

Abstract
Although functional coupling between protein kinase Cε (PKCε) and mitochondria has been implicated in the genesis of cardioprotection, the signal transduction mechanisms that enable this link and the identities of the mitochondrial proteins modulated by PKCε remain unknown. Based on recent evidence that the mitochondrial permeability transition pore may be involved in ischemia/reperfusion injury, we hypothesized that protein-protein interactions between PKCε and mitochondrial pore components may serve as a signaling mechanism to modulate pore function and thus engender cardioprotection. Coimmunoprecipitation and GST-based affinity pull-down from mouse cardiac mitochondria revealed interaction of PKCε with components of the pore, namely voltage-dependent anion channel (VDAC), adenine nucleotide translocase (ANT), and hexokinase II (HKII). VDAC1, ANT1, and HKII were present in the PKCε complex at ≈2%, ≈0.2%, and ≈1% of their total expression, respectively. Moreover, in vitro studies demonstrated that PKCε can directly bind and phosphorylate VDAC1. Incubation of isolated cardiac mitochondria with recombinant PKCε resulted in a significant inhibition of Ca2+-induced mitochondrial swelling, an index of pore opening. Furthermore, cardiac-specific expression of active PKCε in mice, which is cardioprotective, greatly increased interaction of PKCε with the pore components and inhibited Ca2+-induced pore opening. In contrast, cardiac expression of kinase-inactive PKCε did not affect pore opening. Finally, administration of the pore opener atractyloside significantly attenuated the infarct-sparing effect of PKCε transgenesis. Collectively, these data demonstrate that PKCε forms physical interactions with components of the cardiac mitochondrial pore. This in turn inhibits the pathological function of the pore and contributes to PKCε-induced cardioprotection.
Keywords: mitochondria, signal transduction, permeability transition pore, cardioprotection
The mitochondrion has received considerable attention as the target of multiple forms of protection, such as preconditioning (PC), against myocardial ischemia/reperfusion injury. Pharmacological studies have implicated a role for mitochondrial KATP channel opening in cardioprotection,1–4 and PC has been shown to preserve mitochondrial function5,6 and to reduce mitochondrial cytochrome c release during ischemia/reperfusion.7,8 In addition to mitochondria, multiple studies have also demonstrated that activation of protein kinase Cε (PKCε) is critical for the protective phenotype.9–13 Furthermore, cardioprotective stimuli can induce translocation of PKCε to mitochondria.14,15 However, little is known regarding whether this signaling kinase and mitochondria are functionally linked and if so, the specific manner and the molecular mechanisms by which they exert cardioprotection. Recently, we have reported mitochondrial localization of PKCε in the mouse heart, and that mitochondrial PKCε expression and activity were enhanced in mice with cardiac-specific expression of active PKCε.16 Accordingly, we postulate that mitochondrial proteins are targets for PKCε, and that PKCε-dependent modulation of these proteins plays a key role in protection against ischemic injury. However, the molecular identities of the mitochondrial proteins modulated by PKCε have yet to be ascribed.
One potential target is the mitochondrial permeability transition pore. The mitochondrial pore is a multiprotein complex formed at the contact sites between the inner and outer mitochondrial membranes.17–19 Its core components are the voltage-dependent anion channel (VDAC) in the outer membrane and adenine nucleotide translocase (ANT) in the inner membrane. The pore complex also includes hexokinase, which binds to VDAC, and cyclophilin D, which binds to ANT.17–19 The current paradigm is that the VDAC–ANT–cyclophilin D complex is the functional pore, with hexokinase enabling modulation of the pore by glucose.17,18 While the physiological role of the pore is still not clear, pathological opening of the pore is known to cause mitochondrial permeability transition, and consequently plays a critical role in the progression of both apoptotic and necrotic cell death.17–19 Pore opening induces mitochondrial swelling, causing outer membrane rupture and the release of apoptogenic proteins such as cytochrome c and Smac/DIABLO. In addition, the inner membrane potential collapses, thus inhibiting ATP synthesis, which if prolonged will instead induce necrotic death.
The conditions that elicit pore opening are identical to those that exist in the ischemic heart, namely high Ca2+, low glucose and ATP, and high Pi.18,19 Consequently, a role for the mitochondrial pore in ischemia/reperfusion-induced cardiac cell death has been proposed.18,19 Several studies report that pore opening may contribute to ischemic injury and that inhibition of the pore may be involved in cardioprotection.3,8,20–24 Moreover, it has been shown that protein kinase A (PKA) can phosphorylate VDAC,25,26 and that protein kinase G (PKG) can inhibit the pore in brain mitochondria,27 suggesting that kinases can regulate pore opening. Nevertheless, the molecular events by which cardiac protective stimuli or signaling molecules modulate the mitochondrial pore have never been defined.
Building on the concept of signal transduction being mediated by the formation of multiprotein complexes,28,29 we hypothesized that mitochondrial PKCε can directly interact with, and inhibit opening of, the mitochondrial permeability transition pore in the heart, and that this functional coupling contributes to the cardioprotective actions of PKCε. In the present study, we determined that PKCε can interact with and phosphorylate key components of the mitochondrial pore in the mouse heart. Moreover, we found that transgenic activation of PKCε enhances signaling complex formation between PKCε and the pore, concomitant with inhibition of pore opening. Finally, prevention of pore inhibition with atractyloside significantly impairs the cardioprotective effects of PKCε transgenesis against ischemia/reperfusion injury.
Materials and Methods
All procedures were performed in accordance with the University of Louisville IACUC guidelines, which conform with the NIH Guide for the Care and Use of Laboratory Animals.
Antibodies and Chemicals
Anti-PKCε (1:1000) and GST (1:2000) were purchased from BD Pharmingen; anti-VDAC1 (1:2000) from Calbiochem; anti-ANT1 (1:500) and hexokinase II (1:1000) from Santa Cruz Biotechnology; and anti-cyclophilin D (1:1000) from Affinity Bioreagents. Glucose 6-phosphate dehydrogenase (G6PDH) and protease inhibitor cocktail were obtained from Roche Diagnostics, atractyloside from Calbiochem, and recombinant PKCε from BioMol.
