

Abstract
Because of the large size and modest selectivity of the connexin hemichannel aqueous pore, hemichannel opening must be highly regulated to maintain cell viability. At normal resting potentials, this regulation is achieved predominantly by the physiological extracellular Ca2+ concentration, which drastically reduces hemichannel activity. Here, we characterize the Ca2+ regulation of channels formed by wild-type human connexin26 (hCx26) and its human mutations, D50N/Y, that cause aberrant hemichannel opening and result in deafness and skin disorders. We found that in hCx26 wild-type channels, deactivation kinetics are accelerated as a function of Ca2+ concentration, indicating that Ca2+ facilitates transition to, and stabilizes, the closed state of the hemichannels. The D50N/Y mutant hemichannels show lower apparent affinities for Ca2+-induced closing than wild-type channels and have more rapid deactivation kinetics, which are Ca2+ insensitive. These results suggest that D50 plays a role in (a) stabilizing the open state in the absence of Ca2+, and (b) facilitating closing and stabilization of the closed state in the presence of Ca2+. To explore the role of a negatively charged residue at position 50 in regulation by Ca2+, this position was substituted with a cysteine residue, which was then modified with a negatively charged methanethiosulfonate reagent, sodium (2-sulfanoethyl) methanethiosulfonate (MTSES)−. D50C mutant hemichannels display properties similar to those of D50N/Y mutants. Recovery of the negative charge with chemical modification by MTSES− restores the wild-type Ca2+ regulation of the channels. These results confirm the essential role of a negative charge at position 50 for Ca2+ regulation. Additionally, charge-swapping mutagenesis studies suggest involvement of a salt bridge interaction between D50 and K61 in the adjacent connexin subunit in stabilizing the open state in low extracellular Ca2+. Mutant cycle analysis supports a Ca2+-sensitive interaction between these two residues in the open state of the channel. We propose that disruption of this interaction by extracellular Ca2+ destabilizes the open state and facilitates hemichannel closing. Our data provide a mechanistic understanding of how mutations at position 50 that cause human diseases are linked to dysfunction of hemichannel gating by external Ca2+.
INTRODUCTION
Connexins constitute a family of transmembrane proteins, with at least 21 members in humans, and are found in almost all cell types (Willecke et al., 2002). They form gap junction channels (GJCs) that mediate intercellular communication crucial in diverse processes including development and physiology, as well as response to trauma, inflammation, and disease (Sáez et al., 2003; Contreras et al., 2004). Newly synthesized connexins are assembled in the endoplasmic reticulum and/or Golgi apparatus to form hexamers known as hemichannels. The hemichannels are transported to the plasma membrane where they reside until they dock with hemichannels in apposed cells to form GJCs. Hemichannels and GJCs are typically permeable to ions and molecules up to ∼12 Å in diameter (Harris, 2001). Release of small metabolites, such as ATP and glutamate, by unapposed hemichannels at the plasma membrane seems to play an important role in autocrine/paracrine signaling in many cell types and tissues (Bennett et al., 2003; Ebihara, 2003; Sáez et al., 2010; Wang et al., 2013). Exacerbated opening of hemichannels, however, leads to loss of electrochemical gradients and cytoplasmic metabolites, causing cell death. There is compelling evidence that metabolic inhibition, hypoxia, inflammation, and oxidative stress increase the opening of plasma membrane hemichannels, thereby aggravating cell damage and accelerating cell death (Contreras et al., 2002, 2004; Decrock et al., 2009; Orellana et al., 2011a,b). In addition, a large number of human connexin mutations produce exacerbated opening of unapposed hemichannels, causing the cellular dysfunction and death that are pivotal in the development of connexin-associated pathologies (Abrams et al., 2002; Liang et al., 2005; Stong et al., 2006; Dobrowolski et al., 2007; Gerido et al., 2007; Matos et al., 2008; Lee et al., 2009; Minogue et al., 2009; Sánchez et al., 2010; Tong et al., 2011).
Proper control of the opening and closing of unapposed hemichannels is essential and is achieved by physiological levels of extracellular Ca2+ (∼1.8 mM), which drastically reduce hemichannel activity. Opening of unapposed hemichannels was first observed in Xenopus laevis oocytes expressing lens connexins; osmotic swelling rapidly killed the oocytes unless Ca2+ was raised to keep the hemichannels closed (Paul et al., 1991). Reduction of extracellular Ca2+ below physiological levels increases the opening of most, if not all, connexin hemichannels (e.g., Cx26, Cx30, Cx32, Cx43, Cx46, and Cx50) (Paul et al., 1991; Zampighi et al., 1999; Valiunas and Weingart, 2000; Beahm and Hall, 2002; Contreras et al., 2003; Ebihara et al., 2003; Gómez-Hernández et al., 2003; Ripps et al., 2004). Despite the important role of external Ca2+ in regulating hemichannel opening, little is known about the molecular mechanisms underlying this process. Recently, a high resolution structure of the human connexin26 (hCx26) GJC was solved (Maeda et al., 2009), providing crucial information to guide functional studies to elucidate the molecular gating mechanisms of connexin channels. Here, we investigate the mechanism by which extracellular Ca2+ regulates the opening and closing of unpaired hCx26 hemichannels in the plasma membrane. We found that aspartate to asparagine or tyrosine substitutions at position 50 (D50N/Y)–human mutations that cause syndromic deafness– severely compromise the ability of hCx26 hemichannels to be regulated by extracellular Ca2+. Analysis of the kinetic and steady-state data strongly suggests that D50 stabilizes the open state of Cx26 hemichannels by interacting with a positively charged residue (K61) in the adjacent connexin subunit. We propose that disruption of this interaction by extracellular Ca2+ facilitates destabilization of the open state and promotes hemichannel closing.
MATERIALS AND METHODS
Channel expression and molecular biology
cDNA for hCx26 was purchased from OriGene. Wild-type Cx26 was subcloned in the pGEM-HA vector (Promega) for expression in Xenopus oocytes. Mutations of hCx26 were produced with QuikChange II Site-Directed Mutagenesis kits (Agilent Technologies). DNA sequencing performed at the New Jersey Medical School Molecular Resource Facility confirmed the amino acid substitutions. Nhe1-linearized hCx26 wild-type and mutant DNAs were transcribed in vitro to cRNAs using the T7 Message Machine kit (Ambion).
Electrophysiology
Electrophysiological data were collected using the two-electrode voltage-clamp technique. All recordings were made at room temperature (20–22°C). The recording solutions contained (mM) 118 NaCl, 2 KCl, and 5 HEPES, pH 7.4, with a range of Ca2+ concentrations from 0.01 to 20. Currents from oocytes were recorded 1–3 d after cRNA injection using an oocyte clamp (OC-725C; Warner Instruments). Currents were sampled at 2 kHz and low-pass filtered at 0.2 kHz. Microelectrode resistances were between 0.1 and 1.2 MΩ when filled with 3 M KCl. All recordings were performed using agar bridges connecting bath and ground chambers.
Measurement of Ca2+ dose–response curves
In Xenopus oocytes, lowering Ca2+ below ∼0.15 mM activates endogenously expressed Cx38 hemichannels, which can interfere with analysis of heterologously expressed hCx26 currents. To efficiently reduce endogenous Cx38 expression, antisense oligonucleotide against Cx38 (1 mg/ml; using the sequence from Ebihara, 1996) was injected 4 h after harvesting the oocytes. After 1 d, the same oocytes were coinjected with 18–50 nl cRNA (0.5–1 mg/ml) hCx26 or mutants plus the Cx38 (1 mg/ml) antisense. Before performing any Ca2+ dose–response curves in oocytes expressing hCx26 or mutant hemichannels, we tested for the levels of Cx38 endogenous current in oocytes injected only with Cx38 antisense; only batches that showed no or low endogenous Cx38 currents at the lowest extracellular Ca2+ concentration (0.01 mM) were used to perform the Ca2+ dose–response curves. Because of the slow kinetics of hCx26 activation and deactivation, Ca2+ dose–response measurements were obtained by assessing the tail current peaks after reaching current saturation during a depolarizing pulse from −80 to 0 mV. Tail current measurements include the steady-state “holding” currents caused by the opening of hemichannels at −80 mV induced by reduction of extracellular Ca2+ concentrations. The peak tail current thus measured reflects the number of channels that are open at 0 mV, not only those that opened during the pulse to 0 mV. To minimize the compromise of cell viability produced by exacerbated opening of hemichannels under these experimental conditions, no more than three to four different Ca2+ concentrations, including 0.01 mM Ca2+, were assessed per oocyte. This significantly improved oocyte-to-oocyte reproducibility of the currents in response to Ca2+ changes. Deactivation time constants from tail currents were determined by fitting tail current, up to 10 s after reaching steady state, to exponential functions using Clampfit 11 software (Molecular Devices).
