state in determining Pomax (Garneau et al., 2009; Bailey et al., 2010). In the context of a channel gate located at the level of the selectivity filter region, this observation is consistent with a model where the binding of Ca2+ at a high concentration to the CaM N-lobe–KCa3.1 complex would position the S6 transmembrane segment to interact with the selectivity filter region, thus setting the structural bases underlying Pomax. A study was thus undertaken to identify residues of the CaM N-lobe–KCa3.1 complex that either contribute to the channel activation process or control the channel Pomax. Our results show that electrostatic interactions involving residues R362 and E363 are determinant to the activation rate of KCa3.1 at saturating Ca2+ concentration with little changes to Pomax, whereas hydrophobic effects at S367 contribute to Pomax.
MATERIALS AND METHODS
Computer-based homology modeling
The 3-D structures of the C-terminal region of KCa3.1 (D304-K373) with CaM were generated through homology modeling (Sali and Blundell, 1993) using as template the crystal structure (1.6-Å resolution) of the C-terminal region of the rat KCa2.2 channel (rSK2) with CaM (Protein Data Bank accession no. 1G4Y) (Schumacher et al., 2001). Homology modeling is fully justified in this case, as the amino acid sequence of KCa3.1 in this region is 45% identical to template. Sequence alignments were obtained with MUSTER (Wu and Zhang, 2008), and 3-D models were generated using MODELLER 9v4 (Sali and Blundell, 1993). 100 structural models of KCa3.1 CaMBD domain–CaM complex were generated and optimized to minimize possible violations on the spatial restraints. Ranking of the model structures was based on a probability density function (PDF) that accounts for the various structural features of the models. 14 models were selected based on their low PDF and root mean square deviations (RMSDs) of the Cα relative to template. The model with the lowest RMSD (1.7 Å) was kept and used as an initial model structure for the KCa3.1–CaM complex. The overall structural quality of the model was tested with PROCHECK.
Molecular dynamics (MD) simulations
MD simulations were performed using CHARMM-CGENFF with explicit solvent water molecules. The system that comprised two KCa3.1 C-terminal segments (D304-K373) forming a complex with two CaM molecules (A1-A147) was solvated in a 108-Å side cubic cell containing 37 100 TIP3P model water molecules. Altogether, the system consisted of 118 507 atoms including 115 K+ and 97 Cl− ions to ensure electroneutrality at near physiological concentration. Cut-on and cut-off parameters needed to define nonbounded interactions were set to 10 and 12 Å, respectively, and SHAKE constrains were used to fix lengths of bonds involving hydrogen atoms. Trajectories were generated for 10 ns using a time step of 2 fs, and electrostatic plus van der Waals interaction energies computed from trajectories were sampled at 0.2 ns. MD simulations were performed for a system at constant pressure (1 atm) and constant temperature (300°K). Solvent accessibility surface area (SASA) was averaged over a 10-ns trajectory using a standard probe of 1.4 Å.
Channel expression and molecular biology
WT and mutant KCa3.1 channels were expressed in Xenopus laevis oocytes alone or coexpressed with mutant CaM as needed by injection of their RNAs transcribed in vitro from pT7TS vector, which contains 5′ and 3′ untranslated regions of Xenopus β-globin mRNA. The mammalian CaM cDNA was obtained by RT-PCR from human cells (HEK-293) and cloned into the pT7TS vector. Approximately 10 ng KCa3.1 RNA and 10–30 ng CaM (WT or mutant) RNA were injected into each oocyte. Because mutated CaM has to compete with the endogenous CaM (WT), CaM RNA injections were done 6 h before KCa3.1 channel injections to ensure formation of KCa3.1 channel complexes with the mutated CaM. Recordings were typically performed 4–7 d after injection. All the mutants were generated using the Quickchange mutagenesis kit (Agilent Technologies) and verified by sequencing (Banderali et al., 2004). Before patch clamping, the defolliculated oocyte was briefly incubated in a hyperosmotic solution containing (mM) 250 KCl, 1 MgSO4, 1 EGTA, 50 sucrose, and 10 HEPES, buffered at pH 7.4 with KOH; the vitelline membrane was peeled off using fine forceps. The oocyte was then transferred to a superfusion chamber for patch-clamp measurements.
Solutions
The bath and patch pipette solutions contained (mM) 200 K2SO4, 1.8 MgCl2, 0.025 CaCl2, and 25 HEPES, buffered at pH 7.4 with KOH (referred to 200 mM K2SO4). Sulfate salts were used to minimize the contamination from endogenous Ca2+-dependent chloride channels while enabling the chelation of contaminant divalent cations such as Ba2+. Calcium-free solutions were prepared by adding 1–10 mM EGTA to 200-mM K2SO4 solutions without CaCl2. The cysteine-modifying agents [2-(trimethylammonium) ethyl] MTS bromide (MTSET+) and sodium (2-sulfanoethyl) MTS (MTSES−) (Toronto Research Chemicals Inc.) were added directly into the bath solution just before application. Bath solution changes were performed as described previously, using a rapid solution changer system (RSC-160; Biological) (Banderali et al., 2004). The solution exchange time was <20 ms.
Patch-clamp recordings
Multiple- and single-channel inside-out recordings were performed using an amplifier (Axopatch 200A; Molecular Devices). Patch pipettes were pulled from borosilicate capillaries using a pipette puller (model PP-83; Narishige) and used uncoated. The resistance of the patch electrodes ranged from 2 to 5 MΩ. Data acquisition was performed using a Digidata 1320A acquisition system (Molecular Devices) at a sampling rate of 1.0 kHz, unless specified otherwise. Single-channel analysis was performed using the QUB package (Qin et al., 1996, 1997). Dwell-time analysis was performed on data that were idealized according to the segmental-k means method based on a hidden Markov model–type analysis. Channel activation times (ton) were obtained by curve fitting to a single-exponential function the time course of the current increase after a Ca2+ concentration jump from 0 to 25 µM (Origin7; OriginLab Corporation). The exponential increase in channel activity was normally preceded by a short silent period ranging from 50 to 200 ms, most likely reflecting transitions among closed-state configurations before channel opening. This delay was not included in our curve-fitting procedure, but it could be estimated through a first latency analysis, where the time lapse between the Ca2+ concentration change and the first channel opening was directly measured from single-channel recordings. The channel deactivation times (toff) were computed by fitting to a mono-exponential function the time course of the current decrease after the replacement of a 25-µM Ca2+ by a 0-Ca2+ internal solution. Modification rates by MTS reagents (1 mM) of targeted Cys residues were obtained by curve fitting to a single-exponential function the time course of the current variation initiated by MTS application. Algebraic calculations of the proposed kinetic scheme were performed with Mathematica8 (Wolfram Research).
Cysteine cross-bridging experiments
Selected residues were individually mutated into Cys, and spatial proximity was tested using copper phenanthroline (Cu(II)Phe; Sigma-Aldrich) as a zero-length bridging agent. For these experiments, 1 µM of Cu(II)Phe-containing solutions was prepared from a 1-mM stock solution. The presence of disulfide bonds was assayed using a dithiothreitol (DTT; 10 mM; Sigma-Aldrich) containing K2SO4 solution. Each solution was freshly prepared every day.
Membrane preparation and Western blotting
Oocytes expressing KCa3.1–CaM mutants were incubated for 10 min with 10 µM ionomycin in a buffer containing (mM) 200 KCl, 0.5 MgSO4, 25 HEPES, and 2 CaCl2, pH 7.3. After incubation in ionomycin, disulfide cross-linking was performed at 20°C with the addition of 100 µM Cu(II)Phe for 30 min to the same buffer. The oocytes were gently rinsed in PBS, pH 7.4, and homogenized in fresh PBS, pH 7.4, plus a mixture of protease inhibitors (Complete EDTA-free; Roche). Yolk and other oocyte debris were removed by two repeated 5-min low speed centrifugations at 210 g. The supernatants containing crude cell membranes were pelleted for 20 min at 20 800 g, and membranes were solubilized in PBS, pH 7.4, containing 2% sodium deoxycholate for 30 min at 4°C. Insoluble material was removed by centrifugation at 20 800 g at 4°C, and solubilized cell membranes were quantified using the BCA protein assay method (Thermo Fisher Scientific).
For each sample, an aliquot containing 5 µg of total protein was combined with a buffer containing 65 mM Tris-HCl, pH 6.8, 10% glycerol, 2% SDS, and 0.03% (wt/vol) bromophenol blue with or without 1 mM DTT. The samples were separated onto a 10% polyacrylamide gel, and protein was transferred by electroblotting to nitrocellulose membrane (Hybond-C; GE Healthcare). The membranes were blocked in TBS 0.1% Tween 20 (TBS-T buffer) containing 5% nonfat dry milk, and then incubated overnight at 4°C in blocking solution containing a 1:5,000 dilution of a rabbit polyclonal antibody directed against KCa3.1 channel (ab75956; Abcam) or against CaM (ab45689; Abcam). After washing in TBS-T buffer and hybridization with horseradish peroxidase–conjugated goat anti–rabbit IgG at 1:25,000 dilutions (Jackson ImmunoResearch Laboratories, Inc.), proteins were detected using the ECL plus chemiluminescence kit (GE Healthcare).
Statistical analysis
Statistical significance was analyzed using an unpaired Student’s t test. Only experiments with a P of <0.005 were considered for analysis. Data are expressed as mean ± SD.
Online supplemental material
Fig. S1 illustrates results coming from inside-out patch-clamp experiments providing evidence for E84 of CaM being within the electrostatic interaction range of R352 of KCa3.1. Charge-restitution experiments were performed using either the R352C(KCa3.1)–E84(CaM) or R352C(KCa3.1)–E84A(CaM) systems exposed to the MTSET+ reagent. Whereas binding of MTSET+ to R352C(KCa3.1)–E84(CaM) caused channel inhibition, this effect was absent when R352C(KCa3.1) was coexpressed with E84A(CaM). Fig. S2 illustrates the effect of mutating residues of the CaMBD2B (K360 to K373) of KCa3.1 by their KCa2.2 equivalents on the channel activation time. These experiments demonstrate that substituting 7 amino acids of the K360-K373 segment by the KCa2.2 equivalent residues reduced the channel activation time from 306 ± 41 ms (n = 8) for WT KCa3.1 to 91 ± 31 ms (n = 5), a value still five times slower than the activation time measured for the KCa2.2 channel (17 ± 2 ms; n = 3). Figs. S1 and S2 are available at http://www.jgp.org/cgi/content/full/jgp.201210933/DC1.
RESULTS
The CaM–KCa3.1 complex
A topological representation of a single KCa3.1 subunit, with several of the structural features of the channel N-terminal and C-terminal regions, is presented in Fig. 1 B. In addition to the CaMBD1 described as being responsible for the Ca2+-independent binding of the CaM to KCa3.1 (Schumacher et al., 2001), this representation also illustrates the 15RKR17 motif in the N terminus proposed to interact with the channel C-terminal region (Jones et al., 2007) and two leucine zipper domains involved in channel expression at the membrane (Syme et al., 2003). The CaMBD responsible for the Ca2+-dependent binding of CaM to KCa3.1 is extending from residue L344 to Q353 (Fig. 1 B, CaMBD2A, purple) and residue K360 to K373 (CaMBD2B, cyan), with CaMBD2B being in contact with the CaM N-lobe. Fig. 1 C presents a ribbon representation of the resulting Ca2+–CaM–KCa3.1 complex generated through homology modeling, after a 200-ps equilibration period in water. The sequence alignment used for homology modeling is illustrated in Fig. 1 A, revealing a sequence identity of 45% relative to the sequence of the structural template (Protein Data Bank accession no. 1G4Y). The channel C-terminal region covers residues D304 to K373, with a 5–amino acid-long hairpin folding centered at R331. The overall stoichiometry corresponds to one CaM molecule per monomer in accordance with the crystal structure of KCa2.2 (Schumacher et al., 2001). The complex is dimeric, with the CaM C- and N-lobes enfolding two KCa3.1 subunits.