PKCε Transgenic Mice
The cardiac-targeted PKCε transgenic mice have been extensively described.13,16,30 The αMHC promoter was used to drive cardiac expression of either an active (A159E; AE) or a dominant-negative (K436R and A159E; DN) PKCε mutant in Institute for Cancer Research mice. Both lines are free of hypertrophy and display normal cardiac function.13,30 Mice were studied at 9 to 12 weeks.
Constructs and Recombinant Proteins
Mouse heart VDAC1 and ANT1, and rat heart cyclophilin D cDNAs were obtained by RT-PCR and subcloned into pCDNA3. Mouse hexokinase II cDNA was a generous gift from Sami Heikkinen (University of Kuopio, Finland) and was subcloned into pCDNA3. Recombinant [35S]-labeled VDAC1, ANT1, hexokinase II (HKII), and cyclophilin D were generated by in vitro transcription and translation using the TNT rabbit reticulocyte lysate system (Promega). VDAC1 was subcloned into pAcGHLT and recombinant glutathione S-transferase (GST)-VDAC1 fusion protein generated using the baculovirus expression system (BD Pharmingen). The fusion protein (which contains a His6 tag) was then purified from the insect cell lysates on a Ni-NTA column. GST-PKCε was generated using the same system.13,31
Mitochondrial Isolation
Mouse cardiac mitochondria were isolated by differential centrifugation.16 After washing twice, mitochondria were then either directly used for swelling assays or were incubated in lysis buffer (150 mmol/L NaCl, 50 mmol/L Tris-HCl [pH 7.4], 1% NP-40, 1 mmol/L EDTA, 1 mmol/L Na3VO4, and protease inhibitor). Contamination of the mitochondrial fraction by other subcellular compartments was minimal (data not shown), as determined by lactate dehydrogenase activity (cytosol), and Western blotting for nuclear mitotic apparatus protein (nuclei) and the dihydropyridine receptor (sarcolemma). Protein concentrations were determined using the Bradford method.
Western Blotting
Mitochondrial proteins (50 to 100 μg) were resolved on 10% SDS-PAGE gels and transferred onto nitrocellulose membranes. After blocking with 5% milk, the membranes were immunoblotted using the ECL detection system (Amersham). Linearity was maintained by taking and quantifying multiple exposures of each membrane.
Coimmunoprecipitation
Coimmunoprecipitation was carried out as described.13,16 Briefly, 500 μg mitochondrial protein was incubated with 1 μg of either PKCε, VDAC1, ANT1, or hexokinase II antibodies plus 20 μL Protein A/G-Agarose (Santa Cruz) overnight at 4°C. Nonspecific IgG was used as a negative control. After washing three times with lysis buffer, the complexes were subjected to SDS-PAGE and immunoblotting.
GST-Based Affinity Pull-Down Assays
GST-based affinity pull-down assays were performed as previously reported.13,31 Briefly, 1 μg of GST-PKCε or GST-VDAC1 was immobilized on glutathione-Sepharose beads, mixed with either 50 μL of [35S]-labeled in vitro translation mix or 500 μg cardiac mitochondrial protein, and incubated in binding buffer (0.2% Triton X-100, 150 mmol/L NaCl, 20 mmol/L Tris-HCl [pH 7.4], 1 mmol/L EDTA, 1 mmol/L EGTA, 0.2 mmol/L Na3VO4, and protease inhibitor). GST-null proteins were used as negative controls. The GST-protein complexes were then washed, resolved by SDS-PAGE, and analyzed via autoradiography or immunoblotting.
In Vitro Phosphorylation of VDAC
Recombinant PKCε was incubated with GST-VDAC1 in phosphorylation cocktail (50 mmol/L Tris-HCl [pH 7.5], 0.1 mmol/L ATP, 2.3 mmol/L HEPES, 28.8 μg/mL α-phosphatidyl-L-serine, 3 μg/mL PMA, 7.5 mmol/L DTT, 3 mmol/L CaAc, 5.5 mmol/L MgCl2, and 0.5 μCi [γ-32P]-ATP) for 15 minutes at 37°C.13,31 Phosphorylation was then analyzed by SDS-PAGE and autoradiography.
Mitochondrial Hexokinase Activity
Hexokinase activity was determined by analyzing the glucose-6-phosphate–coupled conversion of NADP to NADPH.32 Ten microliters of mitochondrial extract was added to 990 μL of assay buffer (100 mmol/L Tris-HCl [pH 8.0], 0.5 mmol/L EDTA, 10 mmol/L ATP, 10 mmol/L MgCl2, 2 mmol/L glucose, 0.1 mmol/L NADP, and 0.1 U/mL G6PDH). NADPH formation was then followed spectrophotometrically at 340 nm.
Mitochondrial Pore Opening
Opening of the mitochondrial pore was determined by Ca2+-induced swelling of isolated cardiac mitochondria.20–24 Opening of the pore causes mitochondrial swelling, which is measured as a reduction in A520. Isolated cardiac mitochondria were resuspended in swelling buffer (120 mmol/L KCl, 10 mmol/L Tris-HCl [pH 7.4], 20 mmol/L MOPS, and 5 mmol/L KH2PO4) to a final concentration of 0.25 mg/mL. Absorbance was measured spectrophotometrically at 520 nm and pore opening induced by 200 μmol/L CaCl2. To confirm that the absorbance decreases were due to opening of the pore, mitochondria were incubated with either 30 nmol/L cyclosporin A or 10 μmol/L MgCl2, two pore inhibitors, 5 minutes before the addition of CaCl2. In additional experiments, mitochondria were incubated with 2 μg recombinant PKCε plus 1 μmol/L PMA 15 minutes before the addition of CaCl2.
