

Abstract
Purpose
MicroRNAs (miRs) are post-transcriptional gene regulators that may be useful as diagnostic and/or prognostic biomarkers. We aim to study the expression profiles of a high number of miRs and their relationship with clinicopathological and biological relevant features in leukemic mantle cell lymphomas (MCL).
Experimental design
Expression profiling of 664 miRs was investigated using a high-throughput quantitative real-time PCR platform in 30 leukemic MCL. Statistical and bioinformatic analysis were performed to define miRs associated with different clinicopathological parameters. Gene expression profiling was investigated by microarrays in 16 matching cases to study the potential genes and pathways targeted by selected miRs. The prognostic value of miR-34a was investigated in two independent series of 29 leukemic and 50 nodal MCL.
Results
Robust consensus clustering defined two main MCL subgroups with significant differences in the immunoglobulin (IGHV) mutational status, SOX11 expression, genomic complexity and nodal clinical presentation. Supervised analyses regarding IGHV and SOX11 categories identified 17 and 22 miRs differentially expressed, respectively. Enriched targets of these miRs corresponded to relevant pathways in MCL pathogenesis such as DNA stress response, CD40 signaling and chromatin modification. Additionally, we found seven miRs showing prognostic significance independently of IGHV status and SOX11 expression. Among them, miR-34a was also associated with poor prognosis in two independent series of leukemic and nodal MCL, and in cooperation with high expression of the MYC oncogene.
Conclusion
We have identified miRs and target pathways related to clinical and biological variants of leukemic MCL, and validated miR-34a as a prognostic marker in MCL.
INTRODUCTION
MicroRNAs (miRs) are non-coding small RNAs that bind to specific mRNA target transcripts leading to their degradation and/or translational blocking and, consequently, acting as negative regulators of gene expression (1, 2). Posttranscriptional miR regulation seems to be focused on gene transcripts involved in physiological differentiation, proliferation and apoptosis processes (3, 4). Concordantly, miR-expression deregulation has also been described in many types of neoplasm and has proven to be useful as biomarkers for diagnosis (5, 6). Some cancer-related miRs are also causally involved in oncogenesis due to their impact in the deregulation of oncogenes and tumor suppressor genes, and may have prognostic significance as has been previously shown in certain lymphoid neoplasms (7).
Mantle Cell Lymphoma (MCL) is considered an aggressive entity genetically characterized by the t(11;14) (q13;q32) translocation resulting in the overexpression of the CCND1 gene (8). In addition to this primary genetic alteration, most MCL carry a high number of recurrent secondary chromosomal aberrations that contribute to the additional oncogenic events necessary for the progression of the disease (9). These secondary alterations result in alterations of coding genes involved in cell cycle regulation, DNA damage response, and survival signaling pathways among other oncogenic relevant mechanisms (10). Several genes of these pathways may also be deregulated post-transcriptionally by different miRs in MCL and concordantly their altered expression has been related to their biologic and prognostic features (11, 12). Recent studies have identified a MCL subset that tends to present clinically with a leukemic non-nodal disease and an indolent evolution (13–17). These cases have frequently mutated IGHV, low number of chromosomal alterations in addition to the t(11;14) translocation, and a gene expression signature, including SOX11 downregulation, that also differs from conventional MCL with nodal presentation and an aggressive clinical course. These different clinical and biological features suggest that they may correspond to a different subtype of the disease. Recent studies have investigated the miR profile of MCL and have identified subsets of miRs related to the prognosis of the patients and the potential regulation of pathogenetic pathways (11, 12, 18). However, these studies analyzed mainly conventional MCL with nodal presentation and the number of miRs investigated in some of the works was rather low.
In this study, we have investigated a genome-wide profile of human miRs in a series of leukemic MCL samples including the IGHV mutational status and SOX11 expression and analyzed their relationship with the clinical and biological characteristics of the patients, in order to identify potential miRs and their target pathways that may contribute to the pathogenesis of MCL and its clinical and biological variants.
MATERIAL AND METHODS
Primary samples
Three different series of primary MCL samples were studied. An initial series for miR profiling consisted of peripheral blood samples from 30 patients with leukemic MCL. Two independent series of 29 leukemic and 50 nodal MCL were also used for the validation of the prognostic value of selected miR. The main clinical and biological characteristics of these MCL series are summarized in Supplementary Table 1. All samples were obtained from the Departments of Pathology of the Hospital Clinic (Barcelona), Institute of Pathology (Würzburg and Stuttgart), and Hematology Branch of NHLBI, NIH (Bethesda). The leukemic MCL were selected on the basis of sample availability for tumor cell purification, whereas tissue samples were selected on the basis of high content of tumor cells (> 85%). All samples were obtained prior to any treatment and at diagnosis. All MCL cases of the study were positive for cyclin D1 expression. The IGHV mutational status was studied using a previously described method (19). The study was approved by the Hospital Clínic of Barcelona Institutional Review Board.
RNA isolation and miR RT- qPCRs
Total RNA was isolated from all samples using TrizolTM Reagent following the manufacturer’s instructions (Invitrogen, Paisley, UK). A total of 664 human miRs were investigated using a RT-looped qPCR performed with the TaqMan Human A + B microRNA fluidic card system (Applied Biosystems, Darmstadt, Germany) as detailed in Supplementary Material and Methods.