An analysis was next undertaken to determine which residues of the KCa3.1 CaMBD2 contribute to the interface between adjacent KCa3.1 subunits and/or between CaM and individual KCa3.1 subunits. Our approach consisted of computing for each residue of CaMBD2A and CaMBD2B the SASA for a single subunit in the absence of CaM, for two subunits without CaM, and finally for the entire CaM–KCa3.1 dimeric complex. The results of this analysis are presented in Fig. 2. It follows from Fig. 2 A that residues E363 and S367 are the main constituents of the intersubunit interface with, respectively, 76 and 81% of their maximum SASA reduced as a result of the presence of a second subunit. Complementary to these results (Fig. 2 B), residues L345 and N349 in the CaMBD2A show a reduction of 83 and 63% of their maximum SASA in the presence of a second subunit, an effect attributable to L345 and N349 of one subunit forming part of the intersubunit interface with S367 of the second subunit (Fig. 1 F). The results on the CaM–KCa3.1 interface are presented in Fig. 2 (C and D). Drastic changes in SASA caused by the presence of CaM were observed with L361, Q364, V365, N366, S367, M368, V369, and S372. Notably, SASA reductions of >90% were obtained for L361, Q364, S367, M368, V369, and S372 after the addition of CaM to the subunit–subunit complex. With the exception of S367, none of these residues contributes to the intersubunit interface. Similarly, 99 and 98% reductions in SASA caused by CaM were estimated for N349 and F351 of the CaMBD2A. It follows from this analysis that (a) residues S367 and N349 are unique, as they contribute to both the intersubunit and CaM–KCa3.1 interfaces; (b) Q364, M368, and V369 are the main determinants of the CaM–KCa3.1 interface, with >99% of their SASA exposed to the CaM N-lobe; and (c) in contrast to the residues of the CaMBD2A and CaMBD2B, R362 contributes neither to the intersubunit nor to the CaM–KCa3.1–N-lobe interfaces. The results of this analysis are summarized in the horizontal bar graph shown in Fig. 3 A, where buried residues with <10% of the maximum surface exposed to solvent were compared with exposed residues with >30% of their maximum surface exposed to solvent. These calculations were based on 10-ns MD trajectories obtained for the CaM–KCa3.1 complex, a time suitable to capture state transitions over an energy barrier of 4.4 kcal/mol. Buried residues include Q364, S367, M368, V369, and S372, whereas only R362 has >90% of its maximum surface exposed to solvent. As expected from models obtained by homology modeling, those features translate to the KCa3.1 channel the structural constraints described for the KCa2.2 channel (Schumacher et al., 2001).
Figure 2.

Structural analysis of the CaM–CaMBD2 interactions. (A and B) SASA was computed for each residue of CaMBD2B (K360-K373) or CaMBD2A (L344-Q353) for either a single KCa3.1 subunit with no CaM (open boxes) or in the presence of a second subunit with no CaM (filled boxes). Calculations were based on the structural model presented in Fig. 1. A strong decrease in SASA was obtained for E363 and S367, indicating an important contribution to the intersubunit interface. Identical calculations performed for residues in CaMBD2A identified L345 and N349 as contributing to the intersubunit interface. (C and D) Variations in SASA caused by the addition of CaM to the KCa3.1 subunit–subunit complex. A strong decrease in SASA caused by CaM was obtained for Q364, M368, V369, and S372 in agreement with those residues lining the CaMBD2B–CaM N-lobe interface. The most important variation was seen with M368, which is projecting inside the CaM N-lobe core. In contrast, SASA computed for R362 and E363 were not significantly affected by the addition of CaM, in accordance with R362 and E363 not contributing to the CaMBD2B–CaM N-lobe interface. Collectively, R362 appeared as the most solvent accessible residue, with no contribution to either the intersubunit or CaMBD2B–CaM N-lobe interfaces. Results obtained for the CaMBD2A support a strong decrease in SASA caused by CaM for I348, N349, and F351, an indication that these residues are located at the CaMBD2A–CaM interface. Residue R352 was found to be highly solvent accessible, with 50% of its surface in contact with CaM.
Figure 3.

SCAM of the CaM N-lobe–CaMBD2B complex. (A) Percentage of the maximum SASA computed for each residue of the CaMBD2B remaining accessible to solvent after formation of the KCa3.1–CaM complex, as illustrated in Fig. 1. The maximum SASA was taken as the SASA value obtained for a single KCa3.1 subunit in the absence of CaM. Dotted lines indicate areas referring to exposed residues with >30% of their maximum SASA remaining accessible to solvent and buried residues with <10% of their maximum SASA accessible to solvent. Buried residues include Q364, S367, M368, V369, I371, and S372, whereas R362 was confirmed as being the most solvent-exposed residue within the CaMBD2B. Values were obtained from MD trajectories spanning a total of 10 ns. The standard deviations in this case reflect the degree of structural variations during 10-ns simulations. (B) Accessibility of Cys engineered along the CaMBD2B segment to either 1 mM MTSET+ or 1 mM MTSES−. MTS reagents were applied internally according to the pulse application protocol illustrated in C. The ratio IMTS/Ictrl was taken as the ratio of the current amplitude for the Ca2+ pulse after MTS washout over the current amplitude obtained for the Ca2+ pulse before MTS application. Important changes were seen with the R362C, E363C, and Q364C mutants despite the fact that Q364C was predicted to be buried inside the CaM N-lobe–KCa3.1 complex and thus unlikely to be available for MTSET+ binding. The most important changes were seen with residue R362, which is highly solvent accessible. Values not included in the filled area refer to IMTS/Ictrl for which P < 0.005 relative to WT (IMTS/Ictrl (WT) ± 2.5 SD). (C) Examples of inside-out current recordings illustrating the action of MTS reagents on the R362C, E363C, Q364C, and V369C mutant channels. Experiments were performed in symmetrical 200-mM K2SO4 solutions with 25 µM of internal Ca2+ at a pipette potential of 60 mV on channels expressed in Xenopus oocytes. Label “c” refers to zero current measured in Ca2+-free conditions (thick line). Open boxes correspond to perfusion with a Ca2+ (25 µM) plus MTS reagent–containing solution (1 mM). Bars, 50 pA (R362C and E363C) or 200 pA (Q364C and V369C) and 10 s.
Probing the structural model of the Ca2+-CaM N-lobe–KCa3.1 complex
The model presented in Fig. 1 C was obtained by homology modeling and cannot therefore be as accurate as the structure used as template (Protein Data Bank accession no. 1G4Y). Experiments were thus performed to determine to what extent a model based on the crystal structure obtained for the KCa2.2 channel can be used to relate some of the channel gating properties to specific residue–residue interactions. The predicted pattern of solvent accessibility was first examined in SCAM experiments in which the MTSET+ and/or MTSES− reagents were used as probes to measure the accessibility of Cys residue engineered at each position in the CaMBD2B. MTSET+ and MTSES− are small molecules that can fit into a cylinder 10-Å long by 6 Å in diameter, resulting in Cys–MTSET+ and Cys–MTSES− complexes with respective side-chain volumes of 162 and 106 Å3 compared with 116 Å3 for Trp. In addition, the binding of MTS reagents to Cys requires that the thiol group be in a deprotonated form and exposed to water. In fact, the reaction of MTS with protonated cysteine was reported to be 109-fold slower than the reaction with cysteine in a deprotonated form (Karlin and Akabas, 1998). Significant modifications (P < 0.005) were seen with the R362C, E363C, and Q364C mutants despite the prediction of a mean SASA value of 0.3 Å2 for Q364 compared with 95 and 20 Å2 for R362 and E363, respectively. Furthermore, an estimation of the Q364C modification rate by MTSET+ led to a value of 4.1 ± 0.1 × 103 M−1s−1 (n = 3) compared with 1.0 ± 0.2 × 104 M−1s−1 (n = 3) obtained for the solvent-accessible Cys engineered at 362. These values would be compatible with MTSET+ being freely accessible to Cys engineered at position 362, with a reduced accessibility at 364, reflecting either additional stearic constraints as a result of molecular packing or a reduction in deprotonated Cys, or both. However, these SCAM measurements need to be interpreted in light of the MD results indicating maximum SASA values of 160, 83, 14, and 11 Å2 for R362, E363, Q364, and S367 over 10-ns trajectories, suggesting the possibility of structural variations of sufficient amplitudes to allow MTS modifications. In contrast, maximum SASA obtained for M368 was estimated at 4.8 Å2, arguing for strong structural constraints not present for by R362 and E363. Our SCAM data tend therefore to support a dynamic structural model with important variations in surface contact areas between CaM and KCa3.1, and between individual KCa3.1 subunits as well.
The analyses presented in Fig. 3 A indicate that R362 is unique, as it is highly solvent accessible while being potentially in contact with a single CaM residue. The proximity of R362(KCa3.1)–K75(CaM) was thus studied in a series of experiments in which Cu(II)Phe was used as a zero-length bridging agent. KCa3.1 WT contains 13 endogenous Cys residues of which none is located in cystosolic linkers or the C terminus. The production of a Cys-free KCa3.1 led to a channel that was poorly functional with rapid rundown (unpublished data), and KCa3.1 was found to be Cu(II)Phe sensitive, an effect attributable to the presence of four Cys residues located along the channel pore forming the S6 transmembrane helix. Consistent with this interpretation, we observed that the substitution Phe to Trp at position 248 in the C-terminal segment of the channel pore helix resulted in a channel that was Cu(II)Phe insensitive with a Pomax of 0.8. An example of inside-out single-channel recordings illustrating the action of Cu(II)Phe (1 µM) on the F248W-R362C double mutant coexpressed with CaM K75C is presented in Fig. 4. Also included are control recordings where Cu(II)Phe was applied to CaM K75C expressed with F248W(KCa3.1) or to the F248W-R362C(KCa3.1) double mutant. As seen, there were no significant effects of Cu(II)Phe when applied to the F248W-R362C channel or to the K75C CaM mutant coexpressed with the F248W mutant. In contrast, the internal application of 1 µM Cu(II)Phe to the F248W-R362C(KCa3.1)–K75C(CaM) complex resulted in a robust and irreversible inhibition of channel activity that was partly recovered by the subsequent perfusion with a DTT (10 mM) -containing bath solution. These observations argue for the formation of a disulfide bond between R362C of KCa3.1 and K75C of CaM in accordance with the model presented in Fig. 1 D.
Figure 4.

Evidence of cysteine cross-bridging between R362C of KCa3.1 and K75C of CaM catalyzed by Cu(II)Phe. (A) Inside-out current recording illustrating the effect of 1 µM Cu(II)Phe on the F248W-R362C(KCa3.1) channel mutant coexpressed with K75C(CaM) in Xenopus oocytes. Exposure to Cu(II)Phe resulted in a total inhibition of channel activity that was irreversible after Cu(II)Phe washout. Channel activity could be recovered after exposure to 10 mM DDT, confirming disulfide bond formation between K75C of CaM and R362C of KCa3.1. (B) Inside-out single-channel current recording illustrating the action of 1 µM Cu(II)Phe on the K75C(CaM) mutant coexpressed with F248W(KCa3.1). A 1-min application of 1 µM Cu(II)Phe failed under these conditions to affect channel activity. (C) Single-channel recording performed with F248W-R362C(KCa3.1) channel mutant illustrating the absence of the effect of 1 µM Cu(II)Phe when applied internally. This mutant channel was also insensitive to the application of 10 mM DTT. Experiments were performed in symmetrical 200-mM K2SO4 conditions at a pipette potential of 60 mV. Label “c” refers to the channel closed state.
