

Abstract
The mitogen-activated protein kinases (MAPK) have been the subject of many studies to identify signaling pathways that promote cell survival or death. In cultured cardiac myocytes, p38 MAPK promotes cell survival or death depending on whether it is activated by mitogen-activated protein kinase kinase 6 (MKK6) or MKK3, respectively. The objectives of the current study were to examine the effects of MKK6-mediated p38 activation in the heart in vivo. Accordingly, we generated transgenic (TG) mice that overexpress wild type MKK6 in a cardiac-restricted manner. Although p38 was about 17-fold more active in TG than non-transgenic (NTG) mouse hearts, TG mouse hearts were morphologically and functionally similar to those of NTG littermates. However, upon transient ischemia followed by reperfusion, the MKK6 TG mouse hearts exhibited significantly better functional recovery and less injury than NTG mouse hearts. Because MKK6 increases levels of the protective small heat shock protein, αB-crystallin (αBC), in cultured cardiac myocytes, we examined αBC levels in the mouse hearts. The level of αBC was 2-fold higher in MKK6 TG than NTG mouse hearts. Moreover, ischemia followed by reperfusion induced a 6.4-fold increase in αBC levels in the mitochondrial fractions of TG mouse hearts but no increase in αBC levels in any of the other fractions analyzed. These alterations in αBC expression and localization suggest possible mechanisms of cardioprotection in MKK6 TG mouse hearts.
The myocardium can be stressed by chronic increases in blood pressure, ischemia followed by reperfusion (I/R),1 or increases in neurohumoral substances, such as peptide hormones, catecholamine, or cytokines. Numerous signals are activated in stressed cardiac myocytes; in some cases those signals mediate protection, whereas in others they mediate damage (1–5). Because of their potential to serve as targets for the development of therapies aimed at reducing the effects of stress on myocardial damage, the mitogen-activated protein kinases have been the focus of many recent studies. Although all three members of the mitogen-activated protein kinase family are activated during most myocardial stresses, the roles played by each remain unclear. For example, there is strong evidence supporting both protective and damaging roles for p38 in the stressed myocardium. Several recent reviews have described this controversy (4, 6–9); thus, only a brief synopsis is presented here.
Many studies of p38 in the heart have used neonatal rat ventricular myocytes. In neonatal rat ventricular myocytes, overexpression of MKK6, an upstream activator of both the major and minor p38 isoforms in the heart, p38α and -β, respectively, was found to be protective (10–13). Moreover, dominant-negative forms of p38 or the pharmacological p38 inhibitor SB203580 blocked the protective effects of MKK6 (11). However, overexpressing a constitutively active form of MKK3, which preferentially activates p38α, augmented apoptosis (13). These results suggested that p38 can be protective or damaging, depending on the mechanism of activation and the relative levels of p38 isoform activation.
The isolated perfused heart model has also been used to study roles for p38 in preconditioning and/or mediating the damage observed during I/R. Many of these studies demonstrated that p38 is activated during ischemia and/or reperfusion (14, 15). In some reports the inhibition of p38 with SB203580 or SB202190 increased cell survival and improved functional recovery (16). However, others have shown that inhibiting p38 had either no effect or actually increased ischemic damage and reduced functional recovery (17). Although there has been speculation on the reasons for these seemingly paradoxical results (8), in the isolated perfused heart I/R model it remains unclear whether p38 damages or protects.
Several recent studies have used transgenesis or targeted gene disruption to study the function of p38 in the mouse heart in vivo. For example, although complete disruption of p38α in the heart is lethal (18–20), the hearts of p38α +/− mice, which express half the normal amount and activity of p38α, exhibit reduced myocardial injury in response to I/R (21), suggesting a negative role for p38α. In another study, overexpression of constitutively active MKK3 or MKK6 in mouse hearts resulted in cardiac abnormality but did not cause increased myocyte apoptosis (22). Finally, the hearts of mice expressing dominant-negative MKK6 or dominant-negative p38α exhibited reduced damage from I/R in an in vivo model of myocardial infarction (23), again suggesting a damaging role for p38.
In the present study we prepared transgenic mice featuring cardiac-restricted overexpression of wild type MKK6. We found that p38 was activated by about 17-fold in the MKK6 TG mouse hearts, and ventricular morphology and function were similar to non-transgenic (NTG) mouse hearts. However, when exposed to I/R, the MKK6 TG mouse hearts exhibited significantly less tissue damage and functional deficit than the NTG mouse hearts. Combined with findings in other studies, these results suggest that in the heart p38 can either protect or damage; however, the mechanisms underlying these opposing effects are yet to be determined
MATERIALS AND METHODS
Animal Studies
Approximately 200 adult mice (Mus musculus) 6–8 months of age were used in this study. All procedures involving animals were in accordance with institutional guidelines.
Preparation of Transgenic Mice
PCR products of FLAG-human-MKK6 cDNA and FLAG-human-MKK6(Glu) cDNA (R. Davis, University of Massachusetts) with 5′ NotI and 3′ KpnI sites were inserted into the αMHC vector (J. Robbins, University of Cincinnati). The αMHC-MKK6 constructs were linearized by Nru1 digestion. After introduction of the constructs by routine intranuclear injection, transgenic mice were maintained as heterozygotes in a C3H background. All experimental animals were F3 generation or later and were 5–10 months of age. MKK6(wt) and MKK6(Glu) TG mice have been observed for up to 2 years, and compared with NTG, both lines exhibit no difference in life span or any other overt phenotypic characteristic. The biochemical, histological, and physiological results obtained with the MKK6(wt) and MKK6(Glu) mice were essentially the same. Accordingly, this study reports the results from the MKK6(wt) line only.
Genotyping
DNA derived from tail biopsies was used as the template for PCR-based genotyping using the following primers. FLAG-
Molecular Characterization
Cardiac Extracts
For most analyses, mouse hearts were frozen in liquid nitrogen and then pulverized and sonicated in tissue lysis buffer (50 mM Tris, pH 7.5, 20 mM β-glycerophosphate, 250 mM NaCl, 2 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride, 10 μg/ml leupeptin, 3 mM EDTA, 3 mM EGTA, 0.1 mM sodium orthovanadate, 1 mM p-nitrophenyl phosphate, 10 μg/ml aprotinin, 0.1% Triton X-100). When p38 levels were to be assessed, the Triton X-100 in the lysis buffer was replaced with 0.5% SDS, which we found to be required for solubilization of p38β. Lysates were centrifuged at 12,000 × g for 10 min, and the supernatants were precleared with protein G-Sepharose beads for 20 min at 4 °C. The total protein concentration was determined using the DC protein assay kit (Bio-Rad, catalog #500-0112).