Gene expression and genomic alterations
The gene expression profiles of 16 cases with additional RNA available were investigated for further correlation analysis with the miR expression data. The gene expression was studied using hybridization to Affymetrix HG133Plus 2.0 (Affymetrix, Santa Clara, CA) microarrays as previously described (13), and normalized and processed as detailed in Supplementary Material and Methods section. The raw data has been deposited in the Gene Expression Omnibus (GEO) database (GSE36000). In addition, SOX11 mRNA expression measured by qRT-PCR and the genomic profile studied by SNP-arrays were available in all and 23 cases, respectively, from a previous study (16). Expression quantification of additional genes is detailed in Supplementary Material and Methods.
Statistical and bioinformatics analysis
The miR expression data were analyzed with the Consensus Clustering (CC) method as implemented in the Consensus Cluster Plus package from Bioconductor (20). CC is a previously validated approach to improve the classical hierarchical clustering methods in order to obtain reliability in the number of subgroups present in the dataset and the group memberships of cases (21) (see Supplementary Material and Methods for details). To define the miRs differentially expressed in relation to IGHV status and SOX11 expression in the whole series and in relation to the clusters identified in the previous analysis, we used a supervised method based on the empirical Bayes moderated t-statistic from the Bioconductor package limma (http://cran.r-project.org). The potential genes and pathways regulated by the miRs found differentially expressed as well as the associations between miR expression and clinical features were analyzed using different bioinformatic and statistical methods detailed in Supplementary Material and Methods.
RESULTS
miR expression profiling and relationship to IGHV mutational status and SOX11 expression
A total of 583 miRs of the 664 examined showed detectable expression by qRT-PCR in at least one sample of the initial leukemic MCL series. We selected a subset of 286 miRs showing an expression variation > 50th percentile of the variation degree among samples and with detectable expression in more than 25% of cases (Supplementary Table 2 and Figure 1a). An unsupervised consensus clustering analysis was performed trying different partitions (from k = 2 to k = 6). The empirical cumulative distribution function demonstrated that approximate maximum stability of partitioning was reached at k = 3, indicating that the optimal number of robust clusters in this data set was three (Supplementary Figure 1a), as can also be observed in the consensus matrix plots (Supplementary Figure 1b). Only two samples (MCL#29 and MCL#30; Supplementary Table 3a) showed less robust cluster membership even at k = 3 (Supplementary Figure 1c), and therefore were excluded to define the representative samples for each of the core clusters (named as A, B and C) used in subsequent analyses (Figure 1b).
Figure 1.
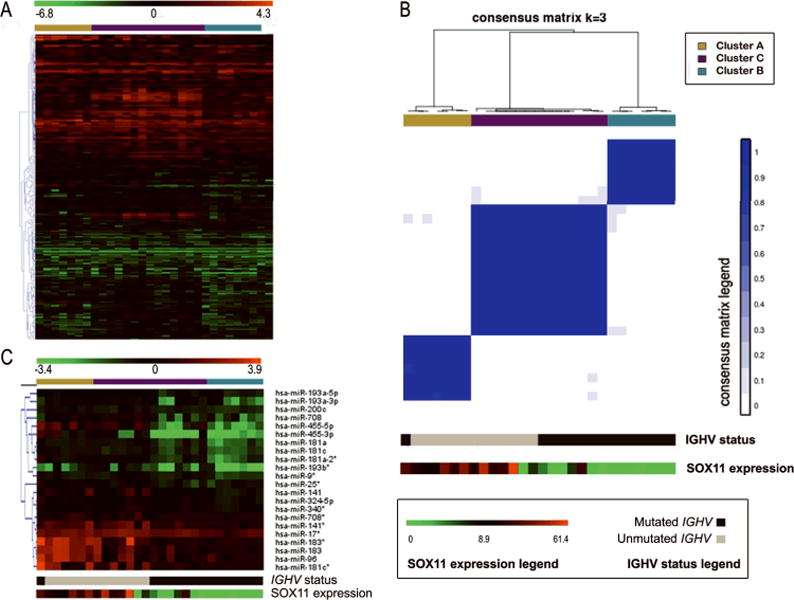
(A) Heat map of the normalized expression values of the 286 miRs showing gene expression variation > 50th percentile of the variation degree among the leukemic MCL samples. Sample order is the same in all panels, as obtained from the consensus clustering analysis, and here also including at the extreme right side the expression values of the two samples excluded from the core clusters. miRs are clustered using a Pearson algorithm. (B) Consensus matrix plot showing the three robust core clusters generated at k = 3 from the miR expression profiling data obtained by qRT-PCR, together with IGHV mutational status and SOX11 expression data. (C) Heat map of the normalized miR expression values of the significant miRs found in the supervised analyses considering either IGHV status as SOX11 expression categories along the MCL series defining the core clusters.
The clinical and biological characteristics of the patients in the three core clusters are summarized in Table 1. Patients in clusters A and B had significant differences in several parameters whereas patients in cluster C had intermediate features between them. Cluster A included patients with nodal presentation (100%) and tumors with unmutated IGHV (86%), high number of chromosomal alterations (mean 8.3; range 2–19) and high levels of SOX11 expression (mean 25.65; range 11.3–42.1 Relative Units (RU)) whereas patients in cluster B had more frequently non-nodal disease (83%) (P = 0.006) and tumors with mutated IGHV (100%) (P = 0.006), low number of chromosomal abnormalities (mean 0.8 range 0–2) (P = 0.041) and low or negative expression of SOX11 (mean 1.55; range 0–8.8 RU) (P = 0.011).
Table 1.