Mutant cycle analysis
Mutant cycle analysis was performed on the apparent affinities of Ca2+ from steady-state currents and on binding rates obtained from deactivation time constants. Macroscopic parameters from apparent affinities have been used previously to support side chain interactions and establish coupling coefficients in ligand-gated channels (Kash et al., 2003; Price et al., 2007; Gleitsman et al., 2008). Coupling energy (ΔΔG) was calculated as:
| (1) |
where R is the ideal gas constant, and T is the absolute temperature. If two mutations are functionally independent with respect to Ca2+ sensitivity, the coupling energy will be close to 0 kcal/mol. Significant coupling is indicated by any value that deviates from zero, but the accepted cutoff for nonadditivity is 0.5 kcal/mol (Laha and Wagner, 2011).
Online supplemental material
The supplemental text and Fig. S1 describe and validate the use of tail current measurements as an indication of hCx26 hemichannel activation in Xenopus oocytes. Fig. S2 demonstrates isolation of hCx26 hemichannel currents from endogenous Ca2+-activated chloride currents. Fig. S3 shows the Ca2+ regulation of D50C mutant hemichannels. Fig. S4 shows the interplay between Ca2+ and voltage regulation of wild-type and D50N/Y mutant hemichannels. Online supplemental material is available at http://www.jgp.org/cgi/content/full/jgp.201210893/DC1.
RESULTS
Ca2+ regulation of hCx26 hemichannels
The Ca2+-regulated gating properties of hCx26 hemichannels expressed in Xenopus oocytes were explored using the two-electrode voltage-clamp technique. We observed that the tail currents that followed depolarizing pulses were more accurate and useful measures of hCx26 hemichannel activation and kinetics than were the outward currents that developed during the pulses (see supplemental text and Figs. S1 and S2 for a complete description). We found that with depolarizing pulses to 0 mV, the peak tail currents increased as a function of pulse duration, reaching a maximum with pulses of 40 s (Fig. S1). Therefore, our standard protocol for assessment of hCx26 hemichannel activation and deactivation was to examine the peak tail currents and their relaxation kinetics after 40-s pulses from −80 to 0 mV. Even though depolarization pulses above 0 mV would activate more hemichannels, limiting depolarizations to 0 mV allowed us to minimize the deleterious effects of massive hemichannel opening during the long pulses required to reach steady-state activation, as well as eliminate contributions from endogenous currents. The tail currents thus measured reflect the steady-state channel activation at 0 mV at any given Ca2+ concentration.
Using this method, the hCx26 hemichannel tail currents in response to changes in external Ca2+ concentration were examined. Fig. 1 A shows current traces obtained at 10, 1.8, 0.5, and 0.1 mM of extracellular Ca2+ from the same oocyte expressing a moderate level of hCx26 currents. The peak tail currents increase with reduction of external Ca2+, showing that external Ca2+ inhibits activation of hCx26 hemichannels. The holding current before depolarization is significantly increased at 0.1 mM Ca2+, indicating that low Ca2+ causes an increase in open hemichannels, even at −80 mV. The activation of the hemichannels as a function of Ca2+ concentration, normalized to the maximal activation of the tail current at the lowest Ca2+ concentration, is shown in Fig. 1 B. The data are fit to a Hill equation of the form:
| (2) |
where the fractional current is I/Imax, I is the tail current at a particular Ca2+ concentration, Imax is the maximal tail current activation at 0.01 mM Ca2+, KD is the apparent affinity, [Ca2+] is the concentration of Ca2+ applied to the bath, and n is the Hill coefficient. The best-fit parameter values for KD and n are 0.33 mM and 1.38, respectively. Fig. 1 C displays the deactivation time constants of tail currents at different Ca2+ concentrations. The deactivation kinetics are accelerated as a function of Ca2+ concentration.
Figure 1.

Ca2+ modulates gating in hCx26 hemichannels. (A) Current traces elicited by a voltage pulse from −80 to 0 mV from oocytes expressing hCx26 hemichannels in the presence of different Ca2+ concentrations. (B) [Ca2+] dose–response relation determined from the peak tail current after a voltage pulse from −80 to 0 mV. The solid line represents the best fits of the data to a Hill equation (Eq. 2). (C) Deactivation time constants as a function of Ca2+ concentration. The solid line corresponds to a linear fit to the data. The data points represent mean ± SEM of at least three independent measurements.
Consistent with previous reports based on currents obtained during depolarizing pulses, the tail current data indicate that external Ca2+ causes a relative stabilization of closed hemichannels. The kinetic data indicate that external Ca2+ also accelerates transitions to the closed state.
Aberrant gating by extracellular Ca2+ in D50N/Y mutant hemichannels
Several hCx26 mutations produce exacerbated opening of hemichannels, the closure of which requires high extracellular Ca2+ (5–10 mM), unlike wild-type channels, for which 1.8 mM of extracellular Ca2+ is sufficient. Using tail current analysis, the effects of extracellular Ca2+ on hCx26 hemichannels containing a single human mutation replacing the aspartate at position 50 with an asparagine (D50N) or a tyrosine (D50Y) were examined. These substitutions remove the negative charge at this position. Fig. 2 A shows current traces in response to depolarizing pulses from −80 to 0 mV in the presence 0.25, 1.8, and 10 mM Ca2+ for oocytes expressing D50N or D50Y mutant hemichannels. For both mutations, the holding currents and peak tail currents significantly increase as extracellular Ca2+ is reduced, and at high Ca2+ concentrations, the tail currents are greater than for wild-type channels. For example, at 10 mM Ca2+, tail currents are completely absent in wild-type channels (Fig. 1 A), but in both D50N and D50Y mutants, they are substantial, suggesting that these mutations decrease the ability of Ca2+ to stabilize the closed state.
Figure 2.

Gating by Ca2+ is altered by D50N/Y mutations in hCx26 hemichannels. (A) Representative current traces elicited by a pulse to 0 mV from a holding potential of −80 mV for an oocyte expressing D50N or D50Y mutant hemichannels. Numbers 1, 2, and 3 correspond to current traces obtained in the presence of 10, 1.8, or 0.25 mM Ca2+, respectively. (B) Ca2+ dose–response curve for oocytes expressing D50N (open circles) or D50Y mutant (open diamonds) hemichannels. The solid and dotted lines represent the best fits to a Hill equation (Eq. 2) for wild-type (from Fig. 1 B) and D50N/Y mutant hemichannels, respectively. (C) Deactivation time constants of the corresponding tail currents at different Ca2+ concentrations for D50N (open circles) or D50Y mutants (fast and slow time constants are shown as two sets of diamonds). Dotted lines are the best linear fit to the D50N/Y mutant hemichannel data. The solid line corresponds to the linear fit of the average data for wild-type hemichannels (from Fig. 1 C). The data represent mean ± SEM of at least three independent measurements.
Given the enhanced hemichannel opening of the mutants at high extracellular Ca2+, we were concerned that Ca2+ influx through the increased number of open hemichannels could activate endogenous Ca2+-activated chloride currents, contaminating the measured macroscopic currents. Ca2+-activated chloride currents can be eliminated by preinjection of oocytes with ∼120 µM BAPTA (see Fig. S2). We obtained [Ca2+] dose–response curves in oocytes expressing the D50N/Y mutants preinjected with BAPTA. No significant differences were observed in the currents in the absence or presence of intracellular BAPTA for oocytes expressing moderate levels of D50N/Y mutants (not depicted), indicating that even with the increased hemichannel activity at high extracellular Ca2+, there was no significant contamination by Ca2+-activated chloride currents.
Fig. 2 B shows the [Ca2+] dose–response relations for D50N and D50Y. The calculated values for KD are 1.5 and 1.3 mM for D50N and D50Y mutants, respectively. There is a significant rightward shift in the KD of D50N/Y mutants with respect to that observed in wild-type hemichannels. At physiological Ca2+ concentration (1.8 mM), although D50N/Y mutant hemichannels reach ≥40% of the maximal response, the wild-type channels reach only ∼15%. As suggested by the traces in Fig. 2 A, these results confirm that the charge-removing substitutions of the aspartate at position 50 with asparagine or tyrosine interfere with the ability of extracellular Ca2+ to favor the closed state of hCx26 hemichannels. There is a decrease in the slope of the relation as well, which could suggest fewer sites of Ca2+ action, reduced cooperativity, or less effective transduction of Ca2+ binding to effect gating changes.
