

Abstract
Activity-based probes (ABPs) are reactive small molecules that covalently bind to active enzymes. When tagged with a fluorophore, ABPs serve as powerful tools to investigate enzymatic activity across a wide variety of applications. In this article, we will provide detailed protocols for using fluorescent ABPs to biochemically characterize the activity of proteases in vitro. Furthermore, we will describe how these probes can be applied to image protease activity in live animals and tissues along with subsequent analysis by histology, flow cytometry, and SDS-PAGE.
Keywords: proteases, activity-based probes, imaging, caspases
INTRODUCTION
Proteases are a large family of enzymes that facilitate the degradation and turnover of proteins within the cell. In addition, they function as tight regulators of numerous signaling pathways responsible for processes such as apoptosis, cell migration, blood coagulation, and antigen presentation, among many others. Proteases also play important roles in the pathogenesis of many diseases, including cancer, atherosclerosis, arthritis, stroke, Alzheimer’s Disease, and Huntington’s Disease.
Proteases function by hydrolyzing peptide bonds of protein substrates and are classified according to their mechanism of catalysis. Cysteine, serine, and threonine proteases use an amino acid to nucleophilically attack the amide bond. Aspartic, glutamic, and metallo proteases use their catalytic amino acid to deprotonate a water molecule for substrate hydrolysis. (For further mechanistic information, see review by Deu et al, 2012) Proteolysis is tightly regulated in a temporal and spatial manner through several mechanisms. The enzymes are synthesized as inactive zymogens that become activated through structural rearrangements mediated by changes in pH, dimerization, or cleavage by upstream proteases (Deu et al., 2012; Edgington et al., 2011; Serim et al., 2012). Once activated, proteases may also be inhibited by endogenous proteins within the cell. As a result, measurements of total protein expression do not often reflect levels of protease activity. To study the function of proteases in normal cellular processes and disease states, tools to directly assess their activity are necessary.
One class of tools that has been developed to meet this need is activity-based probes (ABPs). ABPs are small molecules that bind specifically and irreversibly to active proteases (Deu et al., 2012; Edgington et al., 2011; Serim et al., 2012). Fluorescent ABPs contain a dye that allows for detection and are thus amenable to a wide variety of applications. Probe labeling can be assessed biochemically by SDS-PAGE analysis or by fluorescent imaging in live animals or excised tissues. Furthermore, ABPs can be used to study the localization of active proteases in complex tissues by microscopy as well as in single cells by flow cytometry. In this article, we will describe detailed protocols to study protease activity using ABPs both in vitro and in vivo. Basic Protocol 1 describes a procedure for labeling cell lysates with ABPs while Support Protocol 1 gives instruction on how to perform fluorescent SDS-PAGE to visualize labeled proteases. Basic Protocol 2 describes how to label proteases in live cells. Basic Protocol 3 describes a method to identify ABP targets using an immunoprecipitation assay. Furthermore, we describe the application of ABPs to image protease activity in live mice (Basic Protocol 4) and whole tissues (Basic Protocol 5). We then provide protocols for further ex vivo analysis of ABP-labeled tissues biochemically (Basic Protocol 6), histologically (Basic Protocol 7), and by flow cytometry (Basic Protocol 8).
As animal models designed to recapitulate human disease continue to improve, non-invasive imaging techniques will continue to become more powerful. The ability to monitor an enzyme’s activity in its natural environment can lend significant insight into its disease-promoting functions and aid in the development of therapeutics. Moreover, many proteases have been identified as biomarkers of disease, and functional imaging with activity-based probes has great diagnostic and prognostic potential.
STRATEGIC PLANNING
Of all the classes of proteases, fluorescent ABPs have been developed most broadly for cysteine proteases, including caspases (Edgington et al., 2009; Edgington et al., 2012; Li et al., 2012), cathepsins (Blum et al., 2005; Blum et al., 2007; Gocheva et al., 2010; Joyce et al., 2004; Verdoes et al., 2012), and legumain (Lee and Bogyo, 2010). Probes for the threonine-dependent catalytic subunits of the proteasome also exist (Verdoes et al., 2006), as well for serine proteases (Liu et al., 1999). When planning experiments with ABPs, choose a probe for the protease of interest that has been well characterized. Data regarding dosing/potency, cell permeability, in vivo circulation, off-targets, etc. will be critical to the success of these experiments. If this is a new probe or such data is not available, be prepared to characterize it. The protocols below are written for testing a new probe, so some steps may be skipped if the appropriate characterization data is already available.