Main features of MCL patients according to robust clusters obtained from miR expression profiling.
| Total MCL (N = 28) |
Cluster A (N = 7) |
Cluster B (N = 7) |
Cluster C (N = 14) |
P-value | |
|---|---|---|---|---|---|
| Median age (range) | 67 (45–88) | 67 (52–78) | 70 (61–88) | 66 (45–85) | 0.397 |
| Ratio Male/Female | 21/7 | 7/0 | 4/3 | 10/4 | 0.198 |
| Clinical and pathological data | |||||
| Nodal presentation (lymph nodes > 1cm)(%) | 15/26 (58) | 7/7 (100) | 1/6 (17) | 7/13 (54) | 0.006 |
| Splenomegaly (%) | 13/23 (56) | 4/5 (80) | 3/5 (60) | 6/13 (46) | 0.625 |
| WBC count > 10 × 109/L (%) | 10/17 (59) | 3/6 (50) | 3/4 (75) | 4/7 (57) | 1.000 |
| High serum LDH (%) | 2/17 (12) | 2/6 (33) | 0/4 (0) | 0/7 (0) | 0.154 |
| Evolutive data | |||||
| Median follow-up, months (range) | 48.4 (0.3–129) | 39 (0.3–107.6) | 59 (8–121) | 39.4 (4–129) | 0.755 |
| Chemotherapy at any time (%) | 22/27 (81) | 6/7 (85) | 6/7 (85) | 10/14 (71) | 0.716 |
| Median time to treatment, months | 2.4 | 1.46 | 1.9 | 3.13 | 0.454 |
| 3-year overall survival, % | 68 | 67 | 83 | 60 | 0.636 |
| Molecular data | |||||
| IGHV gene homology ≤ 98 (%) | 15/28 (54) | 1/7 (14) | 7/7 (100) | 7/14 (50) | 0.006 |
| SOX11 high expression ≥ 6.4 (RU) | 15/28 (54) | 7/7 (100) | 1/7 (14) | 7/14 (50) | 0.011 |
| Copy number alterations | |||||
| Median number | 2 | 7 | 1 | 5 | 0.041 |
| 0–1 imbalance | 10/23 (43) | 0/6 (0) | 5/6 (83) | 5/11 (45) | 0.016 |
| 2–4 imbalances | 4/23 (17) | 3/6 (50) | 1/6 (17) | 0/11 (0) | 0.030 |
| ≥ 5 imbalances | 9/23 (39) | 3/6 (50) | 0/6 (0) | 6/11 (54) | 0.094 |
Abbreviations: LDH, lactate dehydrogenase; N, number; RU, relative units of expression; WBC, white blood cell.
Next, we performed a supervised analysis to find differentially expressed miRs between the two IGHV mutational categories. This analysis showed 17 miRs overexpressed in unmutated (U) compared to mutated (M) MCL cases (Supplementary Table 4a and Figure 1c). Interestingly, the top three significant miRs (miR-455-5p/3p and miR-708) were also the only significant miRs found when a supervised analysis was performed between the two different IGHV mutational subgroups in the cluster C (Supplementary Figure 2 and Supplementary Table 5a).
Finally, a total of 22 miRs were differentially expressed in MCL according to SOX11 expression (Supplementary Table 4b and Figure 1c), but 16 of these miRs overlapped with those found differentially expressed in relation to the IGHV status, and the remaining were also found related to IGHV status but only in some comparisons between subgroups with different IGHV status among the core clusters (Supplementary Table 5b). Given these similarities we concentrated in the miRs differentially expressed between the IGHV subsets of MCL in the subsequent analyses.
Pathway enrichment analysis of predicted miR targets differentially expressed among subgroups of leukemic MCL
To determine the potential genes and pathways modulated by the relevant miRs differentially expressed in the MCL with mutated and unmutated IGHV, we compared the expression of these miRs and their putative target coding genes included in high throughput gene expression profiling arrays performed in a common series of 16 MCL, and using the tools described in the material and methods section. The top miRs differentially expressed in this set of 16 cases were representative of the whole series (Supplementary Table 6a). The obtained ranked target genes according to their inverse expression correlation with these miRs are shown in the Supplementary Table 7a. We performed a GSEA analysis on these pre-ranked genes using several gene sets and functional pathways involved in B-cell lymphomagenesis (See Supplementary Material and Methods). This analysis showed that the target genes of the differentially expressed miRs were enriched in several pathways including “Response to DNA stress” (GO:0006950; involving 19 genes) as the most significant (Supplementary Table 7b, Supplementary Figure 3). Noticeably, among the genes in this pathway was ATM, the inactivation of which is associated with increased chromosomal instability in MCL tumors (22). ATM is a validated target of miR-181a/c (23, 24) and in our series was downregulated in U-MCL compared to M-MCL (−1.24 log fold change) (Supplementary Table 6b) and showed a significantly high inverse correlation with miR-181a/c (Supplementary Table 7a), noticeably even in the absence of ATM locus deletions at 11q22.3 (see case MCL#3, Supplementary Table 3b). These results suggest that miR-181a/c upregulation may be an alternative mechanism to 11q deletions to downregulate ATM (Table 1).
The differentially expressed miRs between U- and M-MCL also targeted genes of the CD40 ligand pathway, which are usually expressed at higher levels in memory cells than in germinal center centroblasts or naïve subpopulations of cells in the mantle zone area (25) (Supplementary Table 7b and Supplementary Figure 3). Thirteen of the 27 genes included in this signature were highly expressed in M-MCL compared to U-MCL, and were inversely correlated with 8 out of the 17 miRs differentially expressed between U and M-MCL (Supplementary Table 7c).