Myocardial Infarction
The mouse model of myocardial infarction has been previously described in detail.13,33 Briefly, myocardial infarction was produced in pentobarbital-anesthetized mice by a 30-minute coronary occlusion followed by a 4-hour reperfusion. Physiological variables, including temperature, arterial pressure, heart rate, and blood gases, were maintained within normal limits. At the conclusion of the study, the occluded/reperfused coronary bed and the infarcted region were identified by postmortem perfusion of pthalo blue and tetrazolium chloride, respectively. Infarct size was calculated by videoplanimetry and expressed as a percentage of the ischemic zone. The pore activator atractyloside (25 mg/kg, IV) was administered 15 minutes before ischemia.
Statistical Analysis
Data are expressed as mean±SEM. Biochemical data were compared using unpaired t tests, and infarct size data were compared by one-way ANOVA followed by t test. P<0.05 was considered significant.
Results
PKCε Interacts With the Mitochondrial Pore In Vivo
Examination of mouse cardiac mitochondria demonstrated robust expression of VDAC1, ANT1, hexokinase II, and cyclophilin D, alongside PKCε (Figure 1, n=8). Moreover, immunoprecipitation of mitochondrial PKCε followed by Western blotting revealed the interaction of PKCε with VDAC1, ANT1, and hexokinase II, but not cyclophilin D (Figure 2, n=8). The amounts of VDAC1, ANT1, and HKII in the PKCε complex were estimated to be ≈2%, ≈0.2%, and ≈1% of their total mitochondrial expression, respectively.
Figure 1.

Expression of individual mitochondrial pore proteins in mouse cardiac mitochondria. Mitochondrial lysates from control mouse hearts were subjected to SDS-PAGE followed by Western immunoblotting with (from top to bottom) anti-VDAC1, ANT1, HKII, CypD, and PKCε. Each lane represents an individual heart (n=8). IB indicates immunoblotting.
Figure 2.

PKCε interacts with mitochondrial pore proteins in vivo. Mitochondrial lysates from control mouse hearts were immunoprecipitated with anti-PKCε antibody. The complexes were subjected to SDS-PAGE followed by Western immunoblotting with (from top to bottom) anti-VDAC1, ANT1, HKII, CypD, and PKCε antibodies. Lanes 1 through 3 contain PKCε immunoprecipitates from 3 individual hearts; lane 4 is blank; and lane 5 contains cardiac mitochondrial lysate as positive control (n=8). IP indicates immunoprecipitation; IB, immunoblotting.
PKCε Interacts With and Phosphorylates VDAC In Vitro
In order to ascertain which of the pore proteins bound directly to PKCε, [35S]-labeled recombinant VDAC1, ANT1, hexokinase II, and cyclophilin D were produced by in vitro translation and incubated with either GST or GST-PKCε (Figure 3A, n=3). GST-PKCε was able to bind recombinant VDAC1 more efficiently than GST alone, binding 3.1% of available VDAC1 compared with 0.7%, respectively. In contrast, GST-PKCε and GST bound equally to ANT1, binding 2.6% and 2.1%, respectively. Binding of GST-PKCε to hexokinase II and cyclophilin D was negligible. The relatively high nonspecific binding of ANT1 to GST was somewhat of a concern. However, interaction of ANT with GST-VDAC1 was significantly more robust than GST alone (data not shown), indicating that specific ANT-protein interactions could still be observed with this system. Analysis of the murine VDAC1 sequence revealed 8 putative PKC phosphorylation sites, and recombinant PKCε was able to phosphorylate GST-VDAC1 in a dose-dependent manner (Figure 3B, n=3).
Figure 3.

PKCε interacts with and phosphorylates VDAC in vitro. A, Equal amounts of recombinant GST or GST-PKCε fusion proteins conjugated to glutathione-Sepharose beads were incubated with mixes containing [35S]-labeled VDAC1, ANT1, HKII, or CypD produced by in vitro translation (IVT). The complexes were pulled down and subjected to SDS-PAGE and either autoradiography (top 4 panels) or immunoblotting with anti-GST antibody (bottom panel). Lane 1 contains GST alone plus the respective in vitro translation product; lane 2 contains GST-PKCε plus the respective in vitro translation product; lane 3 is blank; and lane 4 contains the respective in vitro translation product as positive control (n=3). B, Recombinant GST-VDAC1 fusion protein was incubated with γ[32P]-ATP plus increasing amounts of recombinant PKCε and subjected to SDS-PAGE and either autoradiography (top panel) or immunoblotting with anti-GST antibody (bottom panel). Lanes 1 through 4 contain radio-labeled GST-VDAC1 after incubation with 0, 10, 50, and 100 ng of recombinant PKCε, respectively (n=3). IB indicates immunoblotting.
PKCε Inhibits Opening of the Mitochondrial Pore In Vitro
We tested whether binding of PKCε exerts a functional effect on the mitochondrial pore. Mouse cardiac mitochondria were isolated and Ca2+-induced pore opening assessed by measuring mitochondrial swelling. Opening of the pore causes swelling of mitochondria, which is observed as a decrease in absorbance.20–24 In nontreated mitochondria, Ca2+ evoked a large decrease in A520 (Figure 4, n=3). This effect was inhibited by cyclosporin A and Mg2+, two chemically disparate pore inhibitors, confirming that the decrease in absorbance was due to pore opening. In mitochondria treated with recombinant PKCε plus PMA, but neither PKCε nor PMA alone, the absorbance decrease was also significantly attenuated (Figure 4), demonstrating that PKCε can inhibit the mitochondrial pore. Moreover, this effect of PKCε was independent of changes in mitochondrial hexokinase activity (data not shown).
Figure 4.