Cardiac Fractionation
Mouse hearts were homogenized and fractionated into cytosolic, mitochondrial, nuclear, and myofibrillar fractions by differential centrifugation, as previously described (25, 26). Molecular markers of cytosolic, mitochondrial, nuclear, and myofibrillar fractions, i.e. glyceraldehyde-3-phosphate dehydrogenase, cytochrome c oxidase IV (COX IV), c-Jun, and α-actinin, respectively, were used to validate the effectiveness of the fractionation procedure. The putative cytosolic, mitochondrial, nuclear, and myofibrillar fractions contained 93, 22, 66, and 89% of the relevant markers for these fractions (see Table I). The myofibrillar fraction, which was not used further in this study, although enriched in α-actinin as expected, also contained some COX IV, suggesting mitochondrial association with this fraction. The reason for this is not known; however, the putative mitochondrial fraction contained almost all of the remaining COX IV and very little of the other marker proteins. Accordingly, we considered the cytosolic, mitochondrial, and nuclear fractions to be enriched for proteins localized to those cellular compartments. αB-crystallin levels were assessed in each fraction as part of our study of the change in concentration of this small HSP in the mitochondrial fraction after I/R stress.
Table I. Cardiac tissue fractionation.
Hearts from 6–8-month-old mice were submitted to fractionation as described under “Materials and Methods.” Immunoblot analyses were carried out to assess the relative quantities of each subcellular marker. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
| Fraction | % of total protein or marker
|
|||||
|---|---|---|---|---|---|---|
| Protein | GAPDH | COX IV | c-Jun | α-Actinin | αBC | |
| Cytosol | 35.6 | 92.7 | 0.9 | 13 | 8.3 | 39.7 |
| Mitochondrial | 7.1 | 1.1 | 22.2 | 11 | 1.2 | 3.1 |
| Nuclear | 15.1 | 2.8 | 0.9 | 66 | 1.2 | 6.4 |
| Myofibrillar | 42.2 | 3.4 | 76.0 | 10 | 89.3 | 50.8 |
MKK6 primers were GACTATAAGGACGATGATGACAAATCTCAG (5′ primer) and CTAGTGATGTATCCATGAGCTCCATGCAG (3′ primer). β-Globin primers were CCAATCTGCTCACACAGGATAGAGAGGG-CAGG (5′ primer) and CCTTGAGGCTGTCCAAGTGATTCAGGCC-ATCG (3′ primer).
Western Analyses
Unless otherwise stated, SDS-PAGE and Western blot analyses were carried out with 50–100 μg of tissue protein using standard methods.
Antibodies and Antisera
Immunological reagents used to detect the following proteins were obtained from Cell Signaling Technologies, Beverly, MA: ERK (#9102), phospho-ERK (#9101), JNK (#9252), phospho-JNK (#9251), p38α (#9218), phospho-p38 (#9211), phospho-MKK6 antiserum (#92315). Reagents used to detect the following proteins were obtained from Santa Cruz Biotechnology, Santa Cruz, CA: c-Jun (#KM-1), cytochrome c (#sc13156), MKK6 (#sc-6073). Reagents used to detect the following proteins were obtained from Sigma: α-actinin (#A2172), FLAG M2 (#F-3165). Reagents used to detect the following proteins were obtained from Stressgen Biotechnologies Corp. (British Columbia, Canada), Molecular Probes (Eugene, OR), Research Diagnostics, Inc. (Flanders, NJ), and Zymed Laboratories Inc., San Francisco, CA, respectively: Bcl-2 (#AAM-072E), cytochrome c oxidase subunit IV, glyceraldehyde-3-phosphate dehydrogenase (#RDI-TRK564-6C5), and p38β (#33-8700).
Echocardiography
Transthoracic two-dimensional guided M-mode echocardiography was performed on acclimatized conscious mice using an HDI 5000 echocardiograph (ATL, Bothell, WA) as previously described (27).
FLAG Western Analysis and Immunoprecipitation Kinase Assays
FLAG-related material was immunoprecipitated from cardiac extracts and assessed for the ability to phosphorylate the kinase-inactive MKK6-specific substrate, p38β(K53R),2 as previously described (28).
Global Ischemia/Reperfusion
Hearts from NTG and MKK6(wt) mice were equilibrated for 30 min before being subjected to 30 min of no-flow ischemia, which was followed by reperfusion for up to 4 h, as previously described (29). Functional recovery was expressed as a percentage of pre-ischemic left ventricular developed pressure (LVDP).
Myocardial Infarctions
In vivo coronary occlusion followed by reperfusion was carried out essentially as previously described (30, 31). Myocardial infarction was produced in NTG and MKK6(wt) mice by a 30-min coronary occlusion followed by 24 h of reperfusion. At the conclusion of the study, the occluded/reperfused vascular bed and the infarct were identified by postmortem perfusion of the heart with tri-phenyltetrazolium chloride and phthalo blue dye. Infarct size was calculated by using computerized videoplanimetry.
DNA Fragmentation
DNA fragmentation was carried out as previously described (29).
Statistical Analyses
Data that unless otherwise stated are reported as the mean ± S.E. were analyzed with a one-way or two-way repeated-measure ANOVA as appropriate followed by either an unpaired Student’s t test with the Bonferroni correction (myocardial infarcts) or Newman-Keul’s post hoc analysis (all others). The relationship between infarct size and risk region size was compared among groups using an ANOVA. Phospho- and total mitogen-activated protein kinase data were analyzed using a two-tailed Student’s t test.
RESULTS
Molecular Characterization
Immunoblotting and pull-down kinase assays showed that kinase-active FLAG-MKK6 was expressed in the ventricles of the MKK6(wt) mice but not NTG littermates (Fig. 1, A and B). FLAG-MKK6 was not found in TG mouse livers or brains (data not shown). Immunoblotting demonstrated increased levels of phosphorylated MKK3/63 and total MKK6 in the TG mice (Fig. 1C).
Fig. 1. Characterization of FLAG-MKK6 expression and activity in MKK6 transgenic mice.