The previous analysis did not reveal predicted targets for miR-455-5p/3p and miR-708, the three most significant miRs differentially expressed between U- and M-MCL. Therefore, we explored further the potential genes and pathways targeted by these miRs using a Gene Group Functional Profiling tool (g:GOSt-g-Profiler), an improved method for multiple testing correction on complex functional term analysis (26). Using genes with the higher 75% inverse expression mRNA levels with miR-455-5p/3p/miR-708 this method revealed several pathways with significant terms including protein modification by methylation transferase activity, containing 9 genes in common involved in epigenetic modifications of histones (Supplementary Table 8a/8b). Using a more stringent gene set only comprising genes with the higher 30% from the ranked inverse correlation list, four of the previous genes with methyl transferase activity (EZH1, MLL2, SETDB1 and HEMK1) were also present (Supplementary Table 8a/b). All these genes were expressed at lower levels in U-MCL than in M-MCL, and with log fold changes ranging from −0.25 to −0.37 (Supplementary Table 6b). A further validation by qRT-PCR demonstrated a significant inverse expression correlation of miR-455-5p and MLL2 (P = 0.045) (Supplementary Figure 4) suggesting that miR-455-5p expression could contribute to the biological and clinical differences between U- and M-MCL subgroups by modulating this epigenetic regulator of gene expression.
miR expression and clinical outcome
In this series, MCL with unmutated IGHV or high SOX11 expression levels had a worse prognosis than tumors with mutated IGHV or with low or negative SOX11 expression (P = 0.008 and P = 0.039, respectively) (Supplementary Figure 5). To identify miRs that could add prognostic value to these parameters we used Cox regression with these two variables and each individual miR. A total of 7 miRs showed a significant impact on overall survival (OS) with shorter survival associated with high expression in 2, and with low expression in 5 of them (positive and negative Cox coefficients, respectively; Table 2).
Table 2.
Significant results from the survival model including miR expression, IGHV mutational status and SOX11 expression.
| miR |
P-values
|
Cox coefficients
|
||||
|---|---|---|---|---|---|---|
| miR | IGHV | SOX11 | miR | IGHV | SOX11 | |
|
|
|
|||||
| hsa-miR-190 | 0.002 | – | – | 2.892 | – | – |
| hsa-miR-565 | 0.004 | – | – | −1.422 | – | – |
| hsa-miR-34a | 0.005 | 0.007 | – | −2.713 | −3.798 | – |
| hsa-miR-34a* | 0.006 | 0.008 | – | −2.062 | −3.641 | – |
| hsa-miR-149 | 0.007 | – | – | 0.687 | – | – |
| hsa-miR-649 | 0.008 | 0.003 | – | −1.994 | −3.955 | – |
| hsa-miR-483-5p | 0.009 | 0.006 | – | −3.703 | −3.503 | – |
|
| ||||||
As proliferation is one of the most important prognostic factors in MCL we analyzed the possible relationship between miR expression and gene expression proliferation signature (27), in the 16 cases with GEP analysis (Supplementary Table 3b). We found 36 miRs significantly correlated with this signature, some of them with known targets of cell cycle regulation (Supplementary Tables 9/10). None of the 7 miRs that added prognostic value to SOX11 expression or IGHV mutational status were included among them.
Among the miR with most prognostic significant impact (P < 0.01), miR-34a raised our interest because of its association with the control of several relevant genes of MCL pathogenesis, such as CDK4, CDK6, MYC, and CCND1, among others (28). The survival impact of this miR in MCL has not been previously investigated. Interestingly, we categorized miR-34a expression into high and low groups among the 30 leukemic MCL series using the best cutoff identified with Maxstat software (Figure 2), and combined these groups with those previously defined categories of IGHV status and SOX11 expression. Low expression levels of miR-34a were associated with a significant worse outcome in U-MCL (P = 0.037) and SOX11-positive MCL (P = 0.008) subgroups compared to cases with high miR-34a expression included in the same subgroups (Supplementary Figure 6). A similar tendency was observed inside the M-MCL (P = 0.067) and SOX11-negative MCL subgroups (P = 0.085) (Supplementary Figure 6). Finally, the association of miR-34a expression and survival was validated in an independent series of 29 leukemic MCL. In this series, the miR-34a low levels were also associated with worse prognosis (median OS: 25 months) compared to patients with higher expression (median OS: 67 months) (P = 0.041) (Supplementary Figure 7).
Figure 2.
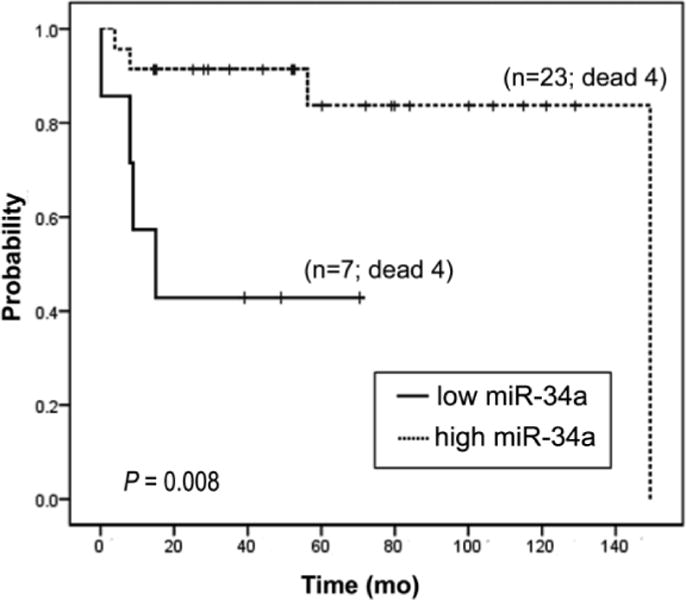
Overall survival of the initial series of 30 leukemic MCL patients according to miR-34a expression level categorized with Maxstat software. Patients with low levels of miR-34a showed worse prognosis (median OS: 15 months) compared to patients with higher miR-34a levels (median OS: 149.3 months) (P = 0.008).