Recombinant PKCε inhibits swelling in mouse cardiac mitochondria. Mitochondria were isolated from nontransgenic mouse hearts and swelling was induced by 200 μmol/L CaCl2 and measured spectrophotometrically as a decrease in A520. In some experiments, mitochondria were incubated with either the pore inhibitors cyclosporin A (30 nmol/L) or MgCl2 (10 μmol/L) 5 minutes before calcium was added, or with recombinant PKCε (2 μg) plus PMA (1 μmol/L) 15 minutes before calcium was added. Figure shows representative traces for control, cyclosporin A–, Mg2+-, and PKCε-treated mitochondria (n=3).
Interaction of PKCε With the Mitochondrial Pore Is Increased in PKCε Transgenic Mice
We next examined the interaction of PKCε with the mitochondrial pore in the active (AE) and dominant-negative (DN) PKCε mouse lines. The AE line exhibits cardioprotection, whereas the DN line does not.13,16 Moreover, while mitochondrial expression of PKCε is increased in both lines, mitochondrial PKCε activity is increased ≈3- to 4-fold in the AE mice and decreased by about half in the DN mice.16 Western blot analysis revealed no significant changes in VDAC1, ANT1, and hexokinase II expression in either AE or DN mitochondria (Figure 5, n=6). Interestingly, cyclophilin D expression was increased in the AE mice and decreased in the DN mice (1.7±0.3-fold and 0.3±0.1-fold versus non-transgenic, respectively). Immunoprecipitation of VDAC1, ANT1, or hexokinase II followed by blotting for PKCε demonstrated increased interaction of PKCε with these pore constituents in the AE mice (Figure 6, n=6). This enhanced complex formation between PKCε and the pore was also evident in the DN mice (Figure 6, n=6), indicating that catalytic activity is not required for these interactions.
Figure 5.

Expression of PKCε and pore proteins in mitochondria from mice with cardiac-specific expression of active and inactive PKCε. Mitochondrial lysates from NTG, AE, and DN mouse hearts were subjected to SDS-PAGE followed by Western immunoblotting with (from top to bottom) anti-VDAC1, ANT1, HKII, CypD, and PKCε antibodies (n=6). IB indicates immunoblotting.
Figure 6.

PKCε interaction with pore proteins is enhanced in mitochondria from mice with cardiac-specific expression of active and inactive PKCε. A, Mitochondrial lysates from NTG and AE mouse hearts were immunoprecipitated with anti-VDAC1 (top), ANT1 (middle), or HKII (bottom) antibodies. The complexes were subjected to SDS-PAGE followed by Western immunoblotting with anti-VDAC1 (top), ANT1 (middle), and HKII (bottom) antibodies (n=6). B, Mitochondrial lysates from NTG, AE, and DN mouse hearts were immunoprecipitated with anti-VDAC1 (top), ANT1 (middle), or HKII (bottom) antibodies. The complexes were subjected to SDS-PAGE followed by Western immunoblotting with anti-PKCε antibody (n=6). IP indicates immunoprecipitation; IB, immunoblotting.
Opening of the Mitochondrial Pore Is Inhibited in PKCε Transgenic Mice
We also examined whether cardiac pore function was modulated in the PKCε transgenic mice. Analysis of mitochondria isolated from the different mouse lines demonstrated a reduction in Ca2+-induced swelling in AE mitochondria when compared with mitochondria isolated from nontransgenic mice (Figure 7, n=4). In contrast, swelling was unaffected in the DN mitochondria. Mitochondrial hexokinase activity was unchanged in both the AE and DN hearts (data not shown).
Figure 7.
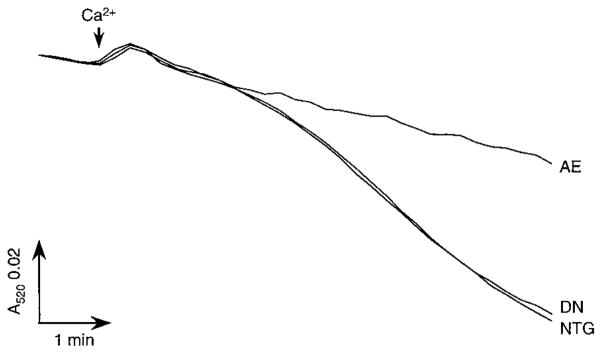
Mitochondrial swelling is inhibited in mice with cardiac-specific expression of active but not inactive PKCε. Mitochondria were isolated from NTG, AE, and DN hearts, and swelling was induced by 200 μmol/L CaCl2 and measured spectrophotometrically as a decrease in A520. Figure shows representative traces for NTG, AE, and DN mitochondria (n=4).
Atractyloside Attenuates Cardioprotection in PKCε Transgenic Mice
Activation of PKCε protects the heart against ischemia/reperfusion injury.9–13 We therefore examined whether inhibition of the mitochondrial pore by PKCε contributes to the cardioprotective phenotype observed in the AE mice. In nontransgenic mice subjected to 30 minutes of regional ischemia/4 hours of reperfusion, myocardial infarction was 42.5±4.7% of the ischemic region (Figure 8, n=5 to 6). In contrast, the degree of infarction was significantly reduced to 21.7±3.6% in the AE mice. Atractyloside is an ANT ligand that places the translocase in a conformation that is more conducive to pore opening.18 Thus, atractyloside would counteract any inhibitory force exerted on the pore. Administration of atractyloside (25 mg/kg IV) 15 minutes before the ischemic period had no significant effect on infarction in nontransgenic animals (47.2±6.8%). However, atractyloside markedly attenuated the protection observed in the AE mice (37.4±2.2%).
Figure 8.