In contrast to wild-type channels, Fig. 2 C shows that these mutations render the deactivation time constants rapid and nearly completely unresponsive to changes in extracellular Ca2+ concentration. Interestingly, even though the mutations favor the open state at high Ca2+ concentrations relative to wild type, the deactivation (i.e., closing) rate of the channels is increased, even at low Ca2+, to that of wild-type channels. Whereas D50N mutant hemichannels show a single-exponential decay component of ∼11 ± 0.4 s, D50Y mutants display two components, a fast component of 2.6 ± 0.5 s and a slow component of 10 ± 0.4 s. Intracellular injection with 120 µM BAPTA did not affect the exponential components for D50Y mutant hemichannels, suggesting that the additional component is not caused by contaminating chloride currents activated by Ca2+ influx, and that it is a component of Cx26 channel closure specifically induced by the D50Y mutation.
These mutations at D50 can be seen to have two effects: (1) from the steady-state measurements, they significantly reduce (but do not entirely eliminate) the ability of Ca2+ to stabilize the closed state relative to the open state (rightward shift in Fig. 2 B); and (2) from the tail current kinetics, they eliminate the Ca2+ effect on the deactivation time constant and increase the deactivation time constant to that seen at high Ca2+ in wild-type channels (at 1.8 mM Ca2+; Fig. 2 C). The more rapid deactivation kinetics suggest that elimination of the negative charge at position 50 decreases the dominant energy barrier for channel closing, just as high Ca2+ does in wild-type channels. The fact that mutations that remove the negative charge at position 50 reduce Ca2+ sensitivity of the currents, accelerate deactivation kinetics, and render them effectively insensitive to Ca2+ lead to the idea that in wild-type channels, Ca2+ may disrupt electrostatic interactions that involve D50.
A negatively charged residue at position 50 is critical to enhance opening and closing of the channel at low and high Ca2+, respectively
To further explore the role of a negatively charged residue at position 50 in Ca2+ regulation, D50 was substituted with a cysteine residue (D50C), which was then chemically modified with negatively or positively charged MTS reagents, sodium (2-sulfanoethyl) MTS (MTSES)− and [2-(trimethylammonium) ethyl] MTS bromide (MTSET)+. As expected, substitution of the Cys residue at this position yielded hemichannels with gating properties similar to those described above for D50N/Y mutants. Fig. S3 shows the [Ca2+] dose–response relation and deactivation time constants for D50C hemichannels. The best-fit parameter values for KD and n were 4 mM and 1.38, respectively. As for D50N/Y, deactivation time constants were fast and essentially unresponsive to changes in extracellular Ca2+ concentration.
Fig. 3 (A and B) shows representative hemichannel current traces from D50C mutant hemichannels obtained at 0.25 and 10 mM of extracellular Ca2+ in the absence (black traces) or the presence (colored traces) of 100 µM MTS reagents. At low extracellular Ca2+, modification of D50C channels with negatively charged MTSES− (Fig. 3 A, red trace) increased the holding and tail currents and slowed the deactivation time constant (Fig. 3, A, top left panel, and B, top trace). Conversely, at high extracellular Ca2+, modification with MTSES− reduced the holding currents and accelerated the deactivation time constant (Fig. 3, A, top right panel, and B, bottom trace). Fig. 3 C shows that modification of D50C with MTSES−, which reinstates the wild-type negative charge at this position, reproduces wild-type Ca2+-sensitive deactivation kinetics (Fig. 3 C, red symbols).
Figure 3.

A negatively charged residue at position 50 is critical for regulation by Ca2+. (A) Hemichannel currents from oocytes expressing D50C mutant hemichannels in low and high extracellular Ca2+ to assess the effects of chemical modification with MTS reagents. Currents were elicited by a pulse to 0 mV from a holding potential of −80 mV before (black trace) or in the presence of MTSES− (red traces) or MTSET+ (blue traces). (B) Holding currents obtained at −80 mV for oocytes expressing D50C mutant hemichannels incubated in low (0.25 mM) or high (10 mM) external Ca2+. The addition of MTSES− increased the holding current in low extracellular Ca2+ (top trace) and decreased the holding current in the presence of high Ca2+ (bottom trace). (C) Deactivation time constants for D50C mutant hemichannels at different Ca2+ concentrations before (dotted line; from Fig. S3) and after chemical modification with MTSES− (red triangles) or MTSET+ (blue triangles). Time constants obtained after MTSES− modification coincide with the solid line (from Fig. 1 C) that corresponds with the linear fit for wild-type hemichannels. Conversely, the time constant obtained after MTSET+ overlaps with the linear fit for D50C mutants with no modification. The data represent mean ± SEM of at least three independent measurements.
These results point to the importance of a negative charge at position D50 to both stabilize the open state relative to the closed state at low Ca2+ and to favor and facilitate transitions to the closed state at high Ca2+. We note that at high Ca2+, the tail currents are larger after modification with MTSES−, rather than smaller as would be expected if full wild-type behavior was recovered. These increased tail currents are not affected by intracellular injection with 120 mM BAPTA, eliminating the potential contribution of endogenous chloride currents (not depicted). Given the clear recapitulation of Ca2+ effects on the deactivation kinetics (Fig. 3 C), and the decrease in holding current at high Ca2+ (Fig. 3 B, bottom trace), we infer that the increase in tail currents at high Ca2+ with MTSES− modification arises from an effect on a different process. A likely effect is on activation of the channels by voltage, perhaps caused by the negative charge of the MTSES− moiety having a different relation to the transmembrane voltage field than that of the native aspartate. This region is known to be involved in voltage-induced rearrangements at the extracellular loop (Tang et al., 2009; Verselis et al., 2009).
D50C mutants modified with the positively charged MTSET+ show a decrease in the holding currents and tail currents at low Ca2+ (opposite of the effect of MTSES−) but no change in the deactivation time constants (Fig. 3 A, bottom panels, and C, blue symbols).
These data show that chemical modification with a negatively charged reagent largely reinstates wild-type behavior in D50C hemichannels, indicating that a negative charge at this position is critical to the effects of Ca2+ on relative stability of the open and closed states, and the transitions between them. When the charge at this position is removed or made positive, the deactivation is independent of the extracellular Ca2+ concentration. We do not know whether all six of the D50C residues in a hemichannel need to be modified to have these effects, or if only a subset is modified by the MTS reagents.
Molecular interactions of D50 that stabilize the open state in low external Ca2+
To better understand the molecular mechanism by which D50 stabilizes the open-channel conformation at low extracellular Ca2+ concentrations, we investigated possible electrostatic interactions between D50 and neighboring residues, using the recent hCx26 crystal structure (Maeda et al., 2009) as a guide. This structure was obtained in the absence of Ca2+ and seems to represent the channel with open gates. D50 is in the outer part of the ion permeation pathway. The only positively charged residue near the carboxyl group of D50 is lysine 61 (K61) in the adjacent connexin subunit, at ∼4.4 Å (Fig. 4). Although this distance is just beyond that of a typical salt bridge interaction (<4 Å), the moderate 3.5-Å resolution of the structure and the proximity of these oppositely charged residues led us to speculate that these residues interact electrostatically in the open conformation to provide the enthalpy that helps to stabilize the open conformation.
Figure 4.

Possible intersubunit salt bridge interaction between positions D50 and K61 in the open conformation. (A) Top (extracellular) view of the hCx26 hemichannel from the crystal structure (Protein Data Bank accession no. 2ZW3; Maeda et al., 2009). Positions D50 and K61 are highlighted in red and blue (in all subunits), respectively. (B) Enlargement showing the proximity between position D50 in one subunit and position K61 in the adjacent subunit. The average distance for the six pairs of D50–K61 residues is ∼4.4 Å.
As a first approach to test this idea, we reasoned that if a salt bridge interaction between D50 and K61 favors the open state of the channels and is involved in regulation by Ca2+, then swapping the positively and negatively charged residues should retain a possible electrostatic interaction between these residues and produce channels sensitive to Ca2+, whereas the individual charge reversals should not (of course, there could be other effects of both the individual and combined mutations). A similar approach was applied successfully to CNG channels to establish the role of salt bridges in gating (Craven and Zagotta, 2004).