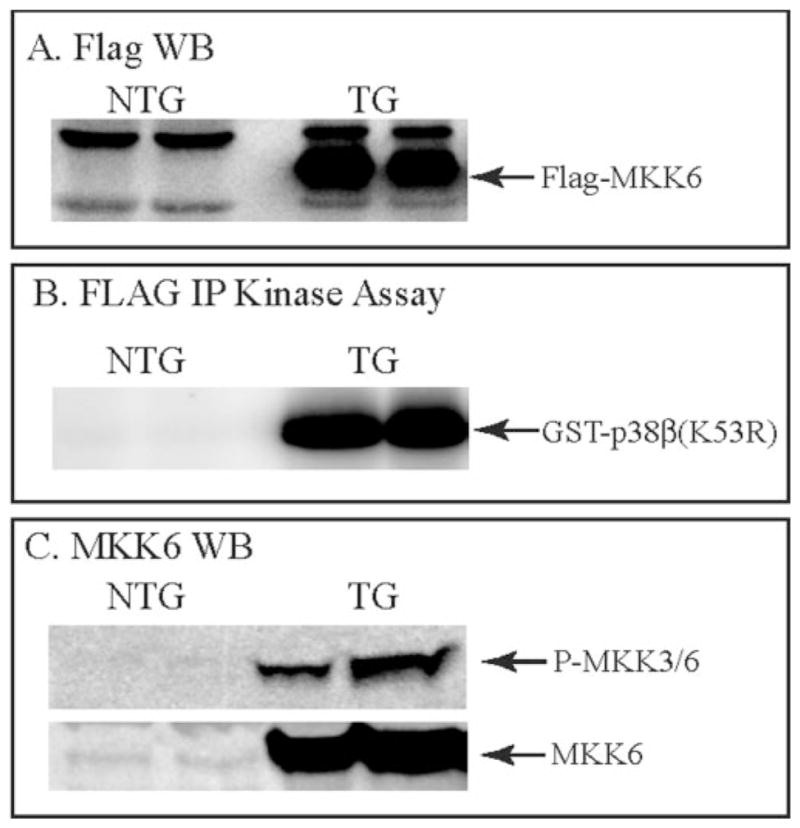
Cardiac extracts from 6–8-month-old NTG or MKK6 TG mice were assessed for FLAG-MKK6 expression (for the FLAG blot 100 and 20 μg of protein from the NTG and TG mouse heart extracts, respectively, were fractionated by SDS-PAGE) (panel A), FLAG-IP kinase activity (panel B), phosphorylated MKK6 (panel C, top), and total MKK6 (panel C, bottom), as described under “Materials and Methods.” Arrows indicate the migration positions of FLAG-MKK6, glutathione S-transferase-p38β(K53R), endogenous phospho-MKK3/6, and endogenous MKK3/6. WB, Western blot.
The effects of MKK6 overexpression on mitogen-activated protein kinase activity were assessed focusing on p38α and -β, the two major isoforms of this mitogen-activated protein kinase in the heart (22, 32, 33). Using isoform-specific antisera (Fig 2A), the relative levels of p38α and -β were determined in various mouse tissues. As expected, more p38α than -β was detected in mouse brain extracts, whereas less p38β was detected in skeletal muscle, kidney, liver, and heart (Fig. 2B).4
Fig. 2. Characterization of p38α and p38β levels.

Panel A, various amounts of recombinant glutathione S-transferase (GST)-p38α or GST-p38β were fractionated by SDS-PAGE and then blotted (WB) with antisera specific for each isoform, as shown. Panel B, extracts of various mouse tissues were analyzed for p38α and p38β expression by immunoblotting using p38α- or p38β-specific antisera. Aliquots of 100 μg of protein were loaded into each lane. sk, skeletal. Panel C, extracts of various mouse heart tissue fractions were analyzed for p38α and p38β expression by immunoblotting using p38α or p38β specific antisera. Aliquots of 100 μg of protein were loaded into each lane.
The activities of p38α and p38β were determined by phospho-p38 immunoblotting. However, because the phospho-specific antiserum cannot distinguish between p38α and p38β, which co-migrate on SDS-PAGE, mouse heart tissue was fractionated (see “Materials and Methods” and Table I) to evaluate whether these two p38 isoforms could be separated into different subcellular fractions. Most of the p38β was localized to the cytosolic fraction, with a small amount in the nuclear fraction, less in the mitochondrial fraction, and none in the myofibrillar fraction (Fig. 2C). In contrast, all of the p38β was localized to the myofibrillar fraction, which required SDS for solubilization (Fig. 2C). Accordingly, the levels of phospho- and total p38α were assessed in the cytosolic and nuclear fraction, whereas the myofibrillar fractions were used to assess phospho- and total p38β levels.
When p38β was assessed, no statistically significant difference was found between the TG and NTG mouse hearts (data not shown). However, greater differences were observed when p38α was assessed. The level of phospho-p38α was increased by 8.7-fold in the cytosol of TG mouse hearts (Fig. 3, A and B). Unexpectedly, in this same fraction the level of total (T)-p38α was decreased by 52% (Fig. 3, A and B). These results indicate that p38α was activated by 16.8-fold in the TG mice (Fig. 3C). The levels of phospho (P)- and total ERK were not significantly different in the two mouse lines (Fig. 3, A–C). P-JNK was undetectable in the cytosolic fractions in both mouse lines (Fig. 3A); however, total JNK was detectible, and although the significance is unknown compared with cyto-NTG, it was significantly reduced by 22% (Fig. 3, A and B). The results in the nuclear extracts qualitatively mirrored those in the cytosolic fractions (Fig. 3, D–F***********).
Fig. 3. Measurement of phosphorylated and total p38, ERK, and JNK.

Cytosolic and nuclear fractions of cardiac extracts were prepared and analyzed for the levels of phosphorylated (P) and total (T) p38, ERK, and JNK by immunoblotting (WB) as described under “Materials and Methods”. Panels A and D, immunoblot analyses of n = 4 NTG and 4 TG hearts from 6–8-month-old mice. MAP, mitogen-activated protein. Panels B and E, densitometry analyses of immunoblots shown in panels A and C as -fold of NTG, which was set to 1.0 for each P and T determination (mean ± S.E.). Panels C and F, P/T ratios (i.e. MAPK, MAP kinase) shown as fold of NTG, which was set to 1.0 for each determination (mean ± S.E.). For statistical analyses p < 0.05 (*) and p < 0.01 (§) different from NTG as determined by a two-tailed t test. ND, not determined.