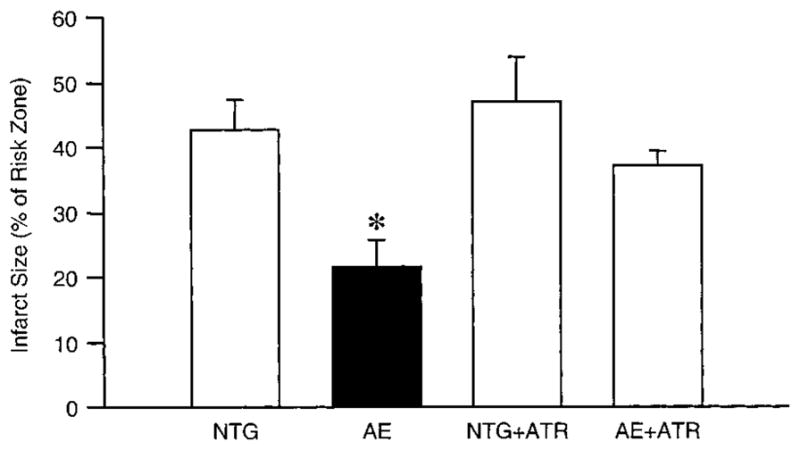
Atractyloside inhibits cardioprotection in mice with cardiac-specific expression of active PKCε. Anesthetized NTG and AE mice were subjected to 30 minutes of coronary occlusion followed by 4 hours reperfusion, and the resultant infarct size was determined by postmortem tetrazolium staining. Atractyloside (ATR) was administered as a bolus injection (25 mg/kg, IV) 15 minutes before the coronary occlusion. Data are represented as mean±SEM. *P<0.05 compared with NTG (n=5 to 6).
Discussion
We have previously demonstrated the constitutive expression of PKCε in cardiac mitochondria,16 and cardioprotective stimuli induce PKCε translocation to the mitochondrial compartment.14,15 Together, these data indicate that PKCε interacts with and modulates multiple mitochondrial proteins. Of these, the mitochondrial permeability transition pore complex was an attractive candidate as emerging evidence indicates that pore opening contributes to myocardial ischemic injury.3,8,20–24 Therefore, inhibition of the mitochondrial pore by PKCε would represent a novel mechanism by which PKCε mediates cardioprotection.
PKCε Interacts With Mitochondrial Permeability Transition Pore
Coimmunoprecipitation from mouse cardiac mitochondria demonstrated interaction of PKCε with VDAC1, ANT1, and hexokinase II in vivo. However, we found that only VDAC1 was a direct binding partner with PKCε in vitro. Consequently, it appears that PKCε is able to bind to the pore in mitochondria via interaction with VDAC, and that the ability of ANT and hexokinase to coprecipitate with PKCε is presumably through interaction with VDAC.17–19 To our knowledge, this is the first time that direct interaction of a signaling kinase with components of the mitochondrial pore has been demonstrated. The amounts of VDAC1, ANT1, and hexokinase II in the PKCε complex were estimated to be very small in relation to their total mitochondrial levels. However, these numbers only reflect the levels of each pore protein in the PKCε complex under basal conditions, and our data would indicate that these amounts would be greatly enhanced under conditions where PKCε is stimulated. Moreover, the numbers of each protein that is physically part of every pore would only be a fraction of the total pool. Consequently, we believe that the amounts of pore-specific VDAC, ANT, and hexokinase II interacting with PKCε are higher than the figures would indicate and are sufficient to be functionally relevant. In addition to interacting with the pore, we found that recombinant PKCε could phosphorylate VDAC in a dose-dependent manner, consistent with previous studies showing that PKA can phosphorylate this protein.25,26
Signal transduction is mediated by the formation of dynamic complexes between signaling elements and their effectors,28,29 and we have demonstrated enhanced interactions of PKCε with multiple proteins within the cardioprotected mouse heart.13,16,31,34 Consequently, if modulation of the mitochondrial pore by PKCε is critical for cardioprotection, then we would expect enhanced PKCε-pore interactions in protected hearts. To address this question, we used a genetic model of cardioprotection engendered by cardiac-specific expression of an active mutant of PKCε,13,30 in which mitochondrial PKCε expression and activity are greatly enhanced.16 Coimmunoprecipitation revealed that mitochondrial PKCε interactions with VDAC, ANT, and hexokinase II were greatly increased in the PKCε transgenic mice when compared with controls. This enhanced complex formation between PKCε and the pore proteins was also present in mouse hearts expressing DN PKCε, consistent with our previous data showing that PKCε interactions are activity-independent.16,31
PKCε Inhibits the Mitochondrial Permeability Transition Pore
A recent study demonstrated that PKG could inhibit the pore in isolated brain mitochondria,27 suggesting that kinases can regulate pore opening. Therefore, by binding to and phosphorylating VDAC, PKCε may exert a modulatory effect on the mitochondrial pore. Incubation of cardiac mitochondria with recombinant PKCε resulted in a small but significant inhibition of Ca2+-induced mitochondrial swelling, an index of mitochondrial pore opening, thus demonstrating a direct inhibitory effect of PKCε on the mitochondrial pore. The small magnitude of the inhibition is presumably a reflection of the artificial conditions under which PKCε interacts with the mitochondria. For example, the phosphorylation reaction is reliant on what mitochondrial ATP is available after isolation, limitations of which would reduce the ability of PKCε to phosphorylate the pore. Unfortunately, addition of exogenous ATP is precluded as ATP is a strong inhibitor of the pore.17 The amount of PKCε may also be an issue, as the study in brain mitochondria used huge amounts of PKG for complete inhibition of the pore.27
To circumvent these issues, pore function was also determined in the PKCε transgenic mice, where phosphorylation will have occurred before mitochondrial isolation. In keeping with the in vitro data, we found that mitochondria isolated from hearts expressing active PKCε exhibited a considerable suppression of Ca2+-induced pore opening. Together, these data demonstrate that both exogenous and endogenous PKCε activation can inhibit the mitochondrial permeability transition pore in the mouse myocardium. Moreover, inhibition of the pore is only associated with the cardioprotective phenotype as pore function in mice expressing the DN PKCε was no different from controls.