The single charge reversal mutations, D50K and K61D, do not yield functional hemichannel currents (Fig. 5 A, top two traces). However, as predicted, hemichannel currents and sensitivity to Ca2+ were rescued in the double mutant D50K/K61D (Fig. 5, A, bottom trace, and C), which swaps the charges at the two positions. The Ca2+ dependence of the deactivation kinetics was only modestly recovered, yet the kinetics were slower at all Ca2+ concentrations than in the D50N/Y mutants, particularly at low Ca2+ (Fig. 5 B, symbols). This suggests that a negatively charged residue is specifically required at position 50 for the full Ca2+ effect on deactivation, and that a negative charge at position 61 cannot serve this function. For the [Ca2+] dose–response relations of D50K/K61D double mutants, the best-fit parameter values for KD and n were 0.5 mM and 0.6, respectively (Fig. 5 C). There is a decrease in the slope of the relation with respect to wild-type hemichannels, suggesting fewer sites of Ca2+ action, less cooperativity, and/or less efficient transduction of Ca2+ binding to effect gating changes. Overall, reversing the polarity of charged residues at positions 50 and 61 produced functional channels and partially rescued the wild-type hCx26 sensitivity to Ca2+, suggesting that having opposite charges at these two positions is important both to stabilize open channels and to respond to Ca2+.
Figure 5.
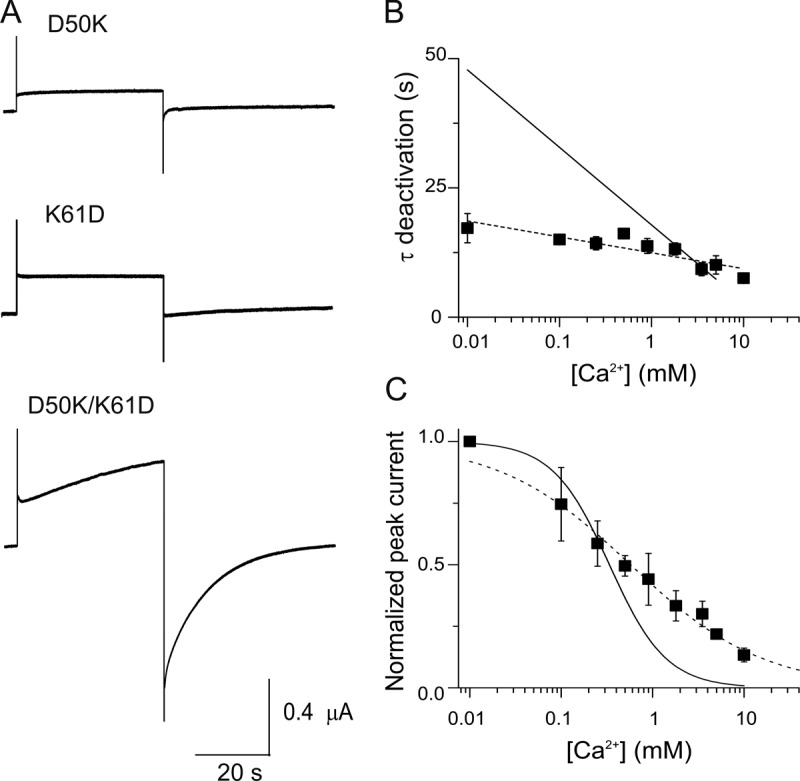
Exchanging the positions of the negative (D50) and positive (K61) residues partially rescues the wild-type hCx26 regulation by Ca2+. (A) Current traces elicited by a pulse to 0 mV from a holding potential of −80 mV are shown for oocytes expressing D50K, K61D, or D50K/K61D mutant hemichannels in 1.8 mM Ca2+. (B) Deactivation time constants for oocytes expressing D50K/K61D mutant hemichannels (closed squares). The solid line corresponds to best fits to the average data for wild-type hemichannels (from Fig. 1 C). The dotted line is the best linear fit to the data from double mutant D50K/K61D hemichannels. (C) [Ca2+] dose–response relations for oocytes expressing D50K/K61D mutant hemichannels (closed squares). The solid and dotted lines correspond to the best fits to the data of a Hill equation (Eq. 1) for wild-type and double mutant D50K/K61D hemichannels. The data represent mean ± SEM of at least three independent measurements.
To obtain further validation of an interaction between D50 and K61 and its relation to regulation of Ca2+, we performed thermodynamic analysis based on mutagenesis, a method known as double mutant cycle analysis, which quantifies the coupling energy between two mutated residues and thus indicates the likelihood that the two residues interact (Faiman and Horovitz, 1996). Ideally, each mutation should eliminate the interaction being studied. For an electrostatic interaction, the full coupling energy can be derived from substitutions that completely eliminate the interaction; however, a partial coupling energy can be derived from mutants that alter but do not eliminate the interaction. For position D50, we used the D50N mutation because it eliminates the electrostatic interaction, yet the channels show robust hemichannel currents and simple kinetics (Fig. 2). However, at position K61, substitution of neutral amino acids (alanine, cysteine, and serine) or histidine did not lead to observable hemichannel currents. However, it has been shown previously that mutant cycle analysis can be performed using a substitution with the same charge that also alters the channel property being studied (e.g., NaChBac channels; Paldi and Gurevitz, 2010). We found that replacing K61 with an arginine (K61R), which retains the charge at this position, results in functional hemichannels. As expected, this channel has kinetic properties similar to those of wild-type channels (Fig. 6), but it has somewhat different parameters of Ca2+ sensitivity, enabling it to be used in the cycle analysis. [Ca2+] dose–response curves in oocytes expressing the K61R mutant channels show values for KD and n of 0.4 mM and 0.85, respectively. Interestingly, the double mutant D50N/K61R shows steady-state currents similar to those of the K61R mutant but with two deactivation time constants nearly insensitive to extracellular Ca2+, similar to the D50Y mutants. The best-fit parameter values for KD and n from the [Ca2+] dose–response curves for double mutant D50N/K61R were 0.51 mM and 0.7, respectively (Fig. 6 B).
Figure 6.
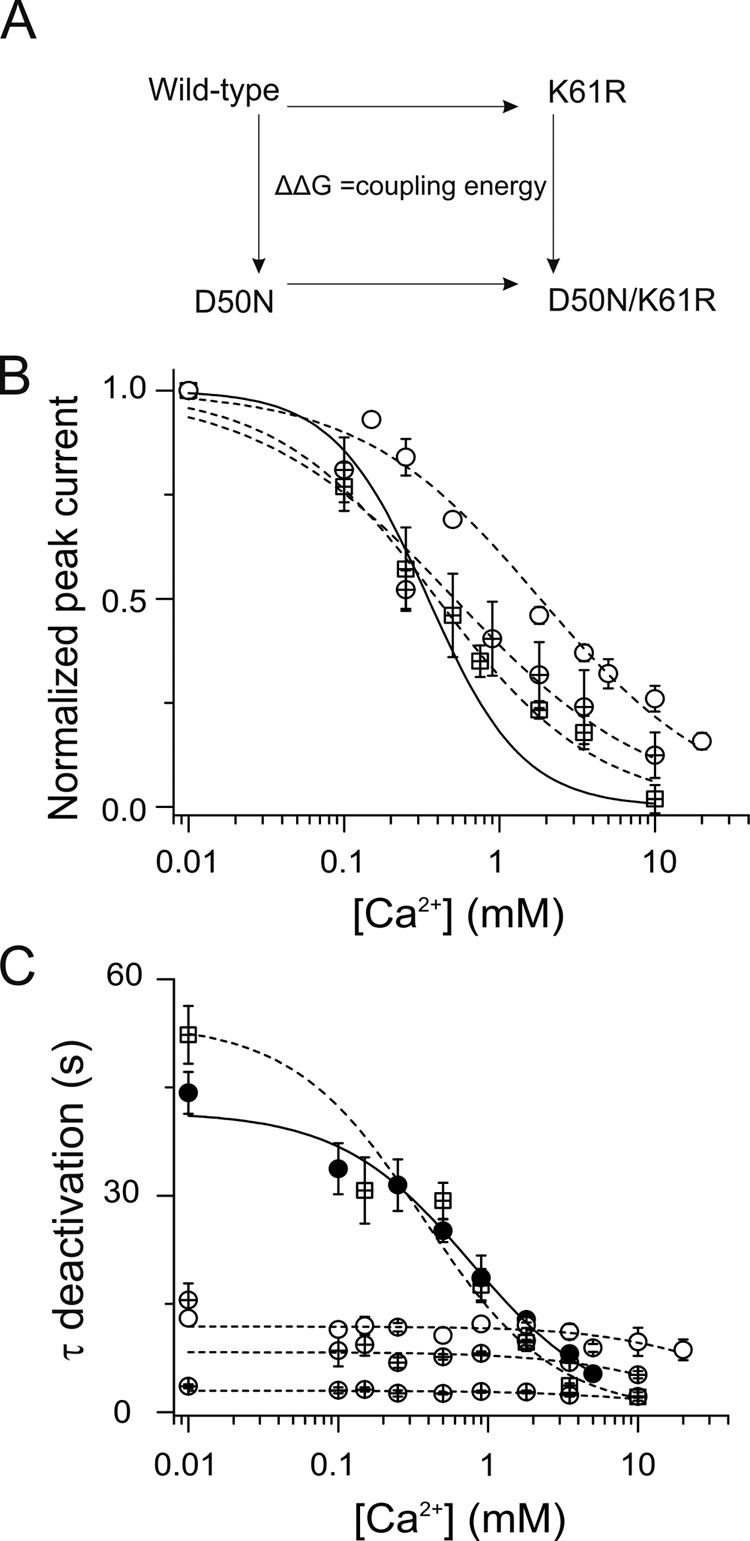
Mutant cycle analysis of KD indicates that D50 and K61 residues are coupled in a Ca2+-sensitive manner. (A) Scheme for mutant cycle analysis of wild-type and mutant hemichannels. (B) Graph shows the [Ca2+] dose–response relations for oocytes expressing D50N (open circles) and K61R (crossed squares) mutants, and D50N/K61R (crossed circles) double mutant hemichannels. The solid and dotted lines represent the best fits to the Hill equation for wild-type (from Fig. 1 B) and mutant channels, respectively. (C) Deactivation time constants for oocytes expressing wild-type (closed circles), D50N (open circles), and K61R (crossed square) mutants, and D50N/K61R (fast and slow time constants are shown as two sets of crossed circles) double mutant hemichannels. The solid line corresponds to the best fits to the average data for wild-type hemichannels using Eq. 3. The dotted line is the best fits to the data to Eq. 3 for single and double mutant hemichannels.