Genetic, Morphological, and Functional Characterization
There was no significant difference in the body or left ventricle weights between the TG and NTG mice (Table II). Two-dimensional guided M-mode echocardiography indicated very little functional difference between the TG and NTG mice (Table III). Compared with NTG, the TG mice did display a lower heart rate, a mildly increased LV end-systolic dimension, and decreased % fractional shortening. The mildly reduced fractional shortening in TG mice is most likely the result of the lower resting heart rate. But as a whole, the morphological and functional evaluation showed little difference between the MKK6 TG and NTG mouse hearts. Histological examination after hematoxylin and eosin staining of paraffin-embedded sections showed no difference in the ventricular morphology or the myocyte size between the NTG and TG mice (data not shown). Moreover, the TG mice exhibited no difference in life span from NTG littermates.
Table II. Body and left ventricle weights.
Results are the mean ± S.E. of n = 10 for NTG and n = 10 for MKK6.
| Parameter | NTG | MKK6 |
|---|---|---|
| Body weight (g) | 28.2 ± 1.1 | 29.6 ± 1.1 |
| LV ( mg) | 99.5 ± 5.4 | 87.7 ± 4.7 |
Table III. Echocardiographic data.
LVEDD, LV end-diastolic dimension; LVESD, LV end-systolic dimension; % FS, % fractional shortening calculated as (LVEDD-LVESD)/LVEDD; SEPth, septal wall thickness; PWth, posterior wall thickness; mean Vcfc, heart rate corrected mean velocity of circumferential fiber shortening calculated as fractional shortening divided by ejection time multiplied by the square root of the R-R interval. Results are mean ± S.E. of n = 10 for NTG and n = 10 for MKK6.
| Parameter | NTG | MKK6 |
|---|---|---|
| Heart rate | 684 ± 12.8 | 630 ± 13.7a |
| LVEDD (mm) | 3.48 ± 0.04 | 3.64 ± 0.08 |
| LVESD (mm) | 1.22 ± 0.08 | 1.59 ± 0.11a |
| %FS | 64.9 ± 2.1 | 56.1 ± 2.9a |
| Ejection Time | 36.67 ± 0.99 | 41.67 ± 1.77 |
| Mean Vcfc(circ/s) | 5.3 ± 0.2 | 4.3 ± 0.3a |
| SEPth (mm) | 0.67 ± 0.04 | 0.66 ± 0.02 |
| PWth (mm) | 0.72 ± 0.04 | 0.73 ± 0.01 |
p < 0.05 compared to NTG.
Ischemia/Reperfusion in Vivo
To determine whether MKK6 overexpression had an effect on the response of the heart to stress, myocardial injury after I/R was assessed in an in vivo model of infarction. Mice were subjected to 30 min of coronary artery occlusion followed by 24 h of reperfusion. The region at risk and the infarcted region were delineated by triphenyltetrazolium chloride staining and pthalo blue staining. Although the region at risk, as a percentage of the left ventricular weight was similar in TG (33.1%) and NTG mice (36.8%), the infarct sizes were significantly different, representing 28.1 and 42.9% that of the region at risk in the TG and NTG mouse hearts, respectively (Table IV). These results indicate that in vivo, overexpression of MKK6 leads to a 35% reduction in infarct size in TG mice subjected to coronary occlusion.
Table IV. In vivo infarction data.
Results are shown as the mean ± S.E. of n = 10 for NTG and n = 10 for MKK6. RR, region at risk.
| Parameter | NTG | MKK6 |
|---|---|---|
| RR (mg) | 37.3 ± 3.4 | 28.1 ± 2.3 |
| Infarct size (mg) | 16.0 ± 2.2 | 7.9 ± 1.1a |
| RR (% of LV) | 36.8 ± 2.0 | 33.1 ± 3.8 |
| Infarct size (% of RR) | 42.9 ± 4.5 | 28.1 ± 3.5a |
| Infarct size (% of LV) | 16.0 ± 2.0 | 9.2 ± 1.2a |
p < 0.05 compared to NTG.
Ischemia/Reperfusion in Vitro
To determine the effects of MKK6 overexpression on myocardial functional recovery after stress, global I/R was performed, and contractile recovery was assessed via Langendorff, retrograde perfusion of isolated hearts. Hearts were submitted to 30 min of no-flow ischemia followed by reperfusion. Within 5 min of reperfusion, the TG mouse hearts exhibited about a 4-fold greater LVDP than NTG hearts (Fig. 4). At 60 min of reperfusion, recovery of LVDP in NTG and TG mouse hearts amounted to 14.9 ± 3.2 and 43.7 ± 2.6% of maximum, respectively. The enhanced LVDP recovery exhibited by the TG mouse hearts continued throughout 4 h of reperfusion. Accordingly, overexpression of MKK6 resulted in an approximate 3-fold increase in LVDP recovery in vitro.
Fig. 4. Effects of MKK6 overexpression on functional recovery after ischemia/reperfusion of isolated perfused mouse hearts.

NTG or TG hearts from 6–8-month-old mice were submitted to 30 min of equilibration (not shown) followed by 30 min of global ischemia and 240 min of reperfusion. LVDP was assessed at 30-min intervals, as described under “Materials and Methods.” Results are shown as the mean ± S.E. of n = 6 (NTG) and n = 6 (TG). For statistical analyses p < 0.05 (*) different from NTG as determined by ANOVA followed by Newman-Keul’s post hoc analysis.
α-BC and Bcl-2 Expression
There are likely to be many pathways through which MKK6 overexpression confers protection from I/R in the TG mice; one of these may involve the small heat shock proteins. Previous studies have shown that MKK6-activated p38 mediates increased expression of the protective small heat shock protein, αB-crystallin (αBC), in cultured cardiac myocytes (28). Moreover, transgenic mice that overexpress αBC in the heart exhibit myocardial protection from I/R (34), whereas the hearts from mice in which αBC expression has been disrupted are more susceptible to I/R-induced damage (29). Accordingly, we assessed αBC levels and found that the MKK6 TG mouse hearts possessed about 2-fold more αBC than NTG mouse hearts (Fig. 5A). Thus, it is possible that at least a portion of the protection observed in the MKK6 TG mouse hearts is the result of increased αBC.