One of the demonstrated miR-34a targets is MYC, the overexpression of which has been associated with poor outcome in MCL (28–30). Moreover, miR-34a has been previously described to affect the MYC transcriptional activity in solid tumors (31–33). To determine whether the poor prognosis of miR-34a downregulation and MYC overexpression are mutually related in MCL we investigated these transcripts in 38 leukemic MCL with additional RNA available and in an independent series of 50 nodal MCL. In both groups of cases, the results showed that low expression of miR-34a was associated with a significantly shorter overall survival (OS) (P = 0.001) in patients who also had high MYC expression (median OS for leukemic and nodal cases: 9 and 21 months, respectively) in comparison with those patients with only one of these factors (median OS for leukemic and nodal: 58 and 49 months, respectively) or none of them (median OS for leukemic and nodal: not reached and 64 months, respectively) (Figure 3). These results support that downregulation of miR-34a and MYC overexpression cooperates in the aggressiveness of these tumors.
Figure 3.
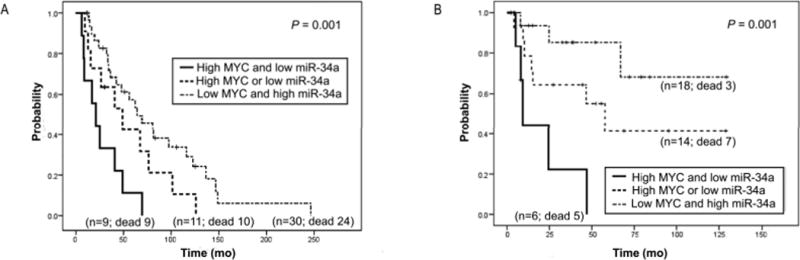
Overall survival of 50 nodal (A) and 38 leukemic (B) MCL patients according to the combined expression of MYC and miR-34a. MCL patients with concomitant high expression of MYC and low expression of these miRs (median OS: 21 months (A); 9 months (B)) have a significant shorter overall survival than patients showing only high expression of MYC or low expression of miR-34a (median OS: 49 months (A); 58 months (B)) or even low MYC expression and high expression of this miR (median OS: 64 months (A); not reached (B)) (P = 0.001).
DISCUSSION
In the present study we have investigated the expression of a high number of miRs (664) in a series of leukemic MCL and have identified three robust subgroups of tumors with a different miR expression profile. Two of these clusters (A and B) included tumors with significant differences in their clinical and biological characteristics whereas the third cluster (cluster C) was composed of tumors with intermediate features. The clinical and biological characteristics of the A and B subgroups are concordant with the two molecular subtypes of MCL that we have recently identified based on the IGHV mutational status and SOX11 expression (14). Tumors in miR cluster A were characterized by unmutated IGHV, high expression of SOX11, complex karyotypes and a nodal clinical presentation whereas tumors in cluster B had mutated IGHV, negative or very low SOX11 expression, low genomic complexity and non-nodal presentation. The differences in the miR expression profile between these two groups would support the hypothesis that they may correspond to different biological subtypes of MCL.
Recently, Iqbal et al have described three miR clusters in lymph node MCL based on the expression profiling of 377 miRs, all included in our study (18). These three clusters are different from the ones identify in our work. One of these clusters (A) was defined by miRs involved in control of cell proliferation and apoptosis, whereas the other two (B and C) included miRs related to growth inhibitory functions depending on the accompanying stroma compartment. Concordant with the different biological significance of the clusters in both studies, we only observed four overlapping miRs between the clusters in the two studies, namely miR-363 that is upregulated in both clusters A, miR-1 that is upregulated in both clusters C, and two miRs associated with stroma inhibitory functions in Iqbal’s cluster B that were upregulated in our cluster C (miR-539 and miR-485-3p). Moreover, miRs found significantly related to the gene expression proliferation signature (27) are poorly overlapped between our leukemic samples and those described in Iqbal et al. (only miR-199a-5p and miR-485-3p). These differences may be due in part to the different subtypes of MCL and type of tumor samples (leukemic vs nodal) investigated in the two works that reflect the biological heterogeneity of MCL and the importance of the microenvironment in modulating the miR expression. In this sense, the differences in miR expression between leukemic and nodal samples are concordant with our previous observation that the expression of some miRs is modulated between peripheral blood and lymph node microenvironment (34).
A supervised analysis of the miRs differentially expressed between MCL with mutated and unmutated IGHV in our study showed 17 miRs overexpressed in U- compared to M-MCL with the top three being miR-708 and miR-455-5p/3p. A similar supervised analysis in the tumors according to SOX11 expression showed that the miRs differentially expressed in these two subgroups virtually overlapped with those found related to the IGHV mutational status, reflecting the close relationship previously described between these two parameters (14). The comparison of the miR expression levels and their paired gene expression profiles followed by a pathway analysis identified that the putative miR targets of the differentially expressed miRs between U- and M-MCL mainly regulated the “response to stress”, CD40 signaling pathway and genes related to epigenetic modifications.