The apparent affinities for Ca2+ from wild-type, D50N, and K61R single mutants, and D50N/K61R double mutant channels (Fig. 6 B), were used to perform mutant cycle analysis. The pairwise interaction energy between positions was estimated using Eq. 1. This analysis yielded a coupling energy (ΔΔG) of −0.75 kcal/mol (cutoff for noninteraction is below ±0.5 kcal/mol). Because this pair of substitutions, in which the charge at one position was preserved, can yield only a portion of the coupling energy of a salt bridge still yields a significant ΔΔG, this result suggests that side chains of residues D50 and K61 interact in a Ca2+-sensitive manner.
Even though the above result indicates a significant Ca2+-sensitive interaction between D50 and K61, an apparent KD can depend on multiple microscopic reactions that involve both ligand affinity and channel gating (Colquhoun, 1998; Gleitsman et al., 2008). These factors may affect the values calculated for the coupling energies that exist between two states (Laha and Wagner, 2011). The fact that charge-eliminating substitutions at position D50 render the deactivation kinetics nearly completely insensitive to Ca2+ suggests that essentially all of the Ca2+ sensitivity of the deactivation time constant is caused by Ca2+-mediated disruption of the D50–K61 interaction that exists in the open state. This idea—the effect of Ca2+ on deactivation kinetics is exclusively on open channels with a D50–K61 interaction—can be assessed by how well the Ca2+ dependence of the deactivation kinetics can be modeled as if they arise from simple open-channel “block” by Ca2+ using an equation of the form:
| (3) |
where kon is a Ca2+-dependent “closing rate,” and koff is a Ca2+-independent “opening rate.” If the system can be accurately described in this way, this analysis can reveal how the apparent Ca2+ affinity of open channels changes with the mutations that preserve and do not preserve the D50–K61 interaction.
In Fig. 6 C, the Ca2+ dependence of deactivation time constants for wild-type, D50N, and K61R single mutants, and the D50N/K61R double mutant, were fit using Eq. 3. The data from the channels that preserve the opposing charges at positions 50 and 61 (wild-type and K61R mutant) were well fit by Eq. 3, strongly suggesting that the Ca2+ effect on deactivation kinetics of channels with this charge pair is on open channels. The derived values for the apparent Ca2+ on-rates (kon) and off-rates (koff) are summarized in Table 1. An apparent affinity (KD) of Ca2+ of open channels can then be calculated by dividing koff by kon. These KD values are independent of those derived from the steady-state data, which include the effects of Ca2+ on the closed state.
Table 1.
Kinetics of Ca2+-dependent deactivations
| Hemichannel | Koff | Kon | KD |
| s | M−1 s−1 | ||
| Wild type | 0.024 | 0.031 | 0.72 |
| D50N | 0.084 | 0.002 | 42 |
| K61R | 0.018 | 0.05 | 0.36 |
| D50N/K61R slow | 0.12 | 0.007 | 17 |
| D50N/K61R fast | 0.338 | 0.021 | 15.7 |
The open-state Ca2+ apparent affinities thus computed for wild-type and K61R mutant channels (which retain the charge interaction between positions 50 and 61) were similar, with KD values of 0.72 and 0.36 mM, respectively, which were close to the apparent KD values derived from steady-state data (0.33 and 0.4 mM, respectively). Conversely, channels in which the interaction was disabled (D50N and D50N/K61R channels) showed substantially lower Ca2+ apparent affinity for open channels (42 mM for D50N and 17 and 15.7 mM for the slow and fast components of D50N/K61R channel deactivation, respectively). These results support the notion that Ca2+ sensitivity of the deactivation kinetics is predominantly accounted for by Ca2+ effects on the D50–K61 interaction that exists in open channels.
To gain information on the thermodynamic linkage between [Ca2+] and the D50–K61 interaction in the open state, we performed mutant cycle analysis based on the apparent rate constants derived from the above calculations. This analysis yielded a coupling energy (ΔΔG) for the on-rate (kon) and off-rate (koff) of approximately −1.0 kcal/mol for the fast kinetic component of D50N/K61R deactivation but lower coupling energy for the on-rate (kon) and off-rate (koff) of −0.46 and −0.38 kcal/mol for the slow component, respectively (Table 2). These results further support a significant thermodynamic linkage between [Ca2+] and the D50–K61 interaction in the open state. Furthermore, the correspondence between the ΔΔGs for this linkage derived from steady-state data (which cannot distinguish effects on open and closed channels) and from the kinetic data strongly suggests that disruption of the D50–K61 interaction occurs in open channels.
Table 2.
Mutant cycle analysis of rates obtained from Ca2+-dependent deactivation
| D50-K61 | ΔΔG coupling |
| kcal | |
| Koff (K61RD50N; slow) | −0.38 |
| Koff (K61RD50N; fast) | −0.99 |
| Kon (K61RD50N; slow) | −0.46 |
| Kon (K61RD50N; fast) | −1.1 |
DISCUSSION
It is widely recognized that extracellular Ca2+ inhibits hemichannel activity. We show here that external Ca2+ also accelerates the deactivation of hCx26 tail currents, facilitating the closing of the channels. Mutation of D50 to uncharged residues (N/Y/C) results in rapid deactivation kinetics that are insensitive to Ca2+ and shifts rightward the steady-state effect of external Ca2+ to close the channels, resulting in more channels being open at physiological Ca2+ levels. Reintroduction of a negative charge at this position by modification with MTSES− in the D50C mutant restores the Ca2+ dependence of the steady-state currents and wild-type Ca2+ dependence of deactivation kinetics. A simple interpretation is that a negative charge at position 50 plays a role in (a) stabilizing the open state, and (b) enabling Ca2+ to facilitate closing and destabilization of the open state. Based on these findings and the crystal structure, we speculated that in wild-type channels, D50 interacts electrostatically with K61 in the adjacent subunit, the only nearby positively charged residue. A mutant channel in which the charges at these two positions are swapped (the D50K/K61D mutant) displays Ca2+ sensitivity of steady-state currents, whereas channels with the corresponding single substitutions were nonfunctional. We therefore propose that the negative charge of D50 interacts with other residues, including K61 via a salt bridge, to stabilize the open state, and that these interactions are disrupted by Ca2+, resulting in destabilization of the open state and Ca2+-dependent closing kinetics. Double mutant cycle analyses support a thermodynamic linkage between Ca2+ and disruption of the D50–K61 interaction in open channels. Analysis of steady state and deactivation kinetics of channels in which the charge at position 50 is removed suggest that in addition to its action to disrupt the D50–K61 interaction in open channels, Ca2+ also acts on closed channels (in which there is no D50–K61 interaction) to stabilize the closed state.