Fig. 5. Effects of MKK6 Overexpression on αBC and Bcl-2.

Panel A, ventricular extracts from NTG and TG mice were fractionated by SDS-PAGE (50 μg of protein/lane) and submitted to immunoblotting (WB) using an αBC-specific antiserum. Panel B, mitochondrial fractions were isolated from hearts treated with 25 min of ischemia followed by 5 min of reperfusion (I/R) on a Langendorff apparatus and then fractionated by SDS-PAGE (50 μg of protein/lane) and submitted to immunoblotting using an αBC-specific antiserum as described under “Materials and Methods.” Panel C, cytosolic fractions were isolated from hearts treated or untreated with I/R on a Langendorff apparatus and then fractionated by SDS-PAGE (50 μg protein/lane) and submitted to immunoblotting using an αBC-specific antiserum, as described under “Materials and Methods.” Panel D, mitochondrial fractions were isolated from hearts treated or untreated with I/R on a Langendorff apparatus and then fractionated by SDS-PAGE (50 μg of protein/lane) and submitted to immunoblotting using a Bcl-2-specific antiserum, as described under “Materials and Methods.” Essentially no Bcl-2 was found in cytosolic fractions, consistent with the constitutive mitochondrial localization of this protein (41).
Further experiments were carried out to pursue a possible mechanism by which αBC might confer myocardial protection. Because the αBC-like small HSP, HSPB2, binds to mitochondria during stress (35), we assessed αBC levels in mitochondrial fractions from the TG and NTG mouse hearts. There was 2.7-fold more αBC in the mitochondrial fractions from TG mouse hearts than NTG mouse hearts (Fig. 5B, lanes 1–4). Unexpectedly, I/R induced an approximate 2–3-fold increase in the levels of mitochondrial αBC in hearts from both mouse lines (Fig. 5B, lanes 5–8) such that compared with untreated NTG mouse hearts, the quantity of αBC localized to the mitochondrial fractions in I/R-treated NTG and TG mouse hearts increased by 3.0- and 6.4-fold, respectively. I/R resulted in no change in αBC in either the cytosolic (Fig. 5C) or nuclear fractions (not shown).5 These results suggest that MKK6 over-expression fosters increased localization of αBC to the mitochondrial fractions of mouse heart extracts and that I/R stimulates a further increase in this localization. Moreover, because the levels of αBC did not change in the other fractions analyzed, this effect was specific to the mitochondrial fraction.
Another pathway that could foster cardioprotection in the MKK6 TG mice involves the anti-apoptotic protein, Bcl-2. For example, overexpression of Bcl-2 in the heart provides cardioprotection against I/R-induced damage in transgenic mice (36). Mitochondrial levels of Bcl-2 were increased by ~5–6-fold in the MKK6 TG mouse hearts (Fig. 5D); Bcl-2 levels did not change after I/R. These results are consistent with a possible role for Bcl-2 in MKK6-mediated cardioprotection in this model system.
Cytochrome c and DNA Fragmentation
Because the MKK6 mice exhibited increased levels of Bcl-2, we examined two hall marks of apoptosis, cytochrome c release and DNA fragmentation. Overall, the levels of cytosolic cytochrome c were lower in the TG than in the NTG mouse hearts (Fig. 6A, lanes 1–2 versus 5–6). Moreover, I/R induced an increase in the cytosolic levels of cytochrome c in the NTG but not the TG mouse hearts (Fig. 6A, lanes 3–4 and 7–8, and B). When DNA fragmentation was assessed we found that compared with NTG, I/R-induced DNA fragmentation was attenuated in the TG mouse hearts (Fig. 6, C and D). Taken together, these results are consistent with reduced apoptosis in MKK6 TG mouse hearts compared with NTG.
Fig. 6. Effects of MKK6 overexpression on cytochrome c release and DNA fragmentation.
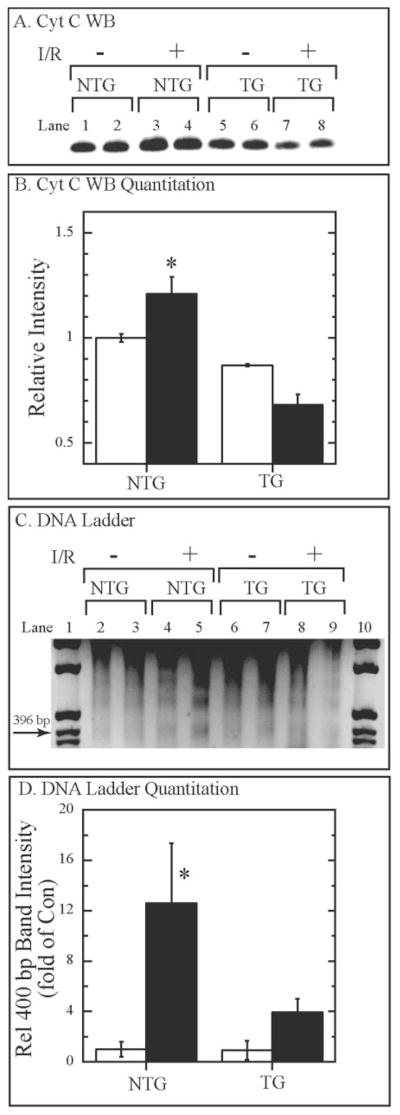
Panel A, hearts were treated with and without I/R as described in Fig. 5. Cytosolic fractions of ventricular extracts were fractionated by SDS-PAGE (50 μg of protein/lane) and then submitted to immunoblotting using a cytochrome c-specific antiserum; n = 2. Panel B, the bands in panel A were quantified by densitometry, and their relative intensities were normalized to untreated NTG mouse hearts. Results are shown as the mean ± S.D.; n = 2. *, p < 0.05 different from all other values as determined by ANOVA followed by Newman-Keul’s post hoc analysis. Panel C, hearts were treated with and without I/R, as described in Fig. 5. DNA was extracted and analyzed as previously described (29). Lanes 1 and 10 contain DNA mass markers; the location of the 396-bp marker is shown. Panel D, the intensities of the ~400-bp bands from each lane in panel C were assessed by densitometry as previously described (28), and the relative intensities of each were normalized to untreated NTG mouse hearts. *, p < 0.05 different from all other values as determined by ANOVA followed by Newman-Keul’s post hoc analysis.