ATM was one of the genes included in the “response to stress” pathway. This gene is a key regulator of the DNA damage response pathway (35), whose inactivation by deletion and/or mutation has been described in MCL in relation to increased chromosomal instability (22). In our leukemic MCL, ATM mRNA showed a highly inverse correlation with their predicted regulators miR-181a/c and a pronounced downregulation in U-MCL compared to M-MCL. As ATM has been validated experimentally as a target of miR-181 family with functional impact (23), and our results show that ATM downregulation was associated to high levels of miR-181c even in absence of ATM locus deletions. These findings suggest that miR-181 upregulation may be an alternative mechanism to 11q deletions to downregulate ATM.
CD40 signaling pathway (25) was also identified as a significant target of the miRs differentially expressed between U- and M-MCL. Interestingly, this pathway was previously described to be targeted by miR-377, miR-378 and miR-204 in nodal MCL samples (11). In our analysis, some genes of this pathway were identified as targets inversely correlated with miRs highly expressed in U-MCL compared to M-MCL. This finding would seem consistent with the activation of the CD40 signaling pathway in memory cells compared to cells in the mantle zone area (25).
miR-708 and miR-455-5p/3p were among the highest miRs differentially expressed between IGHV U- and M-MCL and SOX11-positive and negative MCL. The target genes of these miRs are not well known but recent studies have highlighted their potential role in hematological malignances (36, 37). In our work, the pathway analysis of the inversely correlated predicted targets of these miRs identified genes coding for histone-modifying proteins. Among them MLL2 was validated by qRT-PCR as inversely expressed to miR-455-5p. Interestingly, this gene was found mutated and inactivated in a high proportion of follicular and diffuse large B-cell lymphomas (38). Further studies are required to confirm the relevance of these mechanisms in MCL.
The understanding of the heterogeneity in the clinical presentation and evolution of MCL requires the identification of biological parameters related to the different evolution of the patients. Previous studies have demonstrated the value of IGHV mutational status and SOX11 expression in defining subgroups of MCL patients with different clinical evolution (13–17, 39). We have now found 7 miRs the expression levels of which had a significant survival impact independent of the IGHV and SOX11 alterations, and were not correlated with the gene expression proliferation signature previously described as prognostic factor in MCL (27). None of these miRs had been previously found related with prognosis in MCL (11, 18), but these studies had evaluated only nodal MCL emphasizing the possible biological and clinical differences of our cohort of patients. Among these miRs, miR-34a targets several relevant genes in MCL pathogenesis such as CDK4, CDK6, MYC, and CCND1 among others (28). We observed in two independent series of leukemic MCL that its decreased expression was associated with a poor survival, concordantly with its described role as tumor suppressor (28). Although the number of patients is limited, this prognostic value was also observed in the subgroups of U-MCL and MCL with high SOX11 expression. miR-34a had been previously described as a negative regulator of MYC–dependent transcription (31–33) and MYC overexpression has been associated with poor prognosis in MCL (29, 30). Therefore, we studied the expression of miR-34a and MYC for a possible cooperation between them in independent series of nodal and leukemic MCL. Our results showed that tumors with the concomitant highest levels of MYC and simultaneously the lowest of miR-34a had significant shorter overall survival than cases with high expression of only one or none of these factors. These results would support the cooperation of this miR and one of its targets in tumor progression and suggest that the prognostic value of some miRs could be enhanced by the simultaneous study of their targets.
In summary, we have shown that miR expression profiles identify two main subgroups of leukemic MCL with clear differences in IGHV mutational status, SOX11 expression, nodal presentation, and number of chromosomal alterations. The differentially expressed miRs seem to regulate different pathways relevant for MCL pathogenesis including DNA damage response in U-MCL. On the other hand, a subset of miRs had prognostic implications independently of IGHV mutational status and SOX11 expression. Among them, miR-34a downregulation was validated in two independent series of leukemic and nodal tumors and associated with poor prognosis in cooperation with high MYC expression. All these results highlight the biological and clinical significance of miRs expression in MCL, supporting their involvement in the pathogenesis and prognosis of these tumors.
Supplementary Material
Acknowledgments
L.H. is a researcher from Institut d’Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS) and is supported by FIS and “programa d’estabilització d’investigadors” of Direcció d’Estrategia i Coordinació del Departament de Salut (Generalitat de Catalunya). This work was supported by the Instituto de Salud Carlos III (ISCIII), Fondo Investigaciones Sanitarias” & European Regional Development Fund (ERDF) (Unión Europea. “Una manera de hacer Europa”) (PI12/01302; L.H.), (PI08/0077-PI11/01177;S.B.) and (RD06/0020/0039 and RD12/0036/0036) from Red Temática de Investigación Cooperativa en Cáncer (RTICC); Plan Nacional del MINECO SAF08/03630, and Generalitat de Catalunya AGAUR 2009-SGR-992. A.W. and W.H.W. are supported by the intramural research program of NHLBI and NCI, respectively. The work was carried out at the Centro Esther Koplowitz, Barcelona, Spain.
Footnotes
Author’s disclosure statement: The authors disclose no potential conflicts of interest.