In the absence of a validated kinetic model of connexin hemichannel gating that allows us to obtain reliable microscopic rate constants, mutant cycle analyses were performed using apparent affinity values for extracellular Ca2+ from wild-type and mutant channels, using both steady-state and kinetic data, with the latter focusing on Ca2+ effects on the open state only. These studies were constrained to reveal only part of the total coupling energies because we were unable to identify mutations at position 61 that both removed charge (to eliminate any direct D50–K61 charge interaction) and retained channel function. Instead, we used a charge-preserving mutation (K61R) that nonetheless perturbed Ca2+ sensitivity sufficiently to be useful. Because we had shown that the charges at both positions are involved in regulation by Ca2+, analyses using the K61R mutants could therefore reveal only a small portion of the total coupling energy between positions D51 and K61. The full magnitude of the coupling energy could only be revealed if charge was eliminated at position 61; most charge interactions with position 61 would be retained in the K61R mutant, with the substitution producing only small changes (reflected in the small change in KD). In spite of this limitation, the coupling energies were significant, validating the view that there is interaction between D50 and K61 in the open state and that it is affected by Ca2+. In addition, using KDs derived from deactivation kinetics, we found that essentially all of the meaningful Ca2+ sensitivity of the current deactivation required the D50–K61 linkage.
Our data show that the presence of opposite charges at positions 50 and 61 is a key factor in regulation of the channels by Ca2+, and that there is a thermodynamic linkage between these charges. These results, and the indication of close proximity in the crystal structure, suggest the presence of a salt bridge between positions 50 and 61 (whether D50-K61 or D50K-K61D) that stabilizes the open state of the channel and is required for the ability of Ca2+ to destabilize the open state. The fact that the single mutations (D50K and K61D) do not yield functional channels, and the double mutant (D50K-K61D) does, confirms the specific importance of the interaction between the charges at these residues. However, the swapped charges do not fully restore the ability of Ca2+ to enhance the kinetics of deactivation (closing) of the channels. It appears that a negative charge at position 50 specifically is required for this.
Our experiments showing accessibility by MTS reagents indicate that position D50 is water accessible. Therefore, it is likely that the electrostatic interactions of D50, including that with K61 and linked interactions in this region of the protein, are also accessible by Ca2+. Our data do not bear directly on whether D50 itself is a structural component of the Ca2+-binding site. We favor the view that intrasubunit and intersubunit electrostatic networks are involved in these gating reactions (Kwon et al., 2011, 2012). Thus, we cannot distinguish whether Ca2+ disrupts the D50–K61 interaction directly and/or by acting allosterically elsewhere in the network. There are undoubtedly other open-state stabilizing electrostatic interactions in these channels, some of which may be sensitive to Ca2+ and some not (Tong et al., 2013). The precise mechanism of pore occlusion to ion flux that is induced by interaction of Ca2+ with the channel open state remains elusive.
Our overall view is that in wild-type Cx26 hemichannels, salt bridge formation between positions D50 and K61 enhances the stability of the open state, and that Ca2+ disrupts this salt bridge. In this view, reducing Ca2+ favors formation of the salt bridge, increasing occupancy of open states as well as decreasing the deactivation time constant. Mutations at D50 that prevent formation of this salt bridge mimic the disruption of the salt bridge that occurs at high Ca2+, lowering open-state occupancy and mimicking the deactivation time constant observed at high Ca2+ concentration.
Given this view, what is the explanation for the significant but right-shifted Ca2+ dependence of the steady-state data observed in D50 mutants? We suspect and favor the idea that there are several closed and open states for hemichannels, as is the case for many other types of ion channels, with some of these transitions being Ca2+ sensitive and some Ca2+ insensitive. The persistence of Ca2+ sensitivity in the steady-state data in the face of its elimination in the deactivation kinetics in the charge-removing mutants at position 50 (D50N/Y/C mutations) points to Ca2+-dependent transitions among closed states and/or stabilization of closed states by Ca2+, independent of its disruption of the D50–K61 interaction. That is, in wild-type channels, the Ca2+-dependent disruption of the D50–K61 interaction is the rate-limiting step for deactivation. When it does not exist, as in the D50 mutants, Ca2+-independent transitions become rate limiting for deactivation, yet transitions out of closed states still have a Ca2+ dependence, producing a right-shift in steady-state current dependence on Ca2+. Kinetic analysis of the Ca2+-dependent deactivation also suggests that extracellular Ca2+ can affect both open- and closed-state transitions. The Ca2+ affinity derived from the deactivation data for the D50N mutant was much less than that derived from the steady-state data, compared with wild-type channels (1.5 mM from the steady-state analysis and 42 mM from the kinetic analysis). The kinetic analysis of deactivation excludes Ca2+ effects on closed channels, so the fact that there is a difference in the two apparent affinities points to an effect of Ca2+ on closed channels. Single-channel analysis will be necessary to reveal state-dependent binding/effects of Ca2+. Thus far, we have been unable to obtain single-channel recordings from hCx26 hemichannels under conditions that could provide a reliable kinetic understanding of this process.
Interpreting what happens, at the molecular level, in the closed state(s) is less clear, as we do not have closed-state structural data on which to base an explanation of the effects of mutation at position D50. However, the finding that MTSES− modification of D50C mutants reduces the steady-state current at high Ca2+ (when a D50–K61 salt bridge should be disrupted; Fig. 3 C) supports the idea that in the presence of Ca2+, a negative charge at position 50 contributes to stabilization of the closed state, in the absence of the salt bridge. That is, in the closed salt bridge–disrupted state, D50 could contribute directly or indirectly to Ca2+ stabilization of the closed state. However, we have no experimental information regarding D50 interactions in the Ca2+-closed state that can further support this idea. Experiments to address this will be difficult to design in the absence of a Ca2+-bound closed structure. There is atomic force microscopy evidence that hemichannel gating by Ca2+ in several connexins, including Cx26, is accompanied by large structural changes near the extracellular end of the channel, where these residues are located (Müller et al., 2002; Thimm et al., 2005; Allen et al., 2011). These large conformational rearrangements may result in distinct electrostatic interactions of position D50 in the open and Ca2+-closed states.
Residues at the extracellular end of the pore, the region of transition between the first transmembrane segment (TM1) and the first extracellular loop (E1), where D50 is located, have been shown to undergo voltage-driven conformational changes that close the pore, a process referred to as “loop gating” (Kronengold et al., 2003; Tang et al., 2009; Verselis et al., 2009). It is unknown to what extent the conformational changes induced by voltage stimulation are similar to those triggered by extracellular Ca2+. A recent study suggests that the large conformational changes seen in atomic force microscopy with exposure to Ca2+ may not occur with voltage gating (Kwon et al., 2013). In calcium- and voltage-activated potassium channels, the voltage dependence shifts leftward with increased Ca2+ concentrations (Magleby, 2003; Latorre et al., 2010). In Cx46, the voltage dependence of loop gating shifts leftward with Ca2+ decrease (Ebihara et al., 2003). We do not know whether there is similar allosteric coupling between Ca2+ and voltage-driven loop gating in hCx26 hemichannels. It is possible that mutations at D50 alter voltage sensitivity, thereby indirectly affecting Ca2+ sensitivity. However, the deactivation kinetics of tail currents are only weakly affected by voltage using repolarizing pulses over the range of −120 to −20 mV (Fig. S5). Detailed biophysical analysis of Ca2+ gating, its interplay with voltage gating, and the role of D50 are best explored using single-channel recordings. An exploration of the interplay between Ca2+ and voltage regulation remains elusive in hCx26 hemichannels at this time.
The charges at positions 50 and 61, and the interaction between these positions, are likely to serve similar function in some other connexins, but not all. Human Cx30, which has identical residues at these positions, displays gating behavior similar to Cx26 hemichannels in response to changes in extracellular Ca2+ concentrations when expressed in Xenopus oocytes. Negative (D or E) and positive (R or K) charges at the analogous positions are found in Cx25, Cx30.2, and Cx31.9. Connexins with “swapped” charges at these positions include Cx45 and Cx47. However, residue pairs corresponding to positions D50 and K61 in other connexins strongly regulated by Ca2+ are not always charged or even polar. Lens connexins typically have negatively charged residues at both positions, and have a range of Ca2+ sensitivities (Paul et al., 1991; Zampighi et al., 1999; Beahm and Hall, 2002; Ebihara et al., 2003; Tong and Ebihara, 2006). The fact that these channels are functional and in hCx26 the K61D mutant is not suggests that other factors, such as compensatory sequence/structural changes or voltage sensitivities, or indeed completely distinct mechanisms, may contribute to the Ca2+ sensitivity in those connexins. It remains to be seen whether the general mechanism of Ca2+-mediated disruption of a salt bridge that stabilizes the open state, whether at these particular sites or not, operates in other connexin channels.