Although detection of an intrasubunit disulfide is difficult by biochemical methods, an intersubunit disulfide can be detected, as the size of the cross-species increases when disulfide bonds occur. This proposal was confirmed in experiments where isolated membrane proteins from oocytes expressing F248W-R362C(KCa3.1) plus CaM K75C treated with ionomycin plus 100 µM Cu(II)Phe were subjected to Western blotting using anti-KCa3.1 antibody (Fig. 5). Lane 2 in Fig. 5 (A and B) shows a 57-kD band corresponding to the predicted KCa3.1 molecular weight of linked to CaM (CaM MW of ≈17 kD), which could be reduced to the monomeric form of KCa3.1 (40 kD) with DTT (Fig. 5, A and B, lane 3). No band of 57 kD was detectable in membrane extracts coming from F248W-R362C(KCa3.1)–K75C(CaM) oocytes not treated with Cu(II)Phe (Fig. 5, A and B, lane 1). In addition, the same Cu(II)Phe-based experiments performed with F248W-R362C (Fig. 5, A and B, lane 5) or CaM K75C coexpressed with KCa3.1 F248W (lane 8) failed to yield an equivalent 57-kD band, demonstrating the specificity of the R362C(KCa3.1)–K75C(CaM) interaction. The same samples were also subjected to Western blotting using an anti-CaM antibody (Fig. 5 B) to confirm the nature of the 57-kD band. As illustrated, the antibody anti-CaM reacted only with the cross-species band of 57 kD (Fig. 5 B, lane 2). The specificity of the anti-CaM antibody was shown by hybridization of WT and mutated CaM at 17 kD (Fig. 5 B). These results indicate that Cys engineered at positions 362 of KCa3.1 and 75 of CaM can come close enough to allow formation of disulfide bonds despite prediction of an inter Cβ distance of 9.5 Å according to the model in Fig. 1 D. However, these observations do not allow us to directly conclude that R362 and K75 residues will be in such close proximity under normal conditions, as both residues are charged in the WT CaM–KCa3.1 complex.
Figure 5.

Western blot analysis of KCa3.1 channels coexpressed with CaM mutants subjected to Cu(II)Phe treatment. (A) Membranes were prepared from oocytes expressing either the F248W-R362C(KCa3.1)–K75C(CaM), F248W-R362C(KCa3.1), or the F248W(KCa3.1)–K75C(CaM) mutants. Disulfide cross-linking catalyzed by incubating oocytes in 100 µM Cu(II)Phe and 10 µM ionomycin resulted in a band shift from ≈40 kD for KCa3.1 monomer to ≈57 kD for KCa3.1 linked to CaM (KCa3.1-CaM) (A, lane 2). Samples were subjected to SDS/PAGE (10% gel) under either reducing (+DTT) or nonreducing (−DTT) conditions. Exposing membranes to DTT caused the loss of the ≈57-kD band (A, lane 3). Cu(II)Phe was ineffective in initiating disulfide bond formation using the F248W-R362C(KCa3.1) (A, lane 5) or F248W(KCa3.1)–K75C(CaM) (A, lane 8) systems. (B) Western blotting obtained under the conditions described in A using an antibody directed against CaM. The same band at ≈57 kD could be detected after Cu(II)Phe treatment and disappeared after disulfide bond reduction with DDT (B, lane 3). Also shown is a band at 17 kD corresponding to CaM.
Contributions of residues at the intersubunit interface to Pomax
Experiments were next undertaken to identify residues within CaMBD2 involved in regulating Pomax. Fig. 6 A summarizes the measurements of Pomax for channel mutants generated by substituting each residue along CaMBD2 by a Cys. An important increase in Pomax was observed with the S367C channel mutant, with a value of 0.62 ± 0.12 (n = 5) compared with 0.22 ± 0.07 (n = 8) for WT (P < 0.005). We also noted that the mutation S367C drastically enhanced the channel mean open time from 7 ± 2 ms (n = 5) for WT to 25 ± 7 ms (n = 4) for S367C, while decreasing the channel mean closed time from 30 ± 15 ms (n = 5) for WT to 5 ± 1 ms (n = 4) for S367C. Clearly, the mutation S367C succeeded in modifying the equilibrium constant between the channel open and closed configurations.
Figure 6.

Effect of mutating residues of the CaMBD2 on Pomax. (A) Bar graph summarizing measurements of Pomax, the channel open probability at saturating Ca2+ concentration (25 µM), for channel mutants generated by substituting individual residues along CaMBD2 to a Cys. Values not included in the filled area refer to Pomax for which P < 0.005 relative to WT (Pomax (WT) ± 2.5 SD). A strong increase was seen with the S367C mutant, with Pomax of 0.62 ± 0.12 (n = 5) compared with 0.22 ± 0.07 (n = 8) for WT KCa3.1. (B) Examples of inside-out current traces obtained for the WT, K360C, L361C, R362C, E363C, Q364C, V365C, S367C, M368C, and V369C mutant channels expressed in Xenopus oocytes. Dwell-time analyses showed that the increase in Pomax observed with the S367C mutant resulted in an increase of the channel mean open time from 7 ± 2 ms (n = 5) for WT to 25 ± 7 ms (n = 4) for S367C, and a decrease of the channel mean closed time from 30 ± 15 ms (n = 5) for WT to 5 ± 1 ms (n = 4) for S367C mutant. Also included are single-channel traces illustrating the action of MTSES− or MTSET+ on the R362C, E363C, and Q364C mutants, respectively. These results confirmed that residues identified from SASA calculations as being either buried (Q364) or exposed (R362) can be modified by MTS reagents, suggesting that the targeted Cys residues were accessible to water. Current recordings were performed in symmetrical 200-mM K2SO4 solutions at an internal Ca2+ concentration of 25 µM, with 100 mV applied in the patch electrode. Label “c” refers to the channel closed state. Bars, 2 pA and 500 ms.
The mutation S367C involves substituting to a residue of greater side-chain surface area and thus increasing the residue free energy of transfer to water. The importance of the side-chain surface area was thus tested by substituting S367 to nonpolar amino acids of equivalent (S367A) or greater surface areas (S367T, S367L, and S367W). The results of these experiments are summarized in Fig. 7 A. As seen, the mutations S367T, S367L, and S367W led to significant increases in Pomax, with values of 0.49 ± 0.07 (n = 4), 0.88 ± 0.05 (n = 3), and 0.87 ± 0.01 (n = 3), respectively, compared with 0.19 ± 0.03 (n = 5) for S367A. These observations thus establish a correlation between Pomax and the nonpolar contribution to the hydration free energy of the residue at position 367. As the side-chain surface area of the Glu is equivalent to Leu with the addition of a negative charge, we also used the S367E mutant to investigate the importance of polar interactions to the action of S367 on Pomax. The results presented in Fig. 7 show a decrease in Pomax relative to S367L, with Pomax = 0.24 ± 0.08 (n = 4), indicating a potential destabilizing effect caused by an overall decrease in hydrophobicity. The results of these experiments are summarized in Fig. 7 B.
Figure 7.

Comparing the effect of S367 and M368 mutations on Pomax. (A) Examples of single-channel recordings illustrating the effect on Pomax coming from the substitution of S367 by nonpolar residues of smaller (S367A) or larger side-chain surface areas. These results demonstrate a correlation between the channel Pomax and the free energy of transfer to water for the residue at position 367 for nonpolar amino acids. Also shown is the effect of the substitution S367E, confirming that electrostatic contribution to solvation energy can account in this case for a low Pomax (0.24 ± 0.08; n = 4) despite a side-chain surface area equivalent to Leu (Pomax of 0.88 ± 0.05; n = 3). (B) Bar graph summarizing the effect of mutating S367 on the channel Pomax value. (C) Single-channel recordings illustrating the effects on Pomax of mutating M368 described as being an anchoring point for CaM binding to KCa3.1. Mutating M368 by residues of equivalent or larger side-chain surface areas (M368L and M368W) had a small impact on Pomax, with values of 0.25 ± 0.07 (n = 4) for M368L and 0.27 ± 0.06 (n = 5) for M368W compared with 0.22 ± 0.07 (n = 8) for WT. The mutation M368A resulted in a Pomax of 0.13 ± 0.01 (n = 3), suggesting that decreasing the residue 368 free energy of transfer to water tends to impair the channel activity. Similarly, the mutation to a polar residue M368R led to a lower Pomax value of 0.15 ± 0.02 (n = 6). (D) Bar graph illustrating the effect of mutating M368 on Pomax. A decrease in hydrophobicity tends to result in lower Pomax, as observed for S367, but with a much smaller impact on the Pomax value. Experiments performed in symmetrical 200-mM K2SO4 conditions at 25 µM of internal Ca2+ for a pipette potential of 100 mV. Bars, 2 pA and 0.5 s.
The structural analysis presented in Fig. 2 A indicates that in addition to S367, residue E363 also contributes to the intersubunit interface. In contrast to S367, however, E363 SASA is not affected by the addition of CaM, and the intersubunit interactions in this case are essentially electrostatic in nature, with E363 facing R352 on the adjacent subunit (Fig. 1 D). The results presented in Fig. 6 show that the mutation E363C resulted in a Pomax of 0.12 ± 0.02 (n = 3), a value significantly (P < 0.05) lower than WT. Similarly, single-channel recordings of the E363R mutant led to a Pomax of 0.14 ± 0.04 (n = 4), a value not statistically different from E363C but different from WT. Both mutations are expected to lower the free energy of transfer from water to n-octanol from 3.63 kcal/mol for Glu to −0.02 kcal/mol for Cys and 1.81 kcal/mol for Arg, respectively (Wimley et al., 1996). Such behavior is at variance to the results obtained for the S367 mutants, where more hydrophobic residues are associated with higher Pomax values, and argue for a specific contribution of electrostatic interactions at E363 to the control of Pomax. In this regard, internal pulse applications of the MTSES− reagent to the E363C mutant led to an irreversible 2.0 ± 0.5 (n = 5) –fold current increase, an effect not seen with the positively charged MTSET+ (Fig. 3 C). These observations were confirmed at the single-channel level, where exposure of E363C to MTSES− resulted in an increase of the channel Pomax from 0.12 ± 0.02 (n = 3) to 0.33 ± 0.02 (n = 3) (see Fig. 6). These experiments thus support the requirement of a negatively charged Cys–MTS complex to reestablish Pomax. We cannot rule out that part of the observed increase in Pomax could come from an increase in nonpolar (cavity) hydrophobic interactions, as the side-chain surface area for the Cys–MTSES− complex corresponds to 112 Å2 compared with 32 Å2 for E363C. However, because the positively charged MTS reagent MTSET+ failed to affect channel activity, our data do not support a dominant contribution of nonpolar hydrophobic effects to the observed increase in Pomax. Furthermore, the variations in Pomax obtained by modifying E363 remained overall modest compared with the results described for the S367C, S367L, and S367W mutant channels (Pomax > 0.65), thus limiting the contribution of polar interactions at E363 to Pomax.
Contribution of the residues at the CaM–CaMBD2B interface in setting Pomax
The results presented in Fig. 2 C argue for residues Q364, M368, and V369 as being key determinants to the interface between the CaM N-lobe and the CaMBD2B. Perturbing the interactions between CaM and Q364, M368, or V369 could thus potentially affect the KCa3.1–CaM complex stability, resulting in a modification of Pomax. We found that the mutations Q364C, M368C, and V369C have no significant impact on the channel Pomax, with values of 0.17 ± 0.07 (n = 6), 0.26 ± 0.06 (n = 5), and 0.24 ± 0.09 (n = 3), respectively (Fig. 6, A and B). An analysis of the KCa3.1–CaM complex shows that M368 is projecting inside a hydrophobic cavity formed of residues L32, M36, M51, and M71 of CaM and thus constitutes a unique anchoring point for CaM binding. In this regard, MD simulations identified M368 as the residue with maximum van der Waals interactions with CaM, supporting a prominent role in CaM–KCa3.1 binding. The absence of a significant effect on Pomax with the M368C mutant thus appeared surprising, but it could be the result of substituting to a residue of smaller surface area (32 Å2 compared with 65 Å2) with the associated decrease in hydrophobic effects. However, mutating to residues of equivalent or larger surface areas failed to significantly affect Pomax, with values of 0.25 ± 0.07 (n = 4) for M368L and 0.27 ± 0.06 (n = 5) for M368W, respectively (Fig. 7, C and D). In contrast, the substitutions M368A and M368R involving replacing to a less hydrophobic residue led to Pomax of 0.13 ± 0.01 (n = 3) and 0.15 ± 0.02 (n = 6), respectively (Fig. 7, C and D). Clearly, despite its key contribution to the CaM–KCa3.1 interface, perturbing nonpolar interactions at M368 is less effective on Pomax compared with S367, with Pomax variations ranging from 0.13 to 0.27 only. However, in both cases, a greater energy for transferring the residue to water correlates with higher Pomax values.