References
- 1.Engels BM, Hutvagner G. Principles and effects of microRNA-mediated post-transcriptional gene regulation. Oncogene. 2006;25:6163–9. doi: 10.1038/sj.onc.1209909. [DOI] [PubMed] [Google Scholar]
- 2.Lim LP, Lau NC, Garrett-Engele P, Grimson A, Schelter JM, Castle J, et al. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature. 2005;433:769–73. doi: 10.1038/nature03315. [DOI] [PubMed] [Google Scholar]
- 3.Miska EA. How microRNAs control cell division, differentiation and death. Curr Opin Genet Dev. 2005;15:563–8. doi: 10.1016/j.gde.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 4.Esquela-Kerscher A, Slack FJ. Oncomirs – microRNAs with a role in cancer. Nat Rev Cancer. 2006;6:259–69. doi: 10.1038/nrc1840. [DOI] [PubMed] [Google Scholar]
- 5.Huppi K, Volfovsky N, Mackiewicz M, Runfola T, Jones TL, Martin SE, et al. MicroRNAs and genomic instability. Semin Cancer Biol. 2007;17:65–73. doi: 10.1016/j.semcancer.2006.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Calin GA, Croce CM. MicroRNAs and chromosomal abnormalities in cancer cells. Oncogene. 2006;25:6202–10. doi: 10.1038/sj.onc.1209910. [DOI] [PubMed] [Google Scholar]
- 7.Fabbri M, Croce CM. Role of microRNAs in lymphoid biology and disease. Curr Opin Hematol. 2011;18:266–72. doi: 10.1097/MOH.0b013e3283476012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bosch F, Jares P, Campo E, Lopez-Guillermo A, Piris MA, Villamor N, et al. PRAD-1/cyclin D1 gene overexpression in chronic lymphoproliferative disorders: a highly specific marker of mantle cell lymphoma. Blood. 1994;84:2726–32. [PubMed] [Google Scholar]
- 9.Royo C, Salaverria I, Hartmann EM, Rosenwald A, Campo E, Bea S. The complex landscape of genetic alterations in mantle cell lymphoma. Semin Cancer Biol. 2011;21:322–34. doi: 10.1016/j.semcancer.2011.09.007. [DOI] [PubMed] [Google Scholar]
- 10.Jares P, Colomer D, Campo E. Genetic and molecular pathogenesis of mantle cell lymphoma: perspectives for new targeted therapeutics. Nat Rev Cancer. 2007;7:750–62. doi: 10.1038/nrc2230. [DOI] [PubMed] [Google Scholar]
- 11.Di Lisio L, Gomez-Lopez G, Sanchez-Beato M, Gomez-Abad C, Rodriguez ME, Villuendas R, et al. Mantle cell lymphoma: transcriptional regulation by microRNAs. Leukemia. 2010;24:1335–42. doi: 10.1038/leu.2010.91. [DOI] [PubMed] [Google Scholar]
- 12.Zhao JJ, Lin J, Lwin T, Yang H, Guo J, Kong W, et al. microRNA expression profile and identification of miR-29 as a prognostic marker and pathogenetic factor by targeting CDK6 in mantle cell lymphoma. Blood. 2010;115:2630–9. doi: 10.1182/blood-2009-09-243147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fernandez V, Salamero O, Espinet B, Sole F, Royo C, Navarro A, et al. Genomic and gene expression profiling defines indolent forms of mantle cell lymphoma. Cancer Res. 2010;70:1408–18. doi: 10.1158/0008-5472.CAN-09-3419. [DOI] [PubMed] [Google Scholar]
- 14.Navarro A, Clot G, Royo C, Jares P, Hadzidimitriou A, Agathangelidis A, et al. Molecular subsets of mantle cell lymphoma defined by the IGHV mutational status and SOX11 expression have distinct biological and clinical features. Cancer Res. 2012 doi: 10.1158/0008-5472.CAN-12-1615. (In Press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ondrejka SL, Lai R, Smith SD, Hsi ED. Indolent mantle cell leukemia: a clinicopathological variant characterized by isolated lymphocytosis, interstitial bone marrow involvement, kappa light chain restriction, and good prognosis. Haematologica. 2011;96:1121–7. doi: 10.3324/haematol.2010.036277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Royo C, Navarro A, Clot G, Salaverria I, Gine E, Jares P, et al. Non-nodal type of mantle cell lymphoma is a specific biological and clinical subgroup of the disease. Leukemia. 2012;26:1895–8. doi: 10.1038/leu.2012.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rule SA, Poplar S, Evans PA, O’Connor SJ, Owen RG. Indolent mantle-cell lymphoma: immunoglobulin variable region heavy chain sequence analysis reveals evidence of disease 10 years prior to symptomatic clinical presentation. J Clin Oncol. 2011;29:e437–e439. doi: 10.1200/JCO.2010.31.8006. [DOI] [PubMed] [Google Scholar]
- 18.Iqbal J, Shen Y, Liu Y, Fu K, Jaffe ES, Liu C, et al. Genome-wide MicroRNA profiling of mantle cell lymphoma reveal a distinct subgroup with poor prognosis. Blood. 2012;119:4939–48. doi: 10.1182/blood-2011-07-370122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van Dongen JJ, Langerak AW, Bruggemann M, Evans PA, Hummel M, Lavender FL, et al. Design and standardization of PCR primers and protocols for detection of clonal immunoglobulin and T-cell receptor gene recombinations in suspect lymphoproliferations: report of the BIOMED-2 Concerted Action BMH4-CT98-3936. Leukemia. 2003;17:2257–317. doi: 10.1038/sj.leu.2403202. [DOI] [PubMed] [Google Scholar]
- 20.Wilkerson MD, Hayes DN. ConsensusClusterPlus: a class discovery tool with confidence assessments and item tracking. Bioinformatics. 2010;26:1572–3. doi: 10.1093/bioinformatics/btq170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Monti S, Tamayo P, Mesirov J, Golub T. Consensus Clustering: A Resampling-Based Method for Class Discovery and Visualization of Gene Expression Microarray Data. Machine learning. 2003;52:91–118. [Google Scholar]