Physiological and pathophysiological implications
To date, more than 100 human mutations in hCx26 have been shown to cause sensorineural deafness and dermatological disorders. Many of these mutations result in defective channel biogenesis or trafficking. However, a significant number produce connexin channels that are functional at the plasma membrane yet cause pathology. These mutants likely induce altered gating and/or permeability. Indeed, the lack of associated skin disorders in cases of hCx26-null mutations shows that the epidermal dysfunction is not produced by the simple loss of Cx26 function. D50N/Y mutations are linked to keratitis–ichthyosis–deafness syndrome, which consists mainly of sensorineural hearing loss, keratitis, and severe skin lesions (Martínez et al., 2009; Xu and Nicholson, 2013). Our work suggests that the molecular basis of the pathology is elimination of the salt bridge between positions 50 and 61, with the consequence that the hemichannels are less sensitive to regulation by external Ca2+ and the elimination of the negative charge at position 50 that stabilizes the closed state.
Supplementary Material
Acknowledgments
We thank Drs. Joshua Berlin and Miguel Holmgren for helpful discussions.
This work was supported by National Institutes of Health/National Institute of General Medical Sciences (grant RO1-GM099490 to J.E. Contreras).
Angus C. Nairn served as editor.
Footnotes
Abbreviations used in this paper:
- GJC
- gap junction channel
- hCx26
- human connexin26
- MTSES
- sodium (2-sulfanoethyl) MTS
- MTSET
- [2-(trimethylammonium) ethyl] MTS bromide
References
- Abrams C.K., Bennett M.V., Verselis V.K., Bargiello T.A. 2002. Voltage opens unopposed gap junction hemichannels formed by a connexin 32 mutant associated with X-linked Charcot-Marie-Tooth disease. Proc. Natl. Acad. Sci. USA. 99:3980–3984 10.1073/pnas.261713499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen M.J., Gemel J., Beyer E.C., Lal R. 2011. Atomic force microscopy of Connexin40 gap junction hemichannels reveals calcium-dependent three-dimensional molecular topography and open-closed conformations of both the extracellular and cytoplasmic faces. J. Biol. Chem. 286:22139–22146 10.1074/jbc.M111.240002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beahm D.L., Hall J.E. 2002. Hemichannel and junctional properties of connexin 50. Biophys. J. 82:2016–2031 10.1016/S0006-3495(02)75550-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett M.V., Contreras J.E., Bukauskas F.F., Sáez J.C. 2003. New roles for astrocytes: gap junction hemichannels have something to communicate. Trends Neurosci. 26:610–617 10.1016/j.tins.2003.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colquhoun D. 1998. Binding, gating, affinity and efficacy: the interpretation of structure-activity relationships for agonists and of the effects of mutating receptors. Br. J. Pharmacol. 125:924–947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contreras J.E., Sánchez H.A., Eugenin E.A., Speidel D., Theis M., Willecke K., Bukauskas F.F., Bennett M.V., Sáez J.C. 2002. Metabolic inhibition induces opening of unapposed connexin 43 gap junction hemichannels and reduces gap junctional communication in cortical astrocytes in culture. Proc. Natl. Acad. Sci. USA. 99:495–500 10.1073/pnas.012589799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contreras J.E., Sáez J.C., Bukauskas F.F., Bennett M.V. 2003. Gating and regulation of connexin 43 (Cx43) hemichannels. Proc. Natl. Acad. Sci. USA. 100:11388–11393 10.1073/pnas.1434298100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contreras J.E., Sánchez H.A., Véliz L.P., Bukauskas F.F., Bennett M.V., Sáez J.C. 2004. Role of connexin-based gap junction channels and hemichannels in ischemia-induced cell death in nervous tissue. Brain Res. Brain Res. Rev. 47:290–303 10.1016/j.brainresrev.2004.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craven K.B., Zagotta W.N. 2004. Salt bridges and gating in the COOH-terminal region of HCN2 and CNGA1 channels. J. Gen. Physiol. 124:663–677 10.1085/jgp.200409178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decrock E., Vinken M., De Vuyst E., Krysko D.V., D’Herde K., Vanhaecke T., Vandenabeele P., Rogiers V., Leybaert L. 2009. Connexin-related signaling in cell death: to live or let die? Cell Death Differ. 16:524–536 10.1038/cdd.2008.196 [DOI] [PubMed] [Google Scholar]
- Dobrowolski R., Sommershof A., Willecke K. 2007. Some oculodentodigital dysplasia-associated Cx43 mutations cause increased hemichannel activity in addition to deficient gap junction channels. J. Membr. Biol. 219:9–17 10.1007/s00232-007-9055-7 [DOI] [PubMed] [Google Scholar]
- Ebihara L. 1996. Xenopus connexin38 forms hemi-gap-junctional channels in the nonjunctional plasma membrane of Xenopus oocytes. Biophys. J. 71:742–748 10.1016/S0006-3495(96)79273-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebihara L. 2003. New roles for connexons. News Physiol. Sci. 18:100–103 [DOI] [PubMed] [Google Scholar]
- Ebihara L., Liu X., Pal J.D. 2003. Effect of external magnesium and calcium on human connexin46 hemichannels. Biophys. J. 84:277–286 10.1016/S0006-3495(03)74848-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faiman G.A., Horovitz A. 1996. On the choice of reference mutant states in the application of the double-mutant cycle method. Protein Eng. 9:315–316 10.1093/protein/9.3.315 [DOI] [PubMed] [Google Scholar]
- Gerido D.A., DeRosa A.M., Richard G., White T.W. 2007. Aberrant hemichannel properties of Cx26 mutations causing skin disease and deafness. Am. J. Physiol. Cell Physiol. 293:C337–C345 10.1152/ajpcell.00626.2006 [DOI] [PubMed] [Google Scholar]
- Gleitsman K.R., Kedrowski S.M., Lester H.A., Dougherty D.A. 2008. An intersubunit hydrogen bond in the nicotinic acetylcholine receptor that contributes to channel gating. J. Biol. Chem. 283:35638–35643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez-Hernández J.M., de Miguel M., Larrosa B., González D., Barrio L.C. 2003. Molecular basis of calcium regulation in connexin-32 hemichannels. Proc. Natl. Acad. Sci. USA. 100:16030–16035 10.1073/pnas.2530348100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris A.L. 2001. Emerging issues of connexin channels: biophysics fills the gap. Q. Rev. Biophys. 34:325–472 10.1017/S0033583501003705 [DOI] [PubMed] [Google Scholar]
- Kash T.L., Jenkins A., Kelley J.C., Trudell J.R., Harrison N.L. 2003. Coupling of agonist binding to channel gating in the GABA(A) receptor. Nature. 421:272–275 [DOI] [PubMed] [Google Scholar]
- Kronengold J., Trexler E.B., Bukauskas F.F., Bargiello T.A., Verselis V.K. 2003. Single-channel SCAM identifies pore-lining residues in the first extracellular loop and first transmembrane domains of Cx46 hemichannels. J. Gen. Physiol. 122:389–405 10.1085/jgp.200308861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon T., Harris A.L., Rossi A., Bargiello T.A. 2011. Molecular dynamics simulations of the Cx26 hemichannel: evaluation of structural models with Brownian dynamics. J. Gen. Physiol. 138:475–493 10.1085/jgp.201110679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon T., Roux B., Jo S., Klauda J.B., Harris A.L., Bargiello T.A. 2012. Molecular dynamics simulations of the Cx26 hemichannel: insights into voltage-dependent loop-gating. Biophys. J. 102:1341–1351 10.1016/j.bpj.2012.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon T., Tang Q., Bargiello T.A. 2013. Voltage-dependent gating of the Cx32*43E1 hemichannel: conformational changes at the channel entrances. J. Gen. Physiol. 141:243–259 10.1085/jgp.201210839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laha K.T., Wagner D.A. 2011. A state-dependent salt-bridge interaction exists across the β/α intersubunit interface of the GABAA receptor. Mol. Pharmacol. 79:662–671 10.1124/mol.110.068619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latorre R., Morera F.J., Zaelzer C. 2010. Allosteric interactions and the modular nature of the voltage- and Ca2+-activated (BK) channel. J. Physiol. 588:3141–3148 10.1113/jphysiol.2010.191999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.R., Derosa A.M., White T.W. 2009. Connexin mutations causing skin disease and deafness increase hemichannel activity and cell death when expressed in Xenopus oocytes. J. Invest. Dermatol. 129:870–878 10.1038/jid.2008.335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang G.S., de Miguel M., Gómez-Hernández J.M., Glass J.D., Scherer S.S., Mintz M., Barrio L.C., Fischbeck K.H. 2005. Severe neuropathy with leaky connexin32 hemichannels. Ann. Neurol. 57:749–754 10.1002/ana.20459 [DOI] [PubMed] [Google Scholar]