Finally, the results presented in Fig. 2 C argue for residue Q364 constituting with S367 and M368, a central amino acid to the CaM N-lobe–CaMBD2 interface. The mutation Q364C led to Pomax with values of 0.17 ± 0.07 (n = 6), a value not statistically different from WT despite a decrease of the side-chain surface area from 63 to 32 Å2 (Fig. 6 A). We note, however, that formation of the Cys–MTSET+ complex resulted in an increase in channel activity (Fig. 6 B) associated with a higher Pomax of 0.37 ± 0.08 (n = 3). We conclude that strong perturbations coming either from the addition of a positive charge with the Cys–MTSET+ complex or through an increase in the side-chain surface area could affect Pomax to a lesser degree relative to the S367 mutants.
Nonpolar interactions contribute to the channel open state stability
Pomax measurements do not provide information on the energetics of the CaMBD2B–CaM N-lobe complex formation and stability. The contribution of the residues in CaMBD2B to the channel open state stability was thus investigated by measuring the channel deactivation time. The model presented in Fig. 1 C represents in this regard the initial configuration prevailing after Ca2+ removal. The stability of the CaMBD2B–CaM N-lobe complex was thus tested in a series of experiments in which we measured the effects of mutating residues in CaMBD2B to Cys on the channel deactivation time. The results of these experiments are summarized in Fig. 8. As seen, the mutation S367C appeared unique in affecting the channel deactivation time toff with a value of 234 ± 25 ms (n = 7) compared with 65 ± 6 ms (n = 8) for KCa3.1 WT (P < 0.0001). Less important increases were seen with the V365C (111 ± 9 ms; n = 4) and R352C (106 ± 10 ms; n = 4) mutants (P < 0.005). A slower deactivation time measured for the S367C mutant channel supports our previous findings of Pomax being significantly higher for S367C compared with WT (Fig. 7, A and B). Such correlation was confirmed in experiments where the channel deactivation rate after Ca2+ removal was estimated for the S367A, S367T, S367L, S367W, and S367E mutants. The substitutions S367T, S367L, and S367W resulted in 2.7-, 4.5-, and 13.5-fold increases in toff, with a mean value of 176 ± 7 ms (n = 4), 289 ± 21 ms (n = 3), and 799 ± 22 ms (n = 5), respectively (Fig. 9 B). In contrast, the conservative mutation S367A had no significant impact on toff, with a mean value of 81 ± 7 ms (n = 3), nor did the substitution S367E, with a toff of 74 ± 6 ms (n = 3). It follows that increasing the free energy of transferring the residue at position 367 to water not only impacts the channel Pomax, as already documented, but also affects the channel deactivation rate, indicating that the S367 site is critical to the stabilization of the KCa3.1–CaM N-lobe complex. In this regard, a log plot of the relative variation of the channel deactivation time toff as a function of the free energy of transferring the residue 367 to water revealed a Pearson correlation coefficient of 0.90, supporting a strong correlation between toff and the residue hydrophobicity (Fig. 9 B, inset).
Figure 8.

Effect of mutating residues in the CaMBD2B on channel activation and deactivation times. Horizontal bar graphs summarizing the effect of mutating to Cys residues of CaMBD2B on channel deactivation (toff) (A) and activation (ton) times (B). Values not included in the filled area refer to ton or toff for which P < 0.005 relative to WT (ton or toff [WT] ± 2.5 SD). The mutation D370 did not produce functional channels. (A) Effect on the channel deactivation time toff of substituting to Cys each residue of the CaMBD2B plus residues R352 and Q353 of CaMBD2A. Important variations were seen with S367C (P < 0.0001) and with the mutant channels R352C, L361C, E363C, V365C, and S372C (P < 0.005). (Bottom trace) Inside-out current recording illustrating deactivation of the S367C channel mutant after exchanging a Ca2+-containing solution (25 µM) with a Ca2+-free internal solution (thick line). Deactivation time was obtained by curve fitting to a single exponential the time course of the current decrease initiated by switching to a Ca2+-free internal solution. Measurements were performed in symmetrical K2SO4 conditions at a pipette potential of 60 mV. (B) Effect on the channel activation time ton on substituting to Cys each residue of the CaMBD2B plus residues R352 and Q353 of CaMBD2A. Important variations were seen with E363C (P < 0.0001) and with the mutant channels L361 and M368 (P < 0.005). (Bottom trace) Inside-out current recording illustrating activation of the E363C channel mutant in response to an increase in internal Ca2+ from zero (EGTA; thick line) to 25 µM. Channel activation time was obtained by fitting to a single exponential the time course of the current increase that follows the initial delay observed after adding Ca2+ to the bathing medium.
Figure 9.

Comparing the effect of S367 and M368 mutations on activation and deactivation times. Bar graphs describing the effect of varying the solvation energy of the residue at positions 367 and 368 on the channel activation (ton) and deactivation times (toff). (A) Results demonstrating that mutations at S367 that caused an increase in Pomax and toff (B) tend to accelerate channel activation with smaller ton values relative to WT. The mutation S367E led to a faster ton, despite no significant effect on Pomax or toff compared with WT. (B) Effect on the channel deactivation time toff of mutating S367 by residues of equivalent or higher transfer energy to water. Although the S367A and S367E mutations had no impact on toff, significant increases were seen with the S367T (toff = 176 ± 7 ms; n = 4), S367C (toff = 234 ± 25 ms; n = 7), S367L (toff = 289 ± 21 ms; n = 3), and S367W (toff = 799 ± 22 ms; n = 5) mutants. The inset illustrates the correlation between the relative change in toff computed as ln(toff_mutant/toff_WT) and the solvation energy for water to n-octanol transfer (Wimley et al., 1996). With a Pearson correlation coefficient of 0.90, for nonpolar residues, this analysis confirms that hydrophobic interactions at 367 of KCa3.1 play a critical role in setting Pomax. Substituting to a charged residue (S367E) yielded faster toff values compared with toff measured for nonpolar residues of equivalent surface area (Leu). (C) Bar graph summarizing the effect of mutating the CaM-anchoring residue M368 on the channel activation time ton. With the exception of the M368W mutant, higher ton values correlate with an increase in free energy of transfer to water. (D) Bar graph demonstrating that in contrast to the activation time ton, mutating M368 had no impact on the deactivation time toff and thus on the stability of the channel open configuration.
The model illustrated in Fig. 1 F indicates that S367 should be projecting inside a hydrophobic cavity formed by M36, L39, Q41, and M51 of CaM plus L345 and N349 of the KCa3.1 adjacent subunit. MD simulations revealed in this regard that the strongest nonbonded interactions with S367 involve Q41of CaM and, to a lesser extent, N349 on the adjacent subunit (see Fig. 10). To determine to what extent the interactions between S367 and the CaM residue Q41 contribute to the stability of the channel open conformation, a double mutant cycle analysis was undertaken in which the coupling energy ΔΔG was computed as (ΔGmutation1–mutation2 + ΔGWT–WT) − (ΔGmutation1–WT + ΔGmutation2–WT), with ΔG given by KT ln(toff_mutant/toff_WT) (Horovitz, 1996; Wall-Lacelle et al., 2011). With a deactivation time toff decreasing from 799 ± 22 ms (n = 5) for the S367W mutant to 203 ± 33 ms (n = 4) with the S367W(KCa3.1)–Q41A(CaM) double mutant complex, this procedure led to ΔΔG = −0.69 ± 0.48 kcal/mol, confirming functional coupling between these two residues (Fig. 11). In addition, the mutant cycle diagram presented in Fig. 11 shows that the mutation Q41A in CaM, which involves a small decrease in solvation energy (Wimley et al., 1996), was ineffective at modifying toff when coexpressed with KCa3.1 WT but resulted in a 3.9-fold decrease in toff when coexpressed with the KCa3.1 S367W mutant. The possibility of a Trp at 367 to form a hydrogen bond with Q41 from CaM, as documented for the equivalent T479–Q41 interaction in KCa2.2, could partly account for the large variation in toff, seen when S367W is coexpressed with Q41 compared with Q41A. Finally, our results with the S367T mutant could also indicate that the presence in KCa2.2 of a Thr equivalent to S367 likely contributes to a higher Pomax for KCa2.2 (Fig. S2).
Figure 10.

Predicted interaction coordinates for S367 and E363 residues. (A) Nonbonded electrostatic plus van der Waals energy computed from 10-ns MD trajectories for the E363 and S367 residues. Maximum interactions with S367 involve residue Q41 and to a lesser extent M36 of CaM, plus residues L345 and N349 on the adjacent KCa3.1 subunit. (B) The strongest interactions with E363 involve residue R352 facing E363 on the adjacent subunit, added to weaker interactions with Q41 of CaM.
Figure 11.

Coupling energies determined by double mutant cycle analysis for the channel ton and toff. ΔG was calculated as KT ln(toff_mutant/toff_wt) or KT ln(ton_double_mutant/ton_mutant), where K is the Boltzmann constant and T is the absolute temperature. The coupling energy ΔΔG was computed as (ΔGmutation1–mutation2 + ΔGWT–WT) − (ΔGmutation1–WT + ΔGmutation2–WT). Differences in free energies are given in kcal/mol. (A) The mutation Q41A on CaM strongly affected the potency of the S367W mutation to increase toff. (B) Absence of coupling energy for channel activation between E363 and R352 despite the prediction of strong electrostatic interactions as a result of E363 facing R352 on the adjacent subunit. (C) Importance of the Lys residue at position 75 on CaM to the decrease in activation time caused by the R362E mutation. The charge neutralization K75A resulted in the charge reversal R362E being ineffective in modulating the channel activation process. These results also indicate that the R362 (KCa3.1)–K75 (CaM) interaction contributes to the energy barrier accounting for channel activation, while supporting the structural model presented in Fig. 1. (D) Importance of the Glu residue at position 47 on CaM to the charge neutralization effect on ton seen with the K360C mutation. The mutation E47A on CaM compromised the potency of the mutation K360C to increase ton, suggesting a contribution coming from electrostatic interactions. (E and F) Functional coupling between E363 and S367 in regulating the channel activation (E) and deactivation (F) times. In E, the drastic increase in activation time caused by the charge reversal E363R mutant was significantly reduced when coupled to the mutation S367C, an effect attributable to the substitution S367C stabilizing the channel open configuration thus facilitating channel activation. In F, the mutation S367C can no longer be translated into an increased deactivation time when coupled to the E363R mutation, an effect likely attributable to the mutation E363R increasing the energy barrier to form a functional channel, and thus facilitating the channel exit from a conducting mode (see Eq. 1).