DISCUSSION
This study has demonstrated that overexpression of MKK6 can be cardioprotective. Although this finding contrasts with several other recently published papers that manipulate p38 signaling components in genetically altered mice, it underscores the possibility that in the heart p38 can have protective or damaging effects, most likely depending on the conditions of its activation.
There are many conceivable mechanisms by which MKK6 might confer protection against I/R stress, one of which involves small heat shock proteins, such as αBC. MKK6 overexpression leads to increased αBC expression in cultured cardiac myocytes (28), and αBC overexpression has been shown to protect stressed cardiac myocytes in vitro (37) and stressed hearts in vivo (29, 34). Although it is not known how αBC exerts these protective effects in the heart, in many cell types αBC is known to reduce apoptosis (29, 37, 38), which is responsible for a portion of I/R-induced myocardial damage (39, 40). There is also evidence indicating that small heat shock proteins also function as a cytoprotectant at the mitochondrial level, although the details are not yet understood. For example, HSBP2, an αBC-like small HSP, has been shown to translocate to the mitochondrial outer membrane during stress (35). In the present study we found that the level of HSPB2 in the mitochondrial fraction was increased in TG mouse hearts compared with NTG mouse hearts (data not shown). Moreover, we found that after I/R stress, the quantity of αBC in the mitochondrial fraction increased by as much as 3-fold in NTG mouse hearts and by more than 6-fold in MKK6 TG mouse hearts (Fig. 5). Thus, in analogy to HSPB2, this may represent I/R-induced translocation of αBC to mitochondria from other regions of the cell that serve as reservoirs for αBC (e.g. cytosol). To our knowledge this is the first demonstration of I/R-induced increase of αBC in a mitochondrial fraction in any tissue.
There are other mechanisms by which MKK6 might confer myocardial protection. For example, Bcl-2, a well known regulator of the mitochondrial apoptosis pathway (41), which we found to be increased in MKK6 TG mouse hearts (Fig. 5), fosters cardioprotection from I/R injury in TG mice (36). Also, NFκB, which is known to induce expression of several cardioprotective genes, such as inducible nitric-oxide synthase, cyclooxygenase-2, superoxide dismutase, and interleukin-6 (13, 42–44), might confer cardioprotection in MKK6 mice. It is interesting to note that Bcl-2 fosters NFκB activation in cardiac myocytes (45), providing a potential linkage in the mechanisms by which these two signaling proteins mediate protection.
In contrast to the present study, some reports suggest that p38 damages cardiac myocytes and the myocardium. How can these results be reconciled with those of the present study? Evidence is mounting that like JNK, p38 can mediate cellular protection or damage depending on conditions such as the tissue or cell type under study, the mechanism of activation, and the isoforms that are activated (46, 47). For example, in neonatal rat ventricular myocytes and in human fibroblasts, p38 activation by MKK3 or MKK6 results in apoptosis or protection, respectively (13, 22, 48). And although overexpression of constitutively active MKK3 resulted in lethal heart malformation, overexpression of constitutively active MKK6 resulted in preserved ventricular morphology and no apoptosis (9, 22).
Presumably, MKK3 and MKK6 foster different outcomes at least in part by differentially activating p38 isoforms, which themselves must have different targets with different functions. The major isoforms of p38 in the heart are p38α and -β, although as shown in the present study, the levels of p38α in the mouse heart are considerably greater than p38β. Of these isoforms, MKK3 activates primarily p38α, whereas MKK6 activates both p38α and -β (49). Thus, it is possible that MKK3-mediated p38β activation leads to cellular damage, whereas MKK6-mediated p38α and -β activation leads to protection. This hypothesis is supported by a previous study in neonatal rat ventricular myocytes which shows that dominant negative p38α fosters protection, whereas dominant negative p38β leads to increased damage (13). In the present study we observed very little increase in the level of p38β activation in the MKK6 TG mice, which may at first seem inconsistent with the p38α to -β activation ratio hypothesis. However, when coupled with our finding of reduced levels of total p38α expression in the MKK6 TG mouse hearts, perhaps a p38α to -β activation ratio that is conducive to protection is achieved.
The relative levels of ERK, JNK, and p38 may also contribute to determining the cellular outcome of p38 activation (50, 51). For example, it is conceivable that high ERK to p38 ratios promote cell survival. Consistent with this hypothesis is a study which demonstrates that in addition to activating p38, MKK3 decreases MEK phosphorylation, which leads to a reduction in basal ERK activity (48). In that same study it was shown that by reducing the ERK to p38 activity ratio, MKK3 promoted apoptosis, whereas MKK6, which had no effect on ERK activity, retained an ERK to p38 ratio that was conducive to cell survival. Thus, even though MKK6 does not activate ERK, in order for it to promote cell survival, it is probable that basal ERK activity levels must be maintained. In the present study, although we found that basal ERK activity was unaffected by MKK6 overexpression in transgenic mouse hearts, consistent with the report in fibroblasts (48), we did observe an approximate 50% decrease in the total level of p38α in the MKK6 overexpressing TG mouse hearts. Although the mechanism and functional impact of this decrease remain to be determined, it may represent a compensatory response, which may foster an overall mitogen-activated protein kinase balance favoring cell survival. Consistent with this possibility is a previous study which showed a regulatory linkage between MKK6 and p38α where decreased p38α induced dramatic increases in active MKK6 (24), indicating that p38α can influence MKK6 expression levels. Although untested, it seems plausible that the reciprocal might also be true, that MKK6 may influence p38α levels so as to maintain the activity of p38 in a range conducive to appropriate cellular responses.
In summary, this study showed that overexpression of MKK6 in the hearts of TG mice had no apparent effect under non-stressful conditions; however, upon I/R stress, MKK6 TG mice displayed less myocardial damage and enhanced functional recovery. Further studies will be necessary to understand the molecular mechanisms underlying these apparent conditional effects of p38 in the heart.
Acknowledgments
We thank Holly Hoover and Donna Thuerauf for preparation of some of the DNA constructs.
Footnotes
This work was supported in part by National Institutes of Health Grants HL-63975, NS/HL-25037, and HL-75573 (to C. C. G.), HL-46345 (to H. A. R.), and HL-55757, HL-68088, and HL-70897 (to R. B.). This work was also supported in part by the Jewish Hospital Research Foundation, Louisville, KY.