PKCε-Mediated Pore Inhibition Contributes to Cardioprotection
Opening of the mitochondrial pore has been proposed as a mediator of cardiomyocyte death in response to ischemia/reperfusion.18,19 This is supported by experimental evidence of reperfusion-induced pore opening in isolated rat hearts.21 Moreover, pore inhibitors prevent ischemia/reperfusion-induced cell death in isolated cardiomyocytes or whole hearts.8,20,21,23,24
The present study shows that the cardioprotected phenotype exhibited in the active PKCε transgenic mice correlated with inhibition of the mitochondrial pore. We therefore wanted to test whether these two circumstances were causally linked. The ANT protein exists in two states: the c-state and m-state in which the ATP/ADP binding site faces either the cytosol and the matrix, respectively.35 The specific ANT ligand atractyloside places the translocase into the c-state, facilitating pore opening at high concentrations of the drug.35 Therefore, we reasoned that the conformational changes induced by atractyloside would counteract the inhibition by PKCε. Using a dose of atractyloside that did not itself affect myocardial infarction, protection against ischemia was significantly attenuated in the mice expressing active PKCε. Thus, inhibition of the mitochondrial pore by PKCε appears to mediate cardioprotection in these mice.
Mechanism of PKCε-Mediated Inhibition of the Mitochondrial Pore
Although the specific mechanism by which PKCε influences the VDAC protein and therefore the pore as a whole during cardioprotection is still unknown, several scenarios can be envisioned. One possibility is that PKCε influences hexokinase activity, which can modulate pore opening.36,37 However, we saw no changes in hexokinase activity in mitochondria from PKCε transgenic hearts or those treated with exogenous PKCε. Another possibility is that phosphorylation of VDAC may inhibit the ion channel itself, thus reducing pore conductance. A third possibility is that PKCε phosphorylation reduces the ability of VDAC to interact with ANT and/or hexokinase II, although preliminary studies suggest that this is not the case. A fourth possibility is that PKCε-induced conformational changes may impair the Ca2+ sensitivity of the mitochondrial pore. Halestrap’s group has proposed that binding of cyclophilin D to ANT facilitates opening of the pore by increasing Ca2+ sensitivity.35 As we did not find cyclophilin D in the PKCε-pore complex, PKCε may induce changes within the pore that prevent binding of cyclophilin D and reduce Ca2+ sensitivity. A final possibility is that PKCε recruits inhibitory proteins into the pore complex. For example, we have previously shown that activation of a PKCε-ERK module in mitochondria of the AE transgenic mice leads to inactivation of the proapoptotic protein Bad.16 Inactivation of Bad would release the antiapoptotic protein Bcl-XL, which can bind to and inhibit the mitochondrial pore.38 The parallel phosphorylation of VDAC by PKCε may act to facilitate the binding of Bcl-XL to the pore. Future studies will address these issues.
Experimental Limitations
There are several experimental limitations that should be taken into account. The first is the use of the PKCε transgenic model of protection. Classical PC is an acute phenomenon that relies on posttranslational modifications rather than gene expression. In contrast, the genetic model is associated with chronic changes in protein expression and activity and therefore cannot be directly compared with ischemic PC. Indeed, cyclophilin D was upregulated in the AE mice and down-regulated in the DN mice. While the obvious interpretation is that cyclophilin D expression is controlled by PKCε activity, what effects these changes have on pore function are still not clear. Despite this, PC is known to induce mitochondrial PKCε translocation14,15 and PC’s protection is blocked by atractyloside,8,24 suggesting that PKCε-induced inhibition of the pore may also contribute to PC. A second limitation is that while PKCε can directly bind to and phosphorylate VDAC in vitro, this may not reflect what occurs in vivo. For instance, as PKCε also forms a mitochondrial signaling module with ERK,16 it is feasible that under in vivo conditions ERK phosphorylates VDAC. The third limitation is that although we believe that the dose of atractyloside used is specific for ANT, we cannot completely rule out that its effect in the AE-PKCε mice was due to the metabolic consequences of ANT inhibition rather than the effect on the pore. Further studies using non-ANT binding pore activators are warranted.
Summary
Very little is known about the pore and its regulation in the myocardium. Furthermore, how intracellular signaling networks couple to the mitochondrial pore has only just begun to be examined. Consequently, the present study provides new information as to how a signaling kinase can directly interact with and modulate function of the mitochondrial pore, and consequently contribute to a specific cardiac phenotype. Application of this paradigm to other signaling systems and cardiac phenotypes should enable us to gain novel insight into how mitochondrial dysfunction affects cardiovascular function and disease, and mechanisms by which this can be prevented.
Acknowledgments
This study was supported by AHA Ohio Valley Affiliate Postdoctoral Fellowship 0120412B (C.P.B.), NIH HL-63901 and 65431 (P.P.), HL-43151, 55757, and 68088 (R.B.), University of Louisville Research Foundation, Jewish Hospital Research Foundation, Commonwealth of Kentucky Research Challenge Trust Fund, and by the Laubisch Endowment. We are extremely grateful to Sami Heikkinen, University of Kuopio, Finland, for his very generous gift of the mouse hexokinase II cDNA.
Footnotes
This manuscript was sent to Stephen F. Vatner, Consulting Editor, for review by expert referees, editorial decision, and final disposition.