The results presented in Fig. 8 point to the mutations R352C, L361C, E363C, and V365C as causing a highly significant (P < 0.005) increase in the channel deactivation time toff. However, in contrast to the results obtained with the S367 mutants, a decrease in hydrophobicity with L361C and V365C yielded, in these cases, slower deactivation times (Wimley et al., 1996). The analyses illustrated in Figs. 2 (A and B) and 3 A indicate in this regard that both residues are only partially buried compared with S367 and exclusively interact with CaM. These residues being already exposed to water, the effect of hydrophobic substitution might, under these conditions, be less effective. An increase in toff was also observed by substituting the charged residues R352 and E363 with Cys. In both cases, the substitution procedure involves a decrease in surface area, coupled to charge neutralization. Smaller surface areas are associated with weaker hydrophobic interactions, whereas charge neutralization compromises the electrostatic polarization contribution to solvation energy. The observed increase in toff with R352C and E363C would thus argue for an increase in hydrophobicity coming from charge neutralization. Notably, the CaMBD2 structure illustrated in Fig. 1 D indicates that R352 and E363 are present on adjacent subunits in sufficient close range to feel strong electrostatic interactions. MD simulations confirmed in this regard that E363 interacts essentially with R352, with no significant interactions involving CaM residues (Fig. 10 B). A double mutant cycle analysis performed using the E363C, R352A, and E363C/R352A mutants led to ΔΔG = −0.1 ± 0.2 kcal/mol, indicating an absence of significant functional coupling between these two residues during channel deactivation (unpublished data). However, a small contribution to toff coming from polar interactions between R352 and E363 can be postulated as the ratio toff_E363C/toff_E363 decreased from 1.5 ± 0.2 with R352 to 1.2 ± 0.3 with R352A. These observations suggest a small contribution to toff coming from electrostatic interactions.
Interactions affecting the channel activation rate
The contribution of each individual residue within the CaMBD2B to the KCa3.1 activation process was next investigated by measuring the activation (ton) for Cys mutants engineered along CaMBD2B. The results of this analysis are presented in Fig. 8 B. Although the channel deactivation toff appeared to be regulated by a few residues only, the channel activation time ton was found to be under the control of several CaMBD2B residues including K360, L361, R362, E363, and M368, with the most important effects being observed with E363 and M368. A strong increase in activation time ton was seen with E363C and to a lesser extent K360C, with values of 1,200 ± 100 ms (n = 14) and 577 ± 80 ms (n = 9), respectively, compared with 306 ± 47 ms (n = 8) for the WT KCa3.1 channel. In contrast, the mutations M368C caused a fourfold decrease in ton relative to WT, with a ton value of 77 ± 9 ms (n = 6). Notably, the channel activation time ton was not significantly affected by the mutation S367C, despite a robust effect on Pomax and toff, as described in the previous section.
The results presented in Fig. 8 B show that neutralization of the positive charge at R362C resulted in a decrease in the activation time ton, with a value of 193 ± 37 ms (n = 7), whereas neutralizing the negative charge at E363C caused a strong increase in ton, with an estimated value of 1,200 ± 100 ms (n = 14). This effect could be partly reversed through the binding of MTSES− to E363C, with a ratio of ton_MTSES/ton_ctrl = 0.60 ± 0.04 (n = 6) for E363C. Notably, the results presented in Figs. 2 and 3 support a model of the CaM N-lobe–KCa3.1 complex, where R362 and E363 do not contribute to the CaM N-lobe–CaMBD2B interface. These observations thus suggest that electrostatic interactions involving residues not contributing to CaM–KCa3.1 binding might play a determinant role in the channel activation process. In support of this proposal, charge reversal at 363 with the E363R mutant resulted in a 45-fold increase of the channel activation time ton relative to WT, with a value of 15,000 ± 200 ms (n = 6), whereas substituting a positively charged residue at 362 by a negatively charged amino acid (R362E) caused a fivefold decrease in ton, with a value of 60 ± 10 ms (n = 5) (see Fig. 12, A and D). Swapping the charges at R362 and E363 (R362E/E363R) led to an activation time of 1,700 ± 200 ms (n = 6) compared with 306 ± 47 ms (n = 8) for the WT channel, suggesting that R362 and E363 might work through different mechanisms. The relationship between the total charge of the residues at positions 362 and 363 and the difference in activation energy relative to WT calculated as KT ln(ton_mutant/ton_wt) is illustrated in Fig. 12 E. A linear fit led to a correlation coefficient of 0.90 and a difference in energy of 0.7 kcal/mol per elementary charge. This analysis supports electrostatic interactions involving R362 and E363 as being determinant to the channel formation process.
Figure 12.

Electrostatic interactions determine the channel activation rate. (A) Inside-out current recordings illustrating the effect of charge neutralization and/or charge reversal on KCa3.1 activation kinetics. Measurements were performed in symmetrical 200-mM K2SO4 conditions at a pipette potential of 60 mV. The current records presented in this figure demonstrate that the mutation E363R resulted in a 45-fold increase of the channel activation time constant ton, whereas charge reversal at 362 with the R362E mutant caused a fourfold decreased in ton. (B) First latency analysis for the E363C and E363R mutants. The vertical spike marks the time at which the internal solution was switched from 0 (EGTA; thick line) to 25 µM Ca2+. Double arrow lines refer to latency period before channel opening. Unitary current inside-out recordings were performed in symmetrical 200-mM K2SO4 conditions at 100 mV (E363C) or 60 mV (E363R) of applied pipette potential. Latency of 22 ± 4 s (n = 3) and 1.1 ± 0.2 s (n = 3) was estimated for E363R and E363C, respectively, compared with 0.22 ± 0.07 s (n = 8) for KCa3.1 WT. The channel steady-state open probability once activated was estimated at 0.15 ± 0.08 (n = 4) and 0.12 ± 0.02 (n = 3) for E363R and E363C channels, respectively, compared with 0.22 ± 0.07 (n = 8) for KCa3.1 WT. (C) Gradual decrease in ton resulting from charge modification at position 362 from one positive charge (R362) to one negative charge (R362E). As observed with E363, the presence of a net positive charge at either 362 or 363 tends to increase ton, with the most prominent effect observed with the E363R mutant. Asterisks indicate a significant difference from WT (*, P < 0.05). (D) Horizontal bar graph summarizing the results on the contribution of electrostatic interactions to the action of either E363 or R362 to the channel activation time constant. The double mutation R362E/E363R obtained by swapping the charges on 362 and 363 led to a ton of 1,700 ± 200 ms (n = 6) compared with 306 ± 47 ms (n = 8) for WT. The increase in activation time with the E363R mutation could also be partly reversed by neutralizing the polar and/or charged residues predicted to interact with E363. The most important effects were seen with the mutation S367C with a ton of 4,000 ± 100 ms (n = 5) for the E363R/S367C double mutant, a value significantly different from the single E363R channel. We concluded that there was a strong functional coupling between E363 and S367 during channel activation. Asterisks indicate a significant difference from WT (**, P < 0.001). (E) Plot of the relative channel activation energy as a function of the total charge for the residues at positions 362 and 363. The activation energy relative to WT was taken as KT ln(ton_mutant/ton_wt), where K and T have their usual meanings. The linear fit corresponds to a correlation coefficient of 0.9 and a difference in energy of 0.7 kcal/mol per elementary charge. This analysis supports a model whereby electrostatic interactions involving R362 and E363 play a prominent role in the KCa3.1 activation process.
A strong impact of the R362E and E363R mutations on the channel activation time was unexpected, as both mutations do not significantly (P > 0.05) affect the channel Pomax relative to WT, with values of 0.31 ± 0.03 (n = 3) and 0.14 ± 0.04 (n = 4), respectively. First latency experiments were thus conducted on the E363C and E363R mutants to determine how the channel activation time could be related to Pomax. As illustrated in Fig. 12 B, measuring the channel first opening in response to a change in internal Ca2+ concentration from 0 to 25 µM yielded 22 ± 4 s (n = 3) for the E363R channel and 1.1 ± 0.2 s (n = 3) for the E363C mutant compared with 0.22 ± 0.07 s (n = 8) for KCa3.1 WT. More importantly, the channel steady-state open probability once activated was estimated at 0.14 ± 0.04 (n = 4) and 0.12 ± 0.02 (n = 3) for E363R and E363C, respectively, compared with 0.22 ± 0.07 (n = 8) for KCa3.1 WT. These observations would be compatible with a model whereby E363 would control the channel transition to a closed state from which the channel can access to an open conformation. Collectively, these experiments show that a charge reversal from two positive (R362-E363R) to two negative charges (R362E-E363) at position 362 and 363 caused a 220-fold decrease of the channel activation time, a clear indication of a key functional role of these residues to the channel activation process in response to Ca2+. Notably, mutating both residues had a minor impact on Pomax, suggesting that R362 and E363 contribute to the kinetic underlying the formation of a functional channel, while not being determinant to the end structure responsible for channel gating.
Contribution of R362-based interactions to channel activation
Experiments were next conducted to determine if the structure illustrated in Fig. 1 can account for some of the electrostatic-based actions of R362 and E363 on the channel activation time ton. Cross-bridging experiments have in this regard provided evidence for a close proximity between R362 of KCa3.1 and K75 of CaM (see Figs. 4 and 5). Performing a double mutant cycle analysis on the R362E(KCa3.1)–K75A (CaM) double mutant system led to a ΔΔG of 0.87 ± 0.23 kcal/mol, a clear indication of functional coupling in this case (Fig. 11 C). In addition, the double mutant diagram presented in Fig. 11 C clearly shows that the fivefold decrease in ton from 306 to 60 ms resulting from the mutation R362E requires the presence of a positive charge at K75 of CaM, as the same mutation yielded ton values of 149 and 180 ms when channels were coexpressed with the K75A CaM mutant. Thus, these results would be compatible with a model in which an electrostatic repulsion between R362 of KCa3.1 and K75 of CaM tends to increase to energy barrier for channel formation, whereas electrostatic attraction between E362 and K75 would facilitate the channel activation process.
Contribution of E363- and K360-based interactions to channel activation
On the basis of the MD analysis performed previously, it was proposed that residue E363 essentially interacts with R352 on the adjacent subunit (see Fig. 10 B). However, a double mutant cycle analysis for the E363C–R352A system led to a ΔΔG of 0.1 ± 0.2 kcal/mol, indicating an absence of functional coupling in this case during the channel activation process (Fig. 11 B). The results presented in Fig. 8 B also identified K360 as a contributor to the channel activation kinetic, with ton values of 577 ± 80 ms (n = 9) for the K360C. The structure in Fig. 1 points to K360(KCa3.1) and E47(CaM) as being likely to be involved in electrostatic interactions. Activation time measurements led to coupling energy of ΔΔG = −0.50 ± 0.3 kcal/mol for the K360C(KCa3.1)–E47A(CaM) system, supporting a favorable coupling between K360 and E47 during channel activation (Fig. 11 D). These results suggest that the interaction K360(KCa3.1)–E47(CaM) facilitates channel formation, as ton increased after charge neutralization at 360. In support of this proposal was our observation that the potency of a charge neutralization at position 360 to increase ton was in part dependent on the presence of a negatively charged residue at position 47 on CaM, as the mutation K360C failed to increase ton when coexpressed with K75A.
Hydrophobic interactions at M368 and channel activation
The structure in Fig. 1 also presents residue M368 as being an anchor point to the binding of CaM to CaMBD2. Unexpectedly, mutations of M368 did not affect the channel deactivation time ton, as discussed previously (see Fig. 9 D). A decrease in activation time was, however, observed by substituting M368 to less hydrophobic residues with ton values for the M368A, M368C, and M368R mutants of 95 ± 23 ms (n = 3), 77 ± 9 ms (n = 6), and 72 ± 7 ms (n = 6; P < 0.001), respectively, compared with 306 ± 47 ms (n = 8) for WT. In contrast, substituting to the more hydrophobic Leu residue yielded a ton of 469 ± 89 ms (n = 3), suggesting that increased hydrophobic interactions tend to slow down channel activation. However, this proposal is at variance with the results obtained for the M368W mutant, with a ton of 112 ± 14 (n = 5), despite Trp being more hydrophobic than Met. As the side chain of Trp has been documented to form hydrogen bonds with water, special structural features are likely to be involved in this case.