- 22.Camacho E, Hernandez L, Hernandez S, Tort F, Bellosillo B, Bea S, et al. ATM gene inactivation in mantle cell lymphoma mainly occurs by truncating mutations and missense mutations involving the phosphatidylinositol-3 kinase domain and is associated with increasing numbers of chromosomal imbalances. Blood. 2002;99:238–44. doi: 10.1182/blood.v99.1.238. [DOI] [PubMed] [Google Scholar]
- 23.Wang Y, Yu Y, Tsuyada A, Ren X, Wu X, Stubblefield K, et al. Transforming growth factor-beta regulates the sphere-initiating stem cell-like feature in breast cancer through miRNA-181 and ATM. Oncogene. 2011;30:1470–80. doi: 10.1038/onc.2010.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang Y, Chuang A, Hao H, Talbot C, Sen T, Trink B, et al. Phospho-DeltaNp63alpha is a key regulator of the cisplatin-induced microRNAome in cancer cells. Cell Death Differ. 2011;18:1220–30. doi: 10.1038/cdd.2010.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Basso K, Klein U, Niu H, Stolovitzky GA, Tu Y, Califano A, et al. Tracking CD40 signaling during germinal center development. Blood. 2004;104:4088–96. doi: 10.1182/blood-2003-12-4291. [DOI] [PubMed] [Google Scholar]
- 26.Reimand J, Arak T, Vilo J. g:Profiler–a web server for functional interpretation of gene lists (2011 update) Nucleic Acids Res. 2011;39:W307–W315. doi: 10.1093/nar/gkr378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rosenwald A, Wright G, Wiestner A, Chan WC, Connors JM, Campo E, et al. The proliferation gene expression signature is a quantitative integrator of oncogenic events that predicts survival in mantle cell lymphoma. Cancer Cell. 2003;3:185–97. doi: 10.1016/s1535-6108(03)00028-x. [DOI] [PubMed] [Google Scholar]
- 28.Chen F, Hu SJ. Effect of microRNA-34a in cell cycle, differentiation, and apoptosis: a review. J Biochem Mol Toxicol. 2012;26:79–86. doi: 10.1002/jbt.20412. [DOI] [PubMed] [Google Scholar]
- 29.Hartmann E, Fernandez V, Moreno V, Valls J, Hernandez L, Bosch F, et al. Five-gene model to predict survival in mantle-cell lymphoma using frozen or formalin-fixed, paraffin-embedded tissue. J Clin Oncol. 2008;26:4966–72. doi: 10.1200/JCO.2007.12.0410. [DOI] [PubMed] [Google Scholar]
- 30.Kienle D, Katzenberger T, Ott G, Saupe D, Benner A, Kohlhammer H, et al. Quantitative gene expression deregulation in mantle-cell lymphoma: correlation with clinical and biologic factors. J Clin Oncol. 2007;25:2770–7. doi: 10.1200/JCO.2006.08.7999. [DOI] [PubMed] [Google Scholar]
- 31.Yamamura S, Saini S, Majid S, Hirata H, Ueno K, Chang I, et al. MicroRNA-34a suppresses malignant transformation by targeting c-Myc transcriptional complexes in human renal cell carcinoma. Carcinogenesis. 2012;33:294–300. doi: 10.1093/carcin/bgr286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang X, Liu P, Zhu H, Xu Y, Ma C, Dai X, et al. miR-34a, a microRNA up-regulated in a double transgenic mouse model of Alzheimer’s disease, inhibits bcl2 translation. Brain Res Bull. 2009;80:268–73. doi: 10.1016/j.brainresbull.2009.08.006. [DOI] [PubMed] [Google Scholar]
- 33.Christoffersen NR, Shalgi R, Frankel LB, Leucci E, Lees M, Klausen M, et al. p53-independent upregulation of miR-34a during oncogene-induced senescence represses MYC. Cell Death Differ. 2010;17:236–45. doi: 10.1038/cdd.2009.109. [DOI] [PubMed] [Google Scholar]
- 34.Navarro A, Bea S, Fernandez V, Prieto M, Salaverria I, Jares P, et al. MicroRNA expression, chromosomal alterations, and immunoglobulin variable heavy chain hypermutations in Mantle cell lymphomas. Cancer Res. 2009;69:7071–8. doi: 10.1158/0008-5472.CAN-09-1095. [DOI] [PubMed] [Google Scholar]
- 35.Kastan MB, Lim DS. The many substrates and functions of ATM. Nat Rev Mol Cell Biol. 2000;1:179–86. doi: 10.1038/35043058. [DOI] [PubMed] [Google Scholar]
- 36.Han BW, Feng DD, Li ZG, Luo XQ, Zhang H, Li XJ, et al. A set of miRNAs that involve in the pathways of drug resistance and leukemic stem-cell differentiation is associated with the risk of relapse and glucocorticoid response in childhood ALL. Hum Mol Genet. 2011;20:4903–15. doi: 10.1093/hmg/ddr428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Monk CE, Hutvagner G, Arthur JS. Regulation of miRNA transcription in macrophages in response to Candida albicans. PLoS ONE. 2010;5:e13669. doi: 10.1371/journal.pone.0013669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Morin RD, Mendez-Lago M, Mungall AJ, Goya R, Mungall KL, Corbett RD, et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature. 2011;476:298–303. doi: 10.1038/nature10351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Orchard J, Garand R, Davis Z, Babbage G, Sahota S, Matutes E, et al. A subset of t(11;14) lymphoma with mantle cell features displays mutated IgVH genes and includes patients with good prognosis, nonnodal disease. Blood. 2003;101:4975–81. doi: 10.1182/blood-2002-06-1864. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.