The abbreviations used are: I/R, ischemia followed by reperfusion; HSP, heat shock protein; TG, transgenic; NTG, non-transgenic; ERK, extracellular signal-regulated kinase; JNK, c-Jun NH2-terminal kinase; LVDP, left ventricular (LV) developed pressure; ANOVA, analysis of variance; αBC, αB-crystallin; MKK, mitogen-activated protein kinase kinase; COX, cytochrome c oxidase.
The kinase-inactive form of p38 was used as an MKK6 substrate because we have found that using p38β results in high levels of auto-phosphorylation of p38, which increases the assay background (10).
The phospho-specific antiserum used for these experiments does not distinguish between MKK3 and MKK6; however, the total antiserum is MKK6-specific.
Densitometric analyses indicated that p38α is about 4–5-fold more abundant than p38β in the mouse heart.
It is not known whether the increase in mitochondrial αBC upon I/R is the result of αBC translocation from the cytosolic fraction. However, because there is ~10-fold more αBC in the cytosolic fraction than in the mitochondrial fraction (Table I), the lack of observed change in the cytosolic αBC levels upon I/R is still consistent with the cytosol as the source of the increase in mitochondrial αBC.
References
- 1.Bueno OF, Molkentin JD. Circ Res. 2002;91:776–781. doi: 10.1161/01.res.0000038488.38975.1a. [DOI] [PubMed] [Google Scholar]
- 2.Wang Y. Curr Opin Pharmacol. 2001;1:134–140. doi: 10.1016/s1471-4892(01)00029-7. [DOI] [PubMed] [Google Scholar]
- 3.Jones WK, Brown M, Ren X, He S, McGuinness M. Cardiovasc Toxicol. 2003;3:229–254. doi: 10.1385/ct:3:3:229. [DOI] [PubMed] [Google Scholar]
- 4.Liang Q, Molkentin JD. J Mol Cell Cardiol. 2003;35:1385–1394. doi: 10.1016/j.yjmcc.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 5.Sugden PH, Clerk A. Circ Res. 1998;83:345–352. doi: 10.1161/01.res.83.4.345. [DOI] [PubMed] [Google Scholar]
- 6.Ravingerova T, Barancik M, Strniskova M. Mol Cell Biochem. 2003;247:127–138. doi: 10.1023/a:1024119224033. [DOI] [PubMed] [Google Scholar]
- 7.Abe J, Baines CP, Berk BC. Circ Res. 2000;86:607–609. doi: 10.1161/01.res.86.6.607. [DOI] [PubMed] [Google Scholar]
- 8.Ping P, Murphy E. Circ Res. 2000;86:921–922. doi: 10.1161/01.res.86.9.921. [DOI] [PubMed] [Google Scholar]
- 9.Petrich BG, Wang Y. Trends Cardiovasc Med. 2004;14:50–55. doi: 10.1016/j.tcm.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 10.Zechner D, Thuerauf DJ, Hanford DS, McDonough PM, Glembotski CC. J Cell Biol. 1997;139:115–127. doi: 10.1083/jcb.139.1.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zechner D, Craig R, Hanford DS, McDonough PM, Sabbadini RA, Glembotski CC. J Biol Chem. 1998;273:8232–8239. doi: 10.1074/jbc.273.14.8232. [DOI] [PubMed] [Google Scholar]
- 12.Nemoto S, Sheng Z, Lin A. Mol Cell Biol. 1998;18:3518–3526. doi: 10.1128/mcb.18.6.3518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang Y, Huang S, Sah VP, Ross J, Jr, Brown JH, Han J, Chien KR. J Biol Chem. 1998;273:2161–2168. doi: 10.1074/jbc.273.4.2161. [DOI] [PubMed] [Google Scholar]
- 14.Marais E, Genade S, Huisamen B, Strijdom JG, Moolman JA, Lochner A. J Mol Cell Cardiol. 2001;33:769–778. doi: 10.1006/jmcc.2001.1347. [DOI] [PubMed] [Google Scholar]
- 15.Tanno M, Bassi R, Gorog DA, Saurin AT, Jiang J, Heads RJ, Martin JL, Davis RJ, Flavell RA, Marber MS. Circ Res. 2003;93:254–261. doi: 10.1161/01.RES.0000083490.43943.85. [DOI] [PubMed] [Google Scholar]
- 16.Sato M, Cordis GA, Maulik N, Das DK. Am J Physiol Heart Circ Physiol. 2000;279:901–907. doi: 10.1152/ajpheart.2000.279.3.H901. [DOI] [PubMed] [Google Scholar]
- 17.Mocanu MM, Baxter GF, Yue Y, Critz SD, Yellon DM. Basic Res Cardiol. 2000;95:472–478. doi: 10.1007/s003950070023. [DOI] [PubMed] [Google Scholar]
- 18.Adams RH, Porras A, Alonso G, Jones M, Vintersten K, Panelli S, Valladares A, Perez L, Klein R, Nebreda AR. Mol Cell. 2000;6:109–116. [PubMed] [Google Scholar]
- 19.Wu Z, Woodring PJ, Bhakta KS, Tamura K, Wen F, Feramisco JR, Karin M, Wang JY, Puri PL. Mol Cell Biol. 2000;20:3951–3964. doi: 10.1128/mcb.20.11.3951-3964.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tamura K, Sudo T, Senftleben U, Dadak AM, Johnson R, Karin M. Cell. 2000;102:221–231. doi: 10.1016/s0092-8674(00)00027-1. [DOI] [PubMed] [Google Scholar]
- 21.Otsu K, Yamashita N, Nishida K, Hirotani S, Yamaguchi O, Watanabe T, Hikoso S, Higuchi Y, Matsumura Y, Maruyama M, Sudo T, Osada H, Hori M. Biochem Biophys Res Commun. 2003;302:56–60. doi: 10.1016/s0006-291x(03)00096-2. [DOI] [PubMed] [Google Scholar]