Intrasubunit interactions contribute to the channel activation and deactivation process
Because of the helical structure of the CaMBD2B, residue E363 is expected to interact with S367 on the same subunit. As noted previously, despite an important effect of the S367C mutation on the channel deactivation time toff, the same mutation failed to affect the channel activation process ton (248 ± 33 ms [n = 7] compared with 306 ± 47 ms [n = 8]; see Fig. 9 B). However, a double mutant cycle analysis for the E363R–S367C double mutant revealed a coupling energy ΔΔG of −0.54 ± 0.20 kcal/mol for the channel activation kinetic, indicating a strong influence of S367 on the channel activation process for the E363R–S367C system (see Fig. 11 E). In fact, this observation would be in accordance with a model where stabilizing the channel open state (mutation S367C) accelerates channel activation, a proposal also supported by a faster activation rate for the S367W mutant (153 ± 22 ms; n = 5) compared with WT. Interestingly, we also noted that the channel time deactivation toff, which was drastically increased from 65 ± 6 ms (n = 8) for WT to 234 ± 25 ms (n = 7) with the S367C mutant, decreased to 81 ± 9 ms (n = 5; P < 0.001) with the E363R–S367C double mutant for a ΔΔG of −0.76 ± 0.27 kcal/mol (Fig. 11 F). The double mutant cycle diagram in this case clearly shows that stabilizing effect coming from the mutation S367C, critically, depends on the charge at 363, with a ratio of toff_S367C/toff_S367 = 3.6 with a Glu at 363 compared with 0.72 with E363R (Fig. 11 F). Single-channel measurements showed in this regard that the Pomax decreases from 0.62 ± 0.12 (n = 5) for the S367C mutant to 0.33 ± 0.10 (n = 4) for the E363R–S367C double mutant channel, indicating again a correlation between Pomax and the channel deactivation time toff. These analyses provide clear evidence for a functional link between E363 and S367 on the same subunit, with both residues playing a critical role in the channel activation and deactivation processes.
DISCUSSION
This paper presents a model of the CaM–KCa3.1 N-lobe complex obtained by homology modeling using as template the crystal structure reported for CaM bound to the rat KCa2.2–CaMBD in the presence of Ca2+ (Schumacher et al., 2001). This theoretical model is supported by cysteine cross-bridging experiments that showed disulfide bond formation between R362C of KCa3.1 and K75C of CaM. In addition, our analysis indicated that an increase in the side-chain surface area of nonpolar amino acids engineered at position 367 of the CaMBD2B correlates with an increase in both the channel maximum open probability Pomax and deactivation time toff. A similar behavior was observed by mutating the residue M368, identified as a hydrophobic anchor point projecting directly inside the CaM N-lobe core. However, the mutations at M368 appeared substantially less potent at perturbing either Pomax or the channel deactivation time toff compared with the same mutations at S367. Our work also provides evidence for a channel activation process governed by electrostatic interactions with the substitution of two positive by two negative charges at positions 362 and 363, leading to a 220-fold increase of the channel activation rate. As both residues do not contribute to the CaM N-lobe–CaMBD2B interface, these observations suggest that long-range electrostatic interactions between two CaM N-lobe–CaMBD2B complexes could be determinant to channel activation in response to a Ca2+ increase. Complementary to these observations, substitutions to more hydrophilic residues at M368 were found to accelerate channel activation, indicating the implication of both electrostatic and hydrophobic interactions to functional channel formation.
Validity of the proposed model
Molecular structures derived through homology modeling likely provide an approximate representation of the actual structure. In fact, with sequences differing by 55% between the template (Protein Data Bank accession no. 1G4Y) and the corresponding KCa3.1 C-terminal domain, the resulting homology-based structure could present structural peculiarities not expected from the original template (Protein Data Bank accession no. 1G4Y). However, several observations support the structure proposed for the Ca2+-CaM–KCa3.1 N-lobe complex presented in Fig. 1. Of importance are the results obtained with Cu(II)Phe demonstrating the formation of a disulfide bond between R362C of KCa3.1 and K75C of CaM. These observations are clearly in accordance with the structure shown in Fig. 1 in which the highly solvent accessible residue R362 is expected to interact with the CaM residue K75 only. The same structure also provided the structural bases upon which the results obtained from double mutant cycle experiments involving residues S367 of KCa3.1 and Q41 of CaM, and residues K360 of KCa3.1 and E47 of CaM, can be interpreted. We noted, however, that the modification rate by MTSET+ of the cysteine engineered at the Q364 channel corresponds to 4.1 ± 0.1 × 103 M−1s−1 (n = 3) compared with 1.0 ± 0.2 × 104 M−1s−1 (n = 3) for the highly solvent accessible R262C mutant. As the rate constant of MTSET reaction with free mercaptoethanol was estimated at ∼9.0 × 104 M−1s−1, our results suggests that the side chains of residues at both sites are exposed to an aqueous environment. Our results thus argue for structural fluctuations of the CaM N-lobe–CaMBD2 complex important enough as to allow an MTS reagent with dimensions corresponding to a cylinder 10-Å long and 6 Å in diameter to access residues packed in hydrophobic pockets such as the ones located at Q364. Notably, the crystal structure reported for the rSK2 channel confirmed that the residue equivalent to Q364 (Q476) is in contact with water molecules (Schumacher et al., 2001) and thus could be suitable for binding to MTS reagents. Also, because Cβ1–Cβ2 distance in a disulfide bond can be calculated to vary from 2.9 to 4.6 Å (Hazes and Dijkstra, 1988), the formation of a disulfide bond between Cys engineered at positions 362 in KCa3.1 and 75 in CaM indicates side-chain movements of large amplitudes, as their Cβs are predicted to be 9.5 Å apart. The structure proposed in Fig. 1 can thus be seen as a minimum energy structure essentially reflecting the energy state of the crystal structure of the template (Protein Data Bank accession no. 1G4Y). In fact, the possibility of R362C (KCa3.1) and K75C(CaM) to form a disulfide bond appears to be at variance with recent results obtained for the KCa2.2 channel, as the K75C CaM mutant failed in this case to produce any measurable currents when expressed with KCa2.2 WT (Zhang et al., 2013). Residue K75 was found in this case to form a salt bridge with E404 (E295 KCa3.1), with no evidence of interactions with the residue equivalent to R262 in KCa3.1 (Zhang et al., 2013). In addition, a different x-ray structure has been reported for the CaM–CaMBD complex for a splice variant of the KCa2.2 channel SK2-b, where the residue N477, equivalent to R362 in KCa3.1, is not interacting with K75 of CaM (Zhang et al., 2012). Clearly, our results do not support the conformation obtained for the splice variant of the KCa2.2 channel in which the four EF hands of CaM are occupied by Ca2+ and where the two CaMBD-b peptides are completely separated from each other (Zhang et al., 2012). Finally, evidence of electrostatic interactions between E84 and E87 of CaM with R464 (R352 KCa3.1) and K467 (R355 KCa3.1) has been reported for KCa2.2 (Maylie et al., 2004), a feature not expected for the SK2-b structure (Zhang et al., 2012). Our charge reversal experiments, where the effect of MTSET+ was measured on the R352C(KCa3.1)–E84(CaM), R352E(KCa3.1)–E84C(CaM), and R352C(KCa3.1)–E84A(CaM) double mutant systems, also confirmed functional proximity between these two residues in the CaMBD2A (see Fig. S1).
Determinants of KCa3.1 activation rate
One of the main observations presented in this paper concerns the contribution of R362 and E363 to the channel activation time ton. These observations suggest that an important step in the channel Ca2+-dependent activation process involves electrostatic interactions with R362 and E363 playing a prominent role despite their minimal contribution to Pomax. The various structural changes underlying channel formation and activation in response to Ca2+ remain to be determined. Of interest, our single-channel results for the E363R mutant showed that despite an average initial latency of >22 s in response to a Ca2+ concentration jump, the channel fluctuation pattern corresponded to WT once activated. These observations support a clear dissociation between the channel formation process controlled to a large extent by R362 and E363 and the channel configuration that sets Pomax and gating mode. However, our results do not provide information on the exact mechanism by which R362 and E363 affect channel activation. Double mutant cycle analyses have provided evidence for functional interactions between R362(KCa3.1)–K75(CaM), K360(KCa3.1)–E47(CaM), and E363(KCa3.1)–S367(KCa3.1) during channel activation. In contrast, results obtained for E363(KCa3.1)–Q41(CaM) (unpublished data) and E363(KCa3.1)–R352(KCa3.1) do not support functional coupling despite prediction of structural proximity. The double mutant cycle analysis of the R362(KCa3.1)–K75(CaM) system is of particular interest, as the strong decrease in activation time ton from 306 ± 47 ms (n = 8) to 60 ± 10 ms (n = 5) coming from the charge reversal R362E mutation is abolished by the substitution K75A in CaM, with ton values now corresponding to 149 ± 10 ms (n = 3) and 180 ± 20 ms (n = 3). This analysis thus indicates that repulsive electrostatic interactions involving R362 and K75 contribute to increase the energy barrier controlling channel activation, whereas attractive interactions between R362E and K75 appear to facilitate the channel activation process. Similarly, our results showed that the increase in activation time associated with the charge neutralization K360C mutation in KCa3.1 was abolished in conditions where E47 of CaM was mutated to an Ala. As both residues are expected to be in close proximity, these results support a facilitation effect coming from K360 and E47 interactions in channel formation.
The structural analysis presented in Fig. 1 indicates that R362 and E363 interact minimally with CaM once the complex is formed. It is thus possible that the drastic changes in activation time resulting from charge perturbation at positions 362 and 363 of KCa3.1 arise from R362 and E363 interacting with KCa3.1 constituents other than CaMBD2A and CaMBD2B, or auxiliary proteins yet to be identified. Of interest, dual-color quantum dot measurements have led to the conclusion that KCa3.1 channels are assembled from two homomeric dimers and not from four independent subunits (Waschk et al., 2011). As the structural model presented in Fig. 1 corresponds to a dimeric formation of two adjacent monomers, one possibility to account for the total R362 plus E363 charge dependence of ton would consist of a dimer–dimer assembly process regulated by electrostatic interactions. This particular model does not require the participation of R362 and E363 to the binding of CaMBD2B to the CaM N-lobe, but it supports a control through long-range interactions of the rate at which the dimers coassemble. Notably, recent results have emerged suggesting that, in spite of their fourfold symmetries on the primary sequence, ligand-gated ion channels in fact behave in their ligand-binding domain like dimers of dimers. However, this proposal has been difficult to prove in the absence of crystal structures, and even then, twofold symmetry might be introduced by the crystal lattice (Kusch et al., 2012).
Finally, we also noted that the mutations M368A/C/R led to channel activation times ton that were three to five times faster compared with WT, indicating a facilitation of the channel formation process. Therefore, energy balance seems to favor a faster channel formation in conditions where residue 368A/C/R is more hydrophilic than Met. This proposal would also be in accordance with the mutation of M368 to the more hydrophobic Leu residue, resulting in a slower activation time (ton = 469 ± 89 ms; n = 3). Notably, such behavior is at variance with the ton of 17 ± 2 ms (n = 3) observed for the KCa2.2 channel, where the residue equivalent to M368 is a Leu (Fig. S2). However, substituting seven of the KCa3.1 amino acids of the CaMBD2B (7AA-KCa3.1) by their KCa2.2 equivalents caused an acceleration for the channel activation process from 306 ± 47 ms (n = 8) for the WT KCa3.1 to 91 ± 31 ms (n = 5) for the 7AA-KCa3.1 channel, in line with the CaMBD2 of KCa2.2 contributing to facilitate the CaM–KCa2.2 complex formation. It follows that the effects of M368 we observed on the channel activation kinetic likely depend on the entire structure of the CaMBD2. In fact, the importance of hydrophobic interactions at 368 needs to be considered in light of the observation that the activation time measured for the M368W mutant was not significantly different from the value obtained for the M368A channel, indicating that structural parameters, other than hydrophobicity, must be taken into consideration. As the side chain of Trp is equipped with atoms capable of forming hydrogen bonds with water, this particular feature added to stearic constraints could favor faster channel activation, as observed experimentally. As mentioned previously, M368 corresponds to L480 of KCa2.2, which was described as a hydrophobic anchor point projecting inside the CaM N-lobe core (Schumacher et al., 2001). Hydrophobic anchor points have already been documented in other Ca2+-CaM–peptide structures and are thought to play a pivotal role in binding (Hultschig et al., 2004). Our results thus suggest that perturbing CaM binding at M368 can affect the time course of channel activation. Notably, the proposal that a decrease in hydrophobicity is associated to faster activation times can be extended to other residues, with ton of 133 ± 25 ms (n = 3) and 110 ± 23 ms (n = 3) measured for the L361C and V369C mutants, compared with 306 ± 47 ms (n = 8) for WT. Our results would thus support a general model whereby KCa3.1 formation and activation involve long-range electrostatic interactions dominated by R362 and E363, coupled to a short-range anchoring process in which M368 would represent a key constituent. Once the CaM–KCa3.1 complex was formed, gating would be controlled by residues not necessarily involved in the channel formation process.