Proteases are involved in broad processes and many different biological models may be used to study their function. In some models, proteases are constitutively active while in others they are induced over time by outside stimuli. We could not possibly begin to address each model in this article. Therefore, as an example, we will describe protocols and advice for detecting caspase activity during apoptosis, a highly regulated form of programmed cell death. In general, apoptosis models involve induction of caspase activity through administration of drugs or other stressors. The protocols presented may be easily adapted to detect other proteases simply by changing stimulation methods and buffer conditions. In any case, the more information that is known regarding the timing and localization of protease activation in the preferred model, the better the chances of obtaining success in these protocols, particularly with in vivo imaging. Be prepared for several rounds of optimization, especially if this is a new model.
BASIC PROTOCOL 1
Labeling cell lysates with ABPs
This protocol describes the most basic assay for activity-based labeling of caspases in a complex proteome. Labeling cell lysates will provide a snapshot of the caspase activity at the time the cells are harvested. The timing and degree of caspase activation will depend on the method used to stimulate apoptosis. If this information is not readily available in the literature for the cell line used, a time course and/or dose curve may be performed to determine optimal conditions for caspase activation. There are several advantages of labeling lysates over intact cells. 1) Less probe is required, which may be beneficial if the ABP is of limited supply. This is especially helpful during the early optimization steps of characterizing a new drug/stressor/cell line, etc. 2) Labeling lysates does not require that the ABP be cell permeable; therefore, it is more likely that the total pool of active caspases will be labeled rather than a subset. 3) This assay detects caspases at neutral pH, making it less likely to detect common off-targets of caspase ABPs such as legumain or cathepsins, which are active in the acidic conditions of the lysosome.
Materials
Cells (chosen based on model)
6-well plates
Cell media
Cold PBS
Hypotonic Lysis Buffer (see recipe)
BCA kit or equivalent
ABP (100x stock solution in DMSO)
4x sample buffer
Prepare protein lysates
-
1.
Seed cells in 6-well dishes at 80% confluency (depending on the size of the cells, this will be about 1–1.5 × 106 cells).
-
2.
The following day, refresh media and stimulate apoptosis/caspase activity by preferred method. ‘No-treatment’ controls are also useful to include.
-
3.
Harvest the cells at the optimal time point. In cell death models, some of the cells may have already detached and will be floating in the media. The remaining cells can be dislodged by using cell scrapers or the broad end of a P200 pipette tip.
-
4.
Transfer the cells in media to an eppendorf tube and spin using an eppendorf tabletop centrifuge at 1,000 × g for 1–2 minutes.
-
5.
Aspirate media with care, so as not to disturb the cell pellet and then add 1 mL cold PBS to the cells. Pipette 2–3 times to wash the pellet. Keep tubes on ice from this point to preserve caspase activity.
-
6.
Centrifuge as in step 5 and aspirate PBS. Then add 50 µL of hypotonic lysis buffer, resuspend pellet, and incubate on ice for 10 min. Note: lysis buffer may be adjusted depending on the preference of the protease. For example, legumain and cathepsins prefer a low pH buffer (see recipe).
-
7.
At this point, the sample may be snap frozen in liquid nitrogen and stored at −20°C for later use, or be advanced to the next step.
-
8.
Spin at high speed in a tabletop centrifuge at 4°C for ~15 minutes and then transfer supernatant to a fresh eppendorf tube.
-
9.
Quantify total protein concentration using a BCA kit (or comparable). Sample will likely need to be diluted (typically 1:10). Remember to account for DTT in the buffer by making protein standards in hypotonic lysis buffer at the same dilution as the samples.
Label lysate with ABP
-
10.
Aliquot 50–100 µg lysate into a fresh tube, bringing the volume to 30 µl with hypotonic lysis buffer. (Depending on the size of the wells of the gel used for SDS-PAGE, this volume may need to be decreased.)
-
11.
Add 0.3 µl of a 100x DMSO stock of the ABP (i.e. for 1 µM final concentration, use 100 µM DMSO stock solution. Final DMSO concentration should be no more than 2%). Probe concentration varies by molecule; a dose curve will aid in optimizing if this information is not already available. Note: it is highly recommended to run a ‘no-probe’ control in parallel to account for any background/autofluorescent bands that may appear in the gel. If a protease inhibitor is used to validate the selectivity of the probe, add it before the probe in the same manner as probe addition. The length of the pre-incubation time will vary by inhibitor.
-
12.
Incubate at 37°C for ~30 minutes. Labeling time may vary by ABP.
-
13.
Add 10 µl 4x sample buffer to quench the reaction. Samples may be snap frozen in liquid nitrogen and stored at −20°C or directly analyzed by SDS-PAGE (see Support Protocol 1).
SUPPORT PROTOCOL 1
Fluorescent SDS-PAGE analysis
This support protocol describes how to visualize the proteases that were labeled in Basic Protocols 1, 2, 3, and 6. SDS-PAGE is a widely used technique; however, exact parameters vary by lab. Outlined here are tips that have worked well to detect caspase labeling, but other conditions may work as well.