References
- 1.O’Rourke B. Myocardial KATP channels in preconditioning. Circ Res. 2000;87:845–855. doi: 10.1161/01.res.87.10.845. [DOI] [PubMed] [Google Scholar]
- 2.Baines CP, Liu GS, Birincioglu M, Critz SD, Cohen MV, Downey JM. Ischemic preconditioning depends on interaction between mitochondrial KATP channels and actin cytoskeleton. Am J Physiol. 1999;276:H1361–H1368. doi: 10.1152/ajpheart.1999.276.4.H1361. [DOI] [PubMed] [Google Scholar]
- 3.Murata M, Akao M, O’Rourke B, Marban E. Mitochondrial ATP-sensitive potassium channels attenuate matrix Ca2+ overload during simulated ischemia and reperfusion: possible mechanism of cardioprotection. Circ Res. 2001;89:891–898. doi: 10.1161/hh2201.100205. [DOI] [PubMed] [Google Scholar]
- 4.Garlid KD, Paucek P, Yarov-Yarovoy V, Murray HN, Darbenzio RB, D’Alonzo AJ, Lodge NJ, Smith MA, Grover GJ. Cardioprotective effect of diazoxide and its interaction with mitochondrial ATP-sensitive K+ channels: possible mechanism of cardioprotection. Circ Res. 1997;81:1072–1082. doi: 10.1161/01.res.81.6.1072. [DOI] [PubMed] [Google Scholar]
- 5.Fryer RM, Eells JT, Hsu AK, Henry MM, Gross GJ. Ischemic preconditioning in rats: role of mitochondrial KATP channel in preservation of mitochondrial function. Am J Physiol. 2000;278:H305–H312. doi: 10.1152/ajpheart.2000.278.1.H305. [DOI] [PubMed] [Google Scholar]
- 6.Laclau MN, Boudina S, Thambo JB, Tariosse L, Gouverneur G, Bonoron-Adele S, Saks VA, Garlid KD, Dos Santos P. Cardioprotection by ischemic preconditioning preserves mitochondrial function and functional coupling between adenine nucleotide translocase and creatine kinase. J Mol Cell Cardiol. 2001;33:947–956. doi: 10.1006/jmcc.2001.1357. [DOI] [PubMed] [Google Scholar]
- 7.Liu H, Zhang HY, Zhu X, Shao Z, Yao Z. Preconditioning blocks cardiocyte apoptosis: role of KATP channels and PKCε. Am J Physiol. 2002;282:H1380–H1386. doi: 10.1152/ajpheart.00348.2001. [DOI] [PubMed] [Google Scholar]
- 8.Xu M, Wang Y, Hirai K, Ayub A, Ashraf M. Calcium preconditioning inhibits mitochondrial permeability transition and apoptosis. Am J Physiol. 2001;280:H899–H908. doi: 10.1152/ajpheart.2001.280.2.H899. [DOI] [PubMed] [Google Scholar]
- 9.Ping P, Takano H, Zhang J, Tang XL, Qiu Y, Li RC, Banerjee S, Dawn B, Balafonova Z, Bolli R. Isoform-selective activation of protein kinase C by nitric oxide in the heart of conscious rabbits: a signaling mechanism for both nitric oxide-induced and ischemia-induced preconditioning. Circ Res. 1999;84:587–604. doi: 10.1161/01.res.84.5.587. [DOI] [PubMed] [Google Scholar]
- 10.Ping P, Zhang J, Qiu Y, Tang XL, Manchikalapudi S, Cao X, Bolli R. Ischemic preconditioning induces selective translocation of protein kinase C isoforms ε and η in the heart of conscious rabbits without subcellular redistribution of total protein kinase C activity. Circ Res. 1997;81:404–414. doi: 10.1161/01.res.81.3.404. [DOI] [PubMed] [Google Scholar]
- 11.Dorn GW, 2nd, Souroujon MC, Liron T, Chen CH, Gray MO, Zhou HZ, Csukai M, Wu G, Lorenz JN, Mochly-Rosen D. Sustained in vivo cardiac protection by a rationally designed peptide that causes ε protein kinase C translocation. Proc Natl Acad Sci U S A. 1999;96:12798–12803. doi: 10.1073/pnas.96.22.12798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu GS, Cohen MV, Mochly-Rosen D, Downey JM. Protein kinase Cε is responsible for the protection of preconditioning in rabbit cardiomyocytes. J Mol Cell Cardiol. 1999;31:1937–1348. doi: 10.1006/jmcc.1999.1026. [DOI] [PubMed] [Google Scholar]
- 13.Ping P, Song C, Zhang J, Guo Y, Cao X, Li RC, Wu W, Vondriska TM, Pass JM, Tang XL, Pierce WM, Bolli R. Formation of protein kinase Cε-Lck signaling modules confers cardioprotection. J Clin Invest. 2002;109:499–507. doi: 10.1172/JCI13200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang J, Bolli R, Lalli J, Tang X-L, Li RCX, Zheng Y, Pass J, Ping P. Ischemic preconditioning and phorbol ester redistribute protein kinase Cε to the nucleus, sarcolemmal membranes, and mitochondria in rabbit myocardium. Circulation. 1999;100(suppl I):I-325. Abstract. [Google Scholar]
- 15.Fryer RM, Wang Y, Hsu AK, Gross GJ. Essential activation of PKCδ in opioid-initiated cardioprotection. Am J Physiol. 2001;280:H1346–H1353. doi: 10.1152/ajpheart.2001.280.3.H1346. [DOI] [PubMed] [Google Scholar]
- 16.Baines CP, Zhang J, Wang GW, Zheng YT, Xiu JX, Cardwell EM, Bolli R, Ping P. Mitochondrial PKCε, and MAPK form signaling modules in the murine heart: enhanced mitochondrial PKCε-MAPK interactions and differential MAPK activation in PKCε-induced cardioprotection. Circ Res. 2002;90:390–397. doi: 10.1161/01.res.0000012702.90501.8d. [DOI] [PubMed] [Google Scholar]
- 17.Zamzami N, Kroemer G. The mitochondrion in apoptosis: how Pandora’s box opens. Nat Rev Mol Cell Biol. 2001;2:67–71. doi: 10.1038/35048073. [DOI] [PubMed] [Google Scholar]
- 18.Crompton M, Barksby E, Johnson N, Capano M. Mitochondrial inter-membrane junctional complexes and their involvement in cell death. Biochimie. 2002;84:143–152. doi: 10.1016/s0300-9084(02)01368-8. [DOI] [PubMed] [Google Scholar]