- Maeda S., Nakagawa S., Suga M., Yamashita E., Oshima A., Fujiyoshi Y., Tsukihara T. 2009. Structure of the connexin 26 gap junction channel at 3.5 A resolution. Nature. 458:597–602 10.1038/nature07869 [DOI] [PubMed] [Google Scholar]
- Magleby K.L. 2003. Gating mechanism of BK (Slo1) channels: so near, yet so far. J. Gen. Physiol. 121:81–96 10.1085/jgp.20028721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez A.D., Acuña R., Figueroa V., Maripillan J., Nicholson B. 2009. Gap-junction channels dysfunction in deafness and hearing loss. Antioxid. Redox Signal. 11:309–322 10.1089/ars.2008.2138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matos T.D., Caria H., Simões-Teixeira H., Aasen T., Dias O., Andrea M., Kelsell D.P., Fialho G. 2008. A novel M163L mutation in connexin 26 causing cell death and associated with autosomal dominant hearing loss. Hear. Res. 240:87–92 10.1016/j.heares.2008.03.004 [DOI] [PubMed] [Google Scholar]
- Minogue P.J., Tong J.J., Arora A., Russell-Eggitt I., Hunt D.M., Moore A.T., Ebihara L., Beyer E.C., Berthoud V.M. 2009. A mutant connexin50 with enhanced hemichannel function leads to cell death. Invest. Ophthalmol. Vis. Sci. 50:5837–5845 10.1167/iovs.09-3759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller D.J., Hand G.M., Engel A., Sosinsky G.E. 2002. Conformational changes in surface structures of isolated connexin 26 gap junctions. EMBO J. 21:3598–3607 10.1093/emboj/cdf365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orellana J.A., Figueroa X.F., Sánchez H.A., Contreras-Duarte S., Velarde V., Sáez J.C. 2011a. Hemichannels in the neurovascular unit and white matter under normal and inflamed conditions. CNS Neurol. Disord. Drug Targets. 10:404–414 10.2174/187152711794653869 [DOI] [PubMed] [Google Scholar]
- Orellana J.A., Shoji K.F., Abudara V., Ezan P., Amigou E., Sáez P.J., Jiang J.X., Naus C.C., Sáez J.C., Giaume C. 2011b. Amyloid β-induced death in neurons involves glial and neuronal hemichannels. J. Neurosci. 31:4962–4977 10.1523/JNEUROSCI.6417-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paldi T., Gurevitz M. 2010. Coupling between residues on S4 and S1 defines the voltage-sensor resting conformation in NaChBac. Biophys. J. 99:456–463 10.1016/j.bpj.2010.04.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul D.L., Ebihara L., Takemoto L.J., Swenson K.I., Goodenough D.A. 1991. Connexin46, a novel lens gap junction protein, induces voltage-gated currents in nonjunctional plasma membrane of Xenopus oocytes. J. Cell Biol. 115:1077–1089 10.1083/jcb.115.4.1077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price K.L., Millen K.S., Lummis S.C. 2007. Transducing agonist binding to channel gating involves different interactions in 5-HT3 and GABAC receptors. J. Biol. Chem. 282:25623–25630 [DOI] [PubMed] [Google Scholar]
- Ripps H., Qian H., Zakevicius J. 2004. Properties of connexin26 hemichannels expressed in Xenopus oocytes. Cell. Mol. Neurobiol. 24:647–665 10.1023/B:CEMN.0000036403.43484.3d [DOI] [PubMed] [Google Scholar]
- Sáez J.C., Berthoud V.M., Branes M.C., Martinez A.D., Beyer E.C. 2003. Plasma membrane channels formed by connexins: their regulation and functions. Physiol. Rev. 83:1359–1400 [DOI] [PubMed] [Google Scholar]
- Sáez J.C., Schalper K.A., Retamal M.A., Orellana J.A., Shoji K.F., Bennett M.V. 2010. Cell membrane permeabilization via connexin hemichannels in living and dying cells. Exp. Cell Res. 316:2377–2389 10.1016/j.yexcr.2010.05.026 [DOI] [PubMed] [Google Scholar]
- Sánchez H.A., Mese G., Srinivas M., White T.W., Verselis V.K. 2010. Differentially altered Ca2+ regulation and Ca2+ permeability in Cx26 hemichannels formed by the A40V and G45E mutations that cause keratitis ichthyosis deafness syndrome. J. Gen. Physiol. 136:47–62 10.1085/jgp.201010433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stong B.C., Chang Q., Ahmad S., Lin X. 2006. A novel mechanism for connexin 26 mutation linked deafness: cell death caused by leaky gap junction hemichannels. Laryngoscope. 116:2205–2210 10.1097/01.mlg.0000241944.77192.d2 [DOI] [PubMed] [Google Scholar]
- Tang Q., Dowd T.L., Verselis V.K., Bargiello T.A. 2009. Conformational changes in a pore-forming region underlie voltage-dependent “loop gating” of an unapposed connexin hemichannel. J. Gen. Physiol. 133:555–570 10.1085/jgp.200910207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thimm J., Mechler A., Lin H., Rhee S., Lal R. 2005. Calcium-dependent open/closed conformations and interfacial energy maps of reconstituted hemichannels. J. Biol. Chem. 280:10646–10654 10.1074/jbc.M412749200 [DOI] [PubMed] [Google Scholar]
- Tong J.J., Ebihara L. 2006. Structural determinants for the differences in voltage gating of chicken Cx56 and Cx45.6 gap-junctional hemichannels. Biophys. J. 91:2142–2154 10.1529/biophysj.106.082859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong J.J., Minogue P.J., Guo W., Chen T.L., Beyer E.C., Berthoud V.M., Ebihara L. 2011. Different consequences of cataract-associated mutations at adjacent positions in the first extracellular boundary of connexin50. Am. J. Physiol. Cell Physiol. 300:C1055–C1064 10.1152/ajpcell.00384.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong X., Lopez W., Liu Y., Harris A.L., Contreras J.E. 2013. Disruption of salt bridge interactions modifies gating kinetics of connexin hemichannels. Biophys. J. 104:631a–632a 10.1016/j.bpj.2012.11.3492 [DOI] [Google Scholar]
- Valiunas V., Weingart R. 2000. Electrical properties of gap junction hemichannels identified in transfected HeLa cells. Pflugers Arch. 440:366–379 10.1007/s004240000294 [DOI] [PubMed] [Google Scholar]
- Verselis V.K., Trelles M.P., Rubinos C., Bargiello T.A., Srinivas M. 2009. Loop gating of connexin hemichannels involves movement of pore-lining residues in the first extracellular loop domain. J. Biol. Chem. 284:4484–4493 10.1074/jbc.M807430200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang N., De Bock M., Decrock E., Bol M., Gadicherla A., Vinken M., Rogiers V., Bukauskas F.F., Bultynck G., Leybaert L. 2013. Paracrine signaling through plasma membrane hemichannels. Biochim. Biophys. Acta. 1828:35–50 10.1016/j.bbamem.2012.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willecke K., Eiberger J., Degen J., Eckardt D., Romualdi A., Güldenagel M., Deutsch U., Söhl G. 2002. Structural and functional diversity of connexin genes in the mouse and human genome. Biol. Chem. 383:725–737 10.1515/BC.2002.076 [DOI] [PubMed] [Google Scholar]
- Xu J., Nicholson B.J. 2013. The role of connexins in ear and skin physiology—functional insights from disease-associated mutations. Biochim. Biophys. Acta. 1828:167–178 10.1016/j.bbamem.2012.06.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zampighi G.A., Loo D.D., Kreman M., Eskandari S., Wright E.M. 1999. Functional and morphological correlates of connexin50 expressed in Xenopus laevis oocytes. J. Gen. Physiol. 113:507–524 10.1085/jgp.113.4.507 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.