Materials
Probe-labeled lysate
SDS-PAGE gels (15% acrylamide is ideal for caspase separation)
Gel running apparatus
Appropriate running buffers (tris-glycine works well)
Protein ladders (both colored and fluorescent), such as Seeblue Plus2 prestained marker
(Invitrogen) and ECL Plex Fluorescent Rainbow Markers (Amersham)
Special equipment
Typhoon flatbed laser scanner or comparable device that detects wavelengths appropriate for the probe’s fluorophore
Prepare SDS-PAGE gel for running according to standard protocols (add loading buffer).
Boil protein samples for 5–10 minutes. Then load samples, along with protein ladders on the gel.
Run gel according to standard protocol. For caspases, we have had the most success with running gels at 80 volts until the dye front enters the resolving gel, then turning it up to 120 volts. Running longer typically gives better resolution of the caspase bands, but this depends on the ABP used. It is best to run it long enough that the free, unbound probe runs off the gel, but not so long that the caspases run off, too. We use Seeblue Plus2 prestained marker (Invitrogen) and stop the gel when the pink band is 2 cm from the bottom.
Scan the gel on a Typhoon scanner (or comparable) at the wavelength appropriate for the fluorophore used. For this step, the gel can often be left in the glass plates (once they are cleaned and dried). In this case, the scan depth must be changed from ‘platen’ to +3mm. If the gel is removed from the plates for the scan, rinse it with water to avoid tears and do not touch the gel with ungloved fingers (this causes autofluorescence), and use the platen setting. The PMT setting should be adjusted such that the scan is not overexposed or may be increased to boost signal. Mature, active caspases should appear in the 16–21 kDa range.
At this point, the proteins on the gel may be transferred to a membrane and blotted according to standard western protocols. This is a good way to compare protease activity with total expression.
BASIC PROTOCOL 2
Labeling intact cells with ABPs
Once the optimal conditions for stimulating caspase activity have been determined using lysates, the next step is to label live, intact cells. This protocol depends on the ability of the ABP to freely penetrate cells and enter the cytoplasm (in the case of caspases). If the probe cannot reach its target, it will not label it. Furthermore, one of the major caveats of caspase probes is their varied propensity to label the lysosomal proteases (cathepsins and/or legumain) at acidic pH due to similarities in structure and mechanism of catalysis (Edgington et al., 2009; Edgington et al., 2012). If the probe enters the cell by endocytosis or macropinocytosis, it may pass through the lysosome before reaching the cytosol, labeling these off-targets in addition to caspases. This must be kept in mind when analyzing data. For these reasons, labeling live cells will often give you a more accurate prediction of the labeling profile that will be obtained in vivo.
Materials
Cells (chosen based on model)
Media
Stimulant
Hypotonic Lysis Buffer (see recipe)
ABP (1000x stock solution in DMSO)
Cold PBS
4x sample buffer
Seed cells in 6-well dishes to 80% confluence (depending on the size of the cells, this will be about 1–1.5 × 106 cells).
The following day, refresh media and stimulate apoptosis/caspase activity by preferred method. ‘No-treatment’ controls are also useful to include. To economize probe, it is best if this step is performed in 1 mL media.
The time at which the probe is added to the cells will vary by experiment and probe. To obtain a more cumulative picture of the caspases that become activated over time, the probe can be added as soon as the apoptosis stimulus is added and incubated throughout the course of the experiment. (If the time course is longer than ~4 hours, fresh probe may need to be added, depending on probe stability and the concentration used.) To obtain a snapshot of the particular caspases that are active at a given time point after stimulation, add the probe during the desired window. To do so, add 1 µl of a 1000x DMSO stock solution of ABP directly to the media and mix well. (Use 1 µl of 1 mM stock to make a final concentration of 1 µM. The final DMSO concentration should be no more than 0.2%). As with lysates, the probe concentration will need to be optimized if it has not been already. For most caspase probes, a 30-minute incubation will be more than sufficient, and labeling may be observed after as little as 5 minutes. Timing should be optimized based on the probe and experimental design.
Various protease inhibitors may be used to validate the selectivity of the ABP. In this case, the inhibitor should be added before the addition of the probe. The pre-incubation time and dose will vary by inhibitor.
Harvest the cells at the optimal time point after probe addition (shorter incubation for snapshots, longer for cumulative pictures) by detaching them from the dish. This can be achieved using cell scrapers or the broad end of a P200 pipette tip. Collect the cells and media and pellet the cells by centrifuging at 1,000 × g for 1–2 minutes. Then carefully aspirate the media and wash once with cold PBS, making sure to resuspend the pellet. Remove the PBS. Keep the samples on ice when not in use.
Resuspend the pellet in 50 µl hypotonic lysis buffer and incubate on ice for ten minutes. At this point, the sample can be snap frozen in liquid nitrogen and stored at −20°C, or the protocol may be continued.