- 19.Suleiman MS, Halestrap AP, Griffiths EJ. Mitochondria. a target for myocardial protection. Pharmacol Ther. 2001;89:29–46. doi: 10.1016/s0163-7258(00)00102-9. [DOI] [PubMed] [Google Scholar]
- 20.Clarke SJ, McStay GP, Halestrap AP. Sanglifehrin A acts as a potent inhibitor of the mitochondrial permeability transition and reperfusion injury of the heart by binding to cyclophilin-D at a different site from cyclosporin A. J Biol Chem. 2002;277:34793–34799. doi: 10.1074/jbc.M202191200. [DOI] [PubMed] [Google Scholar]
- 21.Di Lisa F, Menabo R, Canton M, Barile M, Bernardi P. Opening of the mitochondrial permeability transition pore causes depletion of mitochondrial and cytosolic NAD+ and is a causative event in the death of myocytes in postischemic reperfusion of the heart. J Biol Chem. 2001;276:2571–2575. doi: 10.1074/jbc.M006825200. [DOI] [PubMed] [Google Scholar]
- 22.Korge P, Honda HM, Weiss JN. Protection of cardiac mitochondria by diazoxide and protein kinase C: implications for ischemic preconditioning. Proc Natl Acad Sci U S A. 2002;99:3312–3317. doi: 10.1073/pnas.052713199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tsuchida A, Sakamoto J, Miki T, Genda S, Matsumoto T, Ohnuma Y, Miura T. Inhibition of mitochondrial permeability transition pore protects cardiomyocytes from death during ischemia/reperfusion: a possible mechanism of cardioprotection by mitochondrial KATP activation. Circulation. 2001;104(suppl II):II-16. Abstract. [Google Scholar]
- 24.Hausenloy DJ, Maddock HL, Baxter GF, Yellon DM. Inhibiting mitochondrial permeability transition pore opening: a new paradigm for myocardial preconditioning? Cardiovasc Res. 2002;55:534–543. doi: 10.1016/s0008-6363(02)00455-8. [DOI] [PubMed] [Google Scholar]
- 25.Bera AK, Ghosh S. Dual mode of gating of voltage-dependent anion channel as revealed by phosphorylation. J Struct Biol. 2001;135:67–72. doi: 10.1006/jsbi.2001.4399. [DOI] [PubMed] [Google Scholar]
- 26.Bera AK, Ghosh S, Das S. Mitochondrial VDAC can be phosphorylated by cyclic AMP-dependent protein kinase. Biochem Biophys Res Commun. 1995;209:213–217. doi: 10.1006/bbrc.1995.1491. [DOI] [PubMed] [Google Scholar]
- 27.Takuma K, Phuagphong P, Lee E, Mori K, Baba A, Matsuda T. Anti-apoptotic effect of cGMP in cultured astrocytes: inhibition by cGMP-dependent protein kinase of mitochondrial permeable transition pore. J Biol Chem. 2001;276:48093–48099. doi: 10.1074/jbc.M108622200. [DOI] [PubMed] [Google Scholar]
- 28.Schillace RV, Scott JD. Organization of kinases, phosphatases, and receptor signaling complexes. J Clin Invest. 1999;103:761–765. doi: 10.1172/JCI6491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hunter T. Signaling–2000 and beyond. Cell. 2000;100:113–127. doi: 10.1016/s0092-8674(00)81688-8. [DOI] [PubMed] [Google Scholar]
- 30.Pass JM, Zheng Y, Wead WB, Zhang J, Li RC, Bolli R, Ping P. PKCε activation induces dichotomous cardiac phenotypes and modulates PKCε-RACK interactions and RACK expression. Am J Physiol. 2001;280:H946–955. doi: 10.1152/ajpheart.2001.280.3.H946. [DOI] [PubMed] [Google Scholar]
- 31.Song C, Vondriska TM, Wang GW, Klein JB, Cao X, Zhang J, Kang YJ, D’Souza S, Ping P. Molecular conformation dictates signaling module formation: example of PKCε and Src tyrosine kinase. Am J Physiol. 2002;282:H1166–H1171. doi: 10.1152/ajpheart.00830.2001. [DOI] [PubMed] [Google Scholar]
- 32.Riddle SR, Ahmad A, Ahmad S, Deeb SS, Malkki M, Schneider BK, Allen CB, White CW. Hypoxia induces hexokinase II gene expression in human lung cell line A549. Am J Physiol. 2000;278:L407–L416. doi: 10.1152/ajplung.2000.278.2.L407. [DOI] [PubMed] [Google Scholar]
- 33.Guo Y, Wu WJ, Qiu Y, Tang XL, Yang Z, Bolli R. Demonstration of an early and a late phase of ischemic preconditioning in mice. Am J Physiol. 1998;275:H1375–H1387. doi: 10.1152/ajpheart.1998.275.4.H1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ping P, Zhang J, Pierce WM, Bolli R. Functional proteomic analysis of protein kinase Cε signaling complexes in the normal heart and during cardioprotection. Circ Res. 2001;88:59–62. doi: 10.1161/01.res.88.1.59. [DOI] [PubMed] [Google Scholar]
- 35.Halestrap AP, McStay GP, Clarke SJ. The permeability transition pore complex: another view. Biochimie. 2002;84:153–166. doi: 10.1016/s0300-9084(02)01375-5. [DOI] [PubMed] [Google Scholar]
- 36.Pastorino JG, Shulga N, Hoek JB. Mitochondrial binding of hexokinase II inhibits Bax-induced cytochrome c release and apoptosis. J Biol Chem. 2002;277:7610–7618. doi: 10.1074/jbc.M109950200. [DOI] [PubMed] [Google Scholar]
- 37.Gottlob K, Majewski N, Kennedy S, Kandel E, Robey RB, Hay N. Inhibition of early apoptotic events by Akt/PKB is dependent on the first committed step of glycolysis and mitochondrial hexokinase. Genes Dev. 2001;15:1406–1418. doi: 10.1101/gad.889901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shimizu S, Narita M, Tsujimoto Y. Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature. 1999;399:483–487. doi: 10.1038/20959. [DOI] [PubMed] [Google Scholar]