Determinants of the KCa3.1 deactivation rate
Our results showed that increasing the free energy of transfer to water of the residue position 367 in KCa3.1 correlates with slower deactivation times toff and higher Pomax values. S367 is part of the 14–amino acid stretch that has been documented to be essential for KCa3.1 activation by ATP (Gerlach et al., 2001). Our analysis of the CaM N-lobe–CaMBD2B structure indicates that S367 has the unique property of contributing to both the CaM N-lobe–CaMBD2B and intersubunit interfaces. According to the structure proposed in Fig. 1, S367 is projecting in a cavity formed by residues M36, L39, and Q41 on the second helix of the CaM N-lobe and by the KCa3.1 residues L345 and N349. In addition, MD simulations confirmed that the strongest nonbonded interactions with S367 essentially involve residue Q41 of CaM. In support of these conclusions is the observation that the mutation Q41A in CaM markedly impacts the toff increase resulting from the S367W mutation, with toff decreasing from 799 ± 22 ms (n = 5) for the S367W(KCa3.1)–Q41(CaM) system to 203 ± 33 ms (n = 4) when S367W (KCa3.1) is coexpressed with CaM Q41A. The possibility of hydrogen bonding between S367W and Q41 not prevailing with the Q41A mutant could be in part responsible for the observed decrease in deactivation time. This proposal would be in line with results demonstrating the presence of a hydrogen bond between Q41 of CaM and the S367 equivalent T479 residue in KCa2.2 (Schumacher et al., 2001). Notably, toff measurements led to the potency ranking of S367 (60 ms) ≈ S367A (81 ms) < S367T (176 ms) < S367C (234 ms) < S367L (289 ms) << 367W (799 ms), a sequence reflecting the hydrophobic scale reported by Wimley et al. (1996) for the free energies of transfer from water to n-octanol. As n-octanol has been extensively used as a model for the interior of protein, our results argue for hydrophobic interactions at S367 as being determinant to the stability of the CaM N-lobe–CaMBD2B complex. We note, however, that the mutation S367E did not significantly impact either toff or Pomax despite a predicted difference of 3.18 kcal/mol in solvation energy relative to WT (Wimley et al., 1996). This result added to the observation that mutation K360C did not affect toff despite the prediction of K360 of KCa3.1 interacting strongly with E41 of CaM, suggesting that electrostatic interactions might not be determinant to CaM N-lobe–CaMBD2B control of toff. Of interest, S367 in KCa3.1 is replaced by a Thr in channels of the KCa2.x family. As Thr is more hydrophobic than Ser, a higher Pomax value is expected, as observed experimentally (see Figs. S1 and S2). We also observed that the changes in Pomax obtained by mutating residue M368 correlate with the substituting residue hydrophobicity, with a higher free energy of transfer to water leading to higher Pomax values. Compared with S367, however, the potency of these mutations to affect Pomax was low, with values ranging from 0.12 to 0.27, despite M368 being considered a key anchoring point for the binding of CaM to the CaMBD of KCa3.1. A recent structural study has provided evidence that L480 in KCa2.2 (which corresponds to M368 in KCa3.1) is implicated in the formation of the binding pocket for channel potentiators such as 1-EBIO (Zhang et al., 2012). In view of these observations, one would have expected that mutating M368 to residues such as Trp or Arg would affect CaM binding to KCa3.1 in such a way as to affect toff and Pomax. As mentioned previously, in contrast to S367, which contributes to both the CaM N-lobe–CaMBD2B and intersubunit interfaces, M368 interacts with CaM residues exclusively. As such, our results better support a model where S367 but not M368 would contribute to the formation of a binding site for KCa3.1 potentiators.
Connecting channel activation and deactivation processes
Our observations can be qualitatively captured in a simple model:
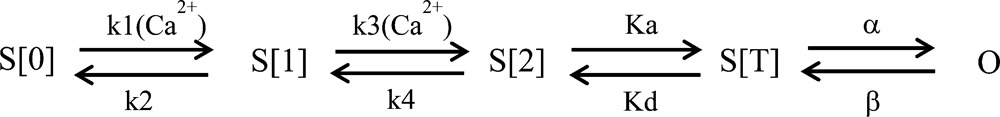 |
(SCHEME 1) |
where S[0], S[1], and S[2] correspond to the channel state with no dimer, one dimer, and two dimers formed; S[T] stands for the dimer–dimer structure, and O is the channel open configuration. The rate constants k1(Ca2+) and k3(Ca2+) account for the Ca2+ dependence of the individual KCa3.1–CaM complex formation. Similar Markovian schemes have already been proposed to describe the kinetic features of both SK and KCa3.1 channels (Hirschberg et al., 1998; Bailey et al., 2010), but their interpretation is not based on a dimer–dimer formation mechanism. This kinetic scheme captures the main features of the conclusions derived from dual-color quantum dot measurements, where KCa3.1 channels appeared to be formed from two homomeric dimers and not from subunits. This model would also be in line with recent results suggesting that ligand-gated ion channels behave in their ligand-binding domain like dimers of dimers (Kusch et al., 2012). According to the proposed Markovian scheme, the deactivation time can be expressed as
| (1) |
This equation was obtained by solving analytically the rate constants for the proposed kinetic scheme with k1(Ca2+) = k3(Ca2+) = 0, in conditions where k4 → ∞. The slowest rate of transition, Kslow, corresponds in this case to the number of times the system will successfully reach state S[2]. The effective exit rate is then obtained by multiplying Kslow by k4/(k4 + Ka), the probability that the system transits to the absorbing state S[1] when reaching state S[2]. Numerical calculations confirmed that the proposed expression can account for toff variations over three orders of magnitude as a function of k4, Ka, Kd, α, and β. This equation accounts for many of the kinetic features described in this paper. In particular, it establishes a direct relationship between toff and the channel opening (α) and closing rates (β) so that an increase of α or a decrease in β should result in slower deactivation times. Results coming from mutating residue S367 showed, for instance, that substitutions to more hydrophobic residues led to higher Pomax values characterized by an increase in both the channel opening rate (α) and mean open time (lower β). According to the proposed equation, such modifications are expected to cause an increase in toff, as we found experimentally. Using typical values of k1(Ca2+) = 105 M−1s−1, k2 = 1.9 s−1, k3(Ca2+) = 1.106 M−1s−1, k4 = 45 s−1, Ka = 46 s−1, and Kd = 64 s−1 obtained from dwell-time analyses of WT KCa3.1, it can be shown that a variation of α from 35 to 280 s−1 and β from 113 to 14 s−1 results in a Pomax increase from 0.22 to 0.87 coupled to a variation of toff from 60 to 790 ms, as observed for the S367W mutation. It follows that the effects observed with the S367 mutants can formally be related to Ca2+-independent processes not related to formation of the CaM–KCa3.1 complex. Notably, evidence has already been presented indicating that a decrease in β caused by parachloromercuribenze sulfonate binding at C276 correlates with slower deactivation times, thus supporting the current analysis (Bailey et al., 2010). The proposed equation indicates that toff should also be dependent on k4, the rate of KCa3.1–CaM complex dissociation, as long as Ka > k4. Changes in the energy barrier accounting for the CaM–KCa3.1 complex formation and dissociation as documented for the R362(KCa3.1)–K75(CaM) interactions could therefore affect the deactivation time toff through modifications of k4. Under such conditions, there could be a strong correlation between an increase in the channel activation time and a slower deactivation process. However, in conditions where Ka << k4, toff should be independent of the CaM–KCa3.1 complex formation process. Finally, the proposed equation predicts that increasing the energy barrier for dimer–dimer formation (S[2] and S[T]), with the resulting decrease in both Ka and Kd at a constant ratio (Ka/Kd), should cause an increase in toff despite the opening (α) and closing (β) rates remaining unchanged. Such behavior could account for mutations affecting the channel activation time (lower Ka and Kd), characterized by higher toff values, as observed with the E363R mutant.
Conclusion
Our results point toward S367 as an important structural determinant to KCa3.1 gating, in particular, in setting the channel Pomax value. Modifications of hydrophobic interactions at this site by small molecules could in principle lead to channel activation, suggesting a potential binding site in the design of KCa3.1 potentiators. In contrast, the channel activation process appears to be governed by long-range electrostatic interactions, with major contributions of R362 and E363, coupled to short-range van der Waals interactions potentially responsible for final docking. It is likely that the contribution of R362 and E363 to channel activation involves residues other than those located in the CaMBD2, or an auxiliary protein yet to be identified. Collectively, our results support a model whereby electrostatic interactions ensure proper formation of the KCa3.1–CaM complex in response to Ca2+, whereas hydrophobic forces are responsible for the control channel Pomax value.
Supplementary Material
Acknowledgments
We acknowledge the contribution of Patrick McNeil from DGTIC at the Université de Montréal and Daniel Stubbs from the Calcul Québec for their help in implementing Charmm. We are quite grateful to the Centre de Calcul de l’Université de Montréal and Calcul Québec for the computer facilities. Finally, we would like to acknowledge the work of Ms. Julie Verner for expert oocyte preparation.
This work was supported by grants from the Canadian Institutes of Health Research (to R. Sauvé) and from Cystic Fibrosis Canada (to R. Sauvé).
Kenton J. Swartz served as editor.