- 22.Liao P, Georgakopoulos D, Kovacs A, Zheng M, Lerner D, Pu H, Saffitz J, Chien K, Xiao RP, Kass DA, Wang Y. Proc Natl Acad Sci U S A. 2001;98:12283–12288. doi: 10.1073/pnas.211086598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kaiser RA, Bueno OF, Lips DJ, Doevendans PA, Jones F, Kimball TF, Molkentin JD. J Biol Chem. 2004;279:15524–15530. doi: 10.1074/jbc.M313717200. [DOI] [PubMed] [Google Scholar]
- 24.Ambrosino C, Mace G, Galban S, Fritsch C, Vintersten K, Black E, Gorospe M, Nebreda AR. Mol Cell Biol. 2003;23:370–381. doi: 10.1128/MCB.23.1.370-381.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baines CP, Zhang J, Wang GW, Zheng YT, Xiu JX, Cardwell EM, Bolli R, Ping P. Circ Res. 2002;90:390–397. doi: 10.1161/01.res.0000012702.90501.8d. [DOI] [PubMed] [Google Scholar]
- 26.Mizukami Y, Yoshida K. Biochem J. 1997;323:785–790. doi: 10.1042/bj3230785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Esposito G, Santana LF, Dilly K, Cruz JD, Mao L, Lederer WJ, Rockman HA. Am J Physiol Heart Circ Physiol. 2000;279:3101–3112. doi: 10.1152/ajpheart.2000.279.6.H3101. [DOI] [PubMed] [Google Scholar]
- 28.Hoover HE, Thuerauf DJ, Martindale JJ, Glembotski CC. J Biol Chem. 2000;275:23825–23833. doi: 10.1074/jbc.M003864200. [DOI] [PubMed] [Google Scholar]
- 29.Morrison LE, Whittaker RJ, Klepper RE, Wawrousek EF, Glembotski CC. Am J Physiol Heart Circ Physiol. 2004;286:847–855. doi: 10.1152/ajpheart.00715.2003. [DOI] [PubMed] [Google Scholar]
- 30.Guo Y, Wu WJ, Qiu Y, Tang XL, Yang Z, Bolli R. Am J Physiol. 1998;275:H1375–H1387. doi: 10.1152/ajpheart.1998.275.4.H1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guo Y, Jones WK, Xuan YT, Tang XL, Bao W, Wu WJ, Han H, Laubach VE, Ping P, Yang Z, Qiu Y, Bolli R. Proc Natl Acad Sci U S A. 1999;96:11507–11512. doi: 10.1073/pnas.96.20.11507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jiang Y, Gram H, Zhao M, New L, Gu J, Feng L, Di Padova F, Ulevitch RJ, Han J. J Biol Chem. 1997;272:30122–30128. doi: 10.1074/jbc.272.48.30122. [DOI] [PubMed] [Google Scholar]
- 33.Lemke LE, Bloem LJ, Fouts R, Esterman M, Sandusky G, Vlahos CJ. J Mol Cell Cardiol. 2001;33:1527–1540. doi: 10.1006/jmcc.2001.1415. [DOI] [PubMed] [Google Scholar]
- 34.Ray PS, Martin JL, Swanson EA, Otani H, Dillmann WH, Das DK. FASEB J. 2001;15:393–402. doi: 10.1096/fj.00-0199com. [DOI] [PubMed] [Google Scholar]
- 35.Nakagawa M, Tsujimoto N, Nakagawa H, Iwaki T, Fukumaki Y, Iwaki A. Exp Cell Res. 2001;271:161–168. doi: 10.1006/excr.2001.5362. [DOI] [PubMed] [Google Scholar]
- 36.Chen Z, Chua CC, Ho YS, Hamdy RC, Chua BH. Am J Physiol Heart Circ Physiol. 2001;280:2313–2320. doi: 10.1152/ajpheart.2001.280.5.H2313. [DOI] [PubMed] [Google Scholar]
- 37.Morrison LE, Hoover HE, Thuerauf DJ, Glembotski CC. Circ Res. 2003;92:203–211. doi: 10.1161/01.res.0000052989.83995.a5. [DOI] [PubMed] [Google Scholar]
- 38.Kamradt MC, Chen F, Sam S, Cryns VL. J Biol Chem. 2002;277:38731–38736. doi: 10.1074/jbc.M201770200. [DOI] [PubMed] [Google Scholar]
- 39.Gottlieb RA, Burleson KO, Kloner RA, Babior BM, Engler RL. J Clin Investig. 1994;94:1621–1628. doi: 10.1172/JCI117504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kajstura J, Cheng W, Reiss K, Clark WA, Sonnenblick EH, Krajewski S, Reed JC, Olivetti G, Anversa P. Lab Investig. 1996;74:86–107. [PubMed] [Google Scholar]
- 41.Green DR, Kroemer G. Science. 2004;305:626–629. doi: 10.1126/science.1099320. [DOI] [PubMed] [Google Scholar]
- 42.Craig R, Wagner M, McCardle T, Craig AG, Glembotski CC. J Biol Chem. 2001;276:37621–37629. doi: 10.1074/jbc.M103276200. [DOI] [PubMed] [Google Scholar]
- 43.Craig R, Larkin A, Mingo AM, Thuerauf DJ, Andrews C, McDonough PM, Glembotski CC. J Biol Chem. 2000;275:23814–23824. doi: 10.1074/jbc.M909695199. [DOI] [PubMed] [Google Scholar]
- 44.Degousee N, Martindale J, Stefanski E, Cieslak M, Lindsay TF, Fish JE, Marsden PA, Thuerauf DJ, Glembotski CC, Rubin BB. Circ Res. 2003;92:757–764. doi: 10.1161/01.RES.0000067929.01404.03. [DOI] [PubMed] [Google Scholar]
- 45.Regula KM, Ens K, Kirshenbaum LA. J Biol Chem. 2002;277:38676–38682. doi: 10.1074/jbc.M206175200. [DOI] [PubMed] [Google Scholar]
- 46.Wada T, Penninger JM. Oncogene. 2004;23:2838–2849. doi: 10.1038/sj.onc.1207556. [DOI] [PubMed] [Google Scholar]
- 47.Nebreda AR, Porras A. Trends Biochem Sci. 2000;25:257–260. doi: 10.1016/s0968-0004(00)01595-4. [DOI] [PubMed] [Google Scholar]
- 48.Li SP, Junttila MR, Han J, Kahari VM, Westermarck J. Cancer Res. 2003;63:3473–3477. [PubMed] [Google Scholar]
- 49.Enslen H, Raingeaud J, Davis RJ. J Biol Chem. 1998;273:1741–1748. doi: 10.1074/jbc.273.3.1741. [DOI] [PubMed] [Google Scholar]
- 50.Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- 51.Ballif BA, Blenis J. Cell Growth Differ. 2001;12:397–408. [PubMed] [Google Scholar]