Footnotes
Abbreviations used in this paper:
- CaM
- calmodulin
- CaMBD
- CaM-binding domain
- Cu(II)Phe
- copper phenanthroline
- DTT
- dithiothreitol
- MD
- molecular dynamics
- MTSES
- sodium (2-sulfanoethyl) MTS
- MTSET
- [2-(trimethylammonium) ethyl] MTS bromide
- SASA
- solvent accessibility surface area
- SCAM
- substituted cysteine accessibility method
References
- Bailey M.A., Grabe M., Devor D.C. 2010. Characterization of the PCMBS-dependent modification of KCa3.1 channel gating. J. Gen. Physiol. 136:367–387 10.1085/jgp.201010430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banderali U., Klein H., Garneau L., Simoes M., Parent L., Sauvé R. 2004. New insights on the voltage dependence of the KCa3.1 channel block by internal TBA. J. Gen. Physiol. 124:333–348 10.1085/jgp.200409145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouhy D., Ghasemlou N., Lively S., Redensek A., Rathore K.I., Schlichter L.C., David S. 2011. Inhibition of the Ca2+-dependent K+ channel, KCNN4/KCa3.1, improves tissue protection and locomotor recovery after spinal cord injury. J. Neurosci. 31:16298–16308 10.1523/JNEUROSCI.0047-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruening-Wright A., Lee W.S., Adelman J.P., Maylie J. 2007. Evidence for a deep pore activation gate in small conductance Ca2+-activated K+ channels. J. Gen. Physiol. 130:601–610 10.1085/jgp.200709828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Félétou M., Vanhoutte P.M. 2007. Endothelium-dependent hyperpolarizations: past beliefs and present facts. Ann. Med. 39:495–516 10.1080/07853890701491000 [DOI] [PubMed] [Google Scholar]
- Garneau L., Klein H., Banderali U., Longpré-Lauzon A., Parent L., Sauvé R. 2009. Hydrophobic interactions as key determinants to the KCa3.1 channel closed configuration. An analysis of KCa3.1 mutants constitutively active in zero Ca2+. J. Biol. Chem. 284:389–403 10.1074/jbc.M805700200 [DOI] [PubMed] [Google Scholar]
- Gerlach A.C., Gangopadhyay N.N., Devor D.C. 2000. Kinase-dependent regulation of the intermediate conductance, calcium-dependent potassium channel, hIK1. J. Biol. Chem. 275:585–598 10.1074/jbc.275.1.585 [DOI] [PubMed] [Google Scholar]
- Gerlach A.C., Syme C.A., Giltinan L., Adelman J.P., Devors D.C. 2001. ATP-dependent activation of the intermediate conductance, Ca2+-activated K+ channel, hIK1, is conferred by a C-terminal domain. J. Biol. Chem. 276:10963–10970 10.1074/jbc.M007716200 [DOI] [PubMed] [Google Scholar]
- Grgic I., Kiss E., Kaistha B.P., Busch C., Kloss M., Sautter J., Müller A., Kaistha A., Schmidt C., Raman G., et al. 2009. Renal fibrosis is attenuated by targeted disruption of KCa3.1 potassium channels. Proc. Natl. Acad. Sci. USA. 106:14518–14523 10.1073/pnas.0903458106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasenau A.L., Nielsen G., Morisseau C., Hammock B.D., Wulff H., Köhler R. 2011. Improvement of endothelium-dependent vasodilations by SKA-31 and SKA-20, activators of small- and intermediate-conductance Ca2+-activated K+-channels. Acta Physiol. (Oxf.). 203:117–126 10.1111/j.1748-1716.2010.02240.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazes B., Dijkstra B.W. 1988. Model building of disulfide bonds in proteins with known three-dimensional structure. Protein Eng. 2:119–125 10.1093/protein/2.2.119 [DOI] [PubMed] [Google Scholar]
- Hirschberg B., Maylie J., Adelman J.P., Marrion N.V. 1998. Gating of recombinant small-conductance Ca-activated K+ channels by calcium. J. Gen. Physiol. 111:565–581 10.1085/jgp.111.4.565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horovitz A. 1996. Double-mutant cycles: a powerful tool for analyzing protein structure and function. Fold. Des. 1:R121–R126 10.1016/S1359-0278(96)00056-9 [DOI] [PubMed] [Google Scholar]
- Hultschig C., Hecht H.J., Frank R. 2004. Systematic delineation of a calmodulin peptide interaction. J. Mol. Biol. 343:559–568 10.1016/j.jmb.2004.08.012 [DOI] [PubMed] [Google Scholar]
- Joiner W.J., Khanna R., Schlichter L.C., Kaczmarek L.K. 2001. Calmodulin regulates assembly and trafficking of SK4/IK1 Ca2+-activated K+ channels. J. Biol. Chem. 276:37980–37985 [DOI] [PubMed] [Google Scholar]
- Jones H.M., Bailey M.A., Baty C.J., Macgregor G.G., Syme C.A., Hamilton K.L., Devor D.C. 2007. An NH2-terminal multi-basic RKR motif is required for the ATP-dependent regulation of hIK1. Channels (Austin). 1:80–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlin A., Akabas M.H. 1998. Substituted-cysteine accessibility method. Methods Enzymol. 293:123–145 10.1016/S0076-6879(98)93011-7 [DOI] [PubMed] [Google Scholar]
- Klein H., Garneau L., Banderali U., Simoes M., Parent L., Sauvé R. 2007. Structural determinants of the closed KCa3.1 channel pore in relation to channel gating: Results from a substituted cysteine accessibility analysis. J. Gen. Physiol. 129:299–315 10.1085/jgp.200609726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein H., Garneau L., Trinh N.T., Privé A., Dionne F., Goupil E., Thuringer D., Parent L., Brochiero E., Sauvé R. 2009. Inhibition of the KCa3.1 channels by AMP-activated protein kinase in human airway epithelial cells. Am. J. Physiol. Cell Physiol. 296:C285–C295 10.1152/ajpcell.00418.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhler R., Ruth P. 2010. Endothelial dysfunction and blood pressure alterations in K+-channel transgenic mice. Pflugers Arch. 459:969–976 10.1007/s00424-010-0819-z [DOI] [PubMed] [Google Scholar]
- Köhler R., Wulff H., Eichler I., Kneifel M., Neumann D., Knorr A., Grgic I., Kämpfe D., Si H., Wibawa J., et al. 2003. Blockade of the intermediate-conductance calcium-activated potassium channel as a new therapeutic strategy for restenosis. Circulation. 108:1119–1125 10.1161/01.CIR.0000086464.04719.DD [DOI] [PubMed] [Google Scholar]
- Kusch J., Thon S., Schulz E., Biskup C., Nache V., Zimmer T., Seifert R., Schwede F., Benndorf K. 2012. How subunits cooperate in cAMP-induced activation of homotetrameric HCN2 channels. Nat. Chem. Biol. 8:162–169 10.1038/nchembio.747 [DOI] [PubMed] [Google Scholar]
- Lam J., Wulff H. 2011. The lymphocyte potassium channels Kv1.3 and KCa3.1 as targets for immunosuppression. Drug Dev. Res. 72:573–584 10.1002/ddr.20467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., Aldrich R.W. 2009. Activation of the SK potassium channel-calmodulin complex by nanomolar concentrations of terbium. Proc. Natl. Acad. Sci. USA. 106:1075–1080 10.1073/pnas.0812008106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., Halling D.B., Hall A.W., Aldrich R.W. 2009. EF hands at the N-lobe of calmodulin are required for both SK channel gating and stable SK–calmodulin interaction. J. Gen. Physiol. 134:281–293 10.1085/jgp.200910295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maylie J., Bond C.T., Herson P.S., Lee W.S., Adelman J.P. 2004. Small conductance Ca2+-activated K+ channels and calmodulin. J. Physiol. 554:255–261 10.1113/jphysiol.2003.049072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanda Kumar N.S., Singh S.K., Rajendran V.M. 2010. Mucosal potassium efflux mediated via Kcnn4 channels provides the driving force for electrogenic anion secretion in colon. Am. J. Physiol. Gastrointest. Liver Physiol. 299:G707–G714 10.1152/ajpgi.00101.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin F., Auerbach A., Sachs F. 1996. Estimating single-channel kinetic parameters from idealized patch-clamp data containing missed events. Biophys. J. 70:264–280 10.1016/S0006-3495(96)79568-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin F., Auerbach A., Sachs F. 1997. Maximum likelihood estimation of aggregated Markov processes. Proc. Biol. Sci. 264:375–383 10.1098/rspb.1997.0054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sali A., Blundell T.L. 1993. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234:779–815 10.1006/jmbi.1993.1626 [DOI] [PubMed] [Google Scholar]
- Schumacher M.A., Rivard A.F., Bächinger H.P., Adelman J.P. 2001. Structure of the gating domain of a Ca2+-activated K+ channel complexed with Ca2+/calmodulin. Nature. 410:1120–1124 10.1038/35074145 [DOI] [PubMed] [Google Scholar]
- Schumacher M.A., Crum M., Miller M.C. 2004. Crystal structures of apocalmodulin and an apocalmodulin/SK potassium channel gating domain complex. Structure. 12:849–860 10.1016/j.str.2004.03.017 [DOI] [PubMed] [Google Scholar]
- Sheng J.Z., Ella S., Davis M.J., Hill M.A., Braun A.P. 2009. Openers of SKCa and IKCa channels enhance agonist-evoked endothelial nitric oxide synthesis and arteriolar vasodilation. FASEB J. 23:1138–1145 10.1096/fj.08-120451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh S., Syme C.A., Singh A.K., Devor D.C., Bridges R.J. 2001. Benzimidazolone activators of chloride secretion: potential therapeutics for cystic fibrosis and chronic obstructive pulmonary disease. J. Pharmacol. Exp. Ther. 296:600–611 [PubMed] [Google Scholar]
- Srivastava S., Li Z., Ko K., Choudhury P., Albaqumi M., Johnson A.K., Yan Y., Backer J.M., Unutmaz D., Coetzee W.A., Skolnik E.Y. 2006. Histidine phosphorylation of the potassium channel KCa3.1 by nucleoside diphosphate kinase B is required for activation of KCa3.1 and CD4 T cells. Mol. Cell. 24:665–675 10.1016/j.molcel.2006.11.012 [DOI] [PubMed] [Google Scholar]
- Syme C.A., Hamilton K.L., Jones H.M., Gerlach A.C., Giltinan L., Papworth G.D., Watkins S.C., Bradbury N.A., Devor D.C. 2003. Trafficking of the Ca2+-activated K+ channel, hIK1, is dependent upon a C-terminal leucine zipper. J. Biol. Chem. 278:8476–8486 10.1074/jbc.M210072200 [DOI] [PubMed] [Google Scholar]
- Szkotak A.J., Murthy M., MacVinish L.J., Duszyk M., Cuthbert A.W. 2004. 4-Chloro-benzo[F]isoquinoline (CBIQ) activates CFTR chloride channels and KCNN4 potassium channels in Calu-3 human airway epithelial cells. Br. J. Pharmacol. 142:531–542 10.1038/sj.bjp.0705846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tharp D.L., Bowles D.K. 2009. The intermediate-conductance Ca2+-activated K+ channel (KCa3.1) in vascular disease. Cardiovasc. Hematol. Agents Med. Chem. 7:1–11 10.2174/187152509787047649 [DOI] [PubMed] [Google Scholar]
- Toyama K., Wulff H., Chandy K.G., Azam P., Raman G., Saito T., Fujiwara Y., Mattson D.L., Das S., Melvin J.E., et al. 2008. The intermediate-conductance calcium-activated potassium channel KCa3.1 contributes to atherogenesis in mice and humans. J. Clin. Invest. 118:3025–3037 10.1172/JCI30836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall-Lacelle S., Hossain M.I., Sauvé R., Blunck R., Parent L. 2011. Double mutant cycle analysis identified a critical leucine residue in the IIS4S5 linker for the activation of the Ca(V)2.3 calcium channel. J. Biol. Chem. 286:27197–27205 10.1074/jbc.M111.237412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waschk D.E., Fabian A., Budde T., Schwab A. 2011. Dual-color quantum dot detection of a heterotetrameric potassium channel (hKCa3.1). Am. J. Physiol. Cell Physiol. 300:C843–C849 10.1152/ajpcell.00053.2010 [DOI] [PubMed] [Google Scholar]
- Wimley W.C., Creamer T.P., White S.H. 1996. Solvation energies of amino acid side chains and backbone in a family of host-guest pentapeptides. Biochemistry. 35:5109–5124 10.1021/bi9600153 [DOI] [PubMed] [Google Scholar]
- Wissmann R., Bildl W., Neumann H., Rivard A.F., Klöcker N., Weitz D., Schulte U., Adelman J.P., Bentrop D., Fakler B. 2002. A helical region in the C terminus of small-conductance Ca2+-activated K+ channels controls assembly with apo-calmodulin. J. Biol. Chem. 277:4558–4564 10.1074/jbc.M109240200 [DOI] [PubMed] [Google Scholar]
- Wu S., Zhang Y. 2008. MUSTER: Improving protein sequence profile-profile alignments by using multiple sources of structure information. Proteins. 72:547–556 10.1002/prot.21945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wulff H., Knaus H.G., Pennington M., Chandy K.G. 2004. K+ channel expression during B cell differentiation: implications for immunomodulation and autoimmunity. J. Immunol. 173:776–786 [DOI] [PubMed] [Google Scholar]
- Zhang M., Pascal J.M., Schumann M., Armen R.S., Zhang J.F. 2012. Identification of the functional binding pocket for compounds targeting small-conductance Ca2+-activated potassium channels. Nat Commun. 3:1021 10.1038/ncomms2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M., Pascal J.M., Zhang J.F. 2013. Unstructured to structured transition of an intrinsically disordered protein peptide in coupling Ca2+-sensing and SK channel activation. Proc. Natl. Acad. Sci. USA. 110:4828–4833 10.1073/pnas.1220253110 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.