Centrifuge at high speed on a tabletop centrifuge at 4°C for 15 minutes. Transfer the supernatant to a fresh eppendorf tube. Reserve 5 µl for protein quantification by BCA (see Basic Protocol 1 step 9). To the remaining 45 µl, add 15 µl 4x sample buffer. The sample may then be frozen at −20°C, or boiled and loaded onto a gel according to Supporting Protocol 1.
BASIC PROTOCOL 3
Immunoprecipitations with APBs to confirm targets
Once ABP-labeled species have been visualized by SDS-PAGE, it may be necessary to confirm their identity. This can usually be achieved by pre-treating samples with selective inhibitors to compete for probe labeling; however, in the case of caspases, truly selective inhibitors do not exist. They tend to react broadly with members of the caspase family as well as lysosomal cysteine proteases like legumain and cathepsins. For this reason, immunoprecipitation may be the ideal way to validate probe-labeled species. If enough material remains, the sample used for SDS-PAGE analysis may be used directly in the immunoprecipitation assay.
Materials
Probe-labeled lysate
Protein A/G agarose beads
Specific antibody towards protease of interest
IP Buffer
0.9% Sodium Chloride
Acetone
Sample buffer
Reserve 50 µg of probe-labeled protein lysate to load as an input sample (or the original amount loaded on the first gel). Store at −20°C.
For the immunoprecipitation, aliquot probe-labeled lysate into an eppendorf tube (100+ µg total protein, depending on intensity of labeling in the previous gel. If weak, use more). It is usually acceptable if the sample has already been boiled in sample buffer, though there may be exceptions. For caspases, the IPs generally work better on denatured samples.
Dilute lysate with 500 µl IP Buffer. Add 5–10 µl antibody and incubate on ice for 10 min. This antibody should be carefully chosen such that it binds selectively to the protease of interest. It is critical that the antibody be raised towards the subunit to which the probe binds. In the case of caspases, the probes bind to the active site cysteine, which is found within the large subunit.
Aliquot a 40-µl slurry of protein A/G agarose beads. Add 500 µl IP buffer to wash beads and centrifuge at high speed on a table top centrifuge for 30 seconds. Remove buffer, add 50 µl fresh IP buffer, and transfer beads to the tube containing the protein + antibody. Nutate tube overnight at 4°C.
The next morning, transfer supernatant to a 2-mL eppendorf tube. To precipitate and concentrate the proteins in the supernatant, add enough acetone to fill the tube and freeze at −80°C for at least two hours. Then spin the tube at high speed at 4°C for 15 minutes. Aspirate the acetone, taking great care not to disturb the protein pellet. Allow the pellet to dry completely before dissolving it in 30 µl of 1x sample buffer.
While the supernatant is precipitating, wash the agarose beads four times with IP buffer and then once with 0.9% sodium chloride. After the last wash, remove all of the remaining supernatant. An insulin syringe is helpful to remove the liquid without aspirating the beads. Then add 30 µl 2x sample buffer and boil the beads to elute the immunoprecipitated proteins.
Analyze immunoprecipitation by SDS-PAGE as described in Supporting Protocol 1. Load the input sample first, followed by the pulldown and the supernatant. (Sometimes it is helpful to leave a blank lane in between each sample.) Bands in the pulldown lane confirm that the labeled protease corresponds to the target of the immunoprecipitating antibody. When immunoprecipitation is complete, this band should be absent from the ‘supernatant’ sample.
BASIC PROTOCOL 4
Non-invasive in vivo imaging with ABPs
Once the activity-based probe has been characterized in vitro, it can then be tested in vivo. Protocol details will largely depend on the mouse model and probes used, but there are certain guidelines to follow that will help increase the success rate of non-invasive imaging experiments. Rather than going through a stepwise protocol, we will outline these guidelines here. A scheme of a typical imaging experiment is outlined in Figure 1.
Figure 1.

Scheme of a typical non-invasive imaging experiment to detect protease activity with activity-based probes.
Materials
Mice (chosen based on model)
20 nmol ABP per mouse
DMSO
PBS
Isoflurane
Non-fluorescent (alfalfa-free) chow
Beard trimmer
Nair hair removal lotion
Insulin syringes
Mouse restrainer for tail vein injections
Special equipment
Anesthesia vaporizer
Fluorescent mouse imager such as the IVIS or FMT (both from Perkin Elmer)
First and foremost, all protocols using live animals must be reviewed and approved by an Institutional Animal Care and Use Committee (IACUC) and must conform to government regulations regarding the care and use of laboratory animals.
Choose ABPs with fluorophores that are compatible with the filter sets of the imager. In general, near infrared dyes work better for in vivo imaging since there is reduced tissue autofluorescence in this region of the spectrum compared to lower wavelengths. Most imagers have filter sets for ~670 nm and ~750 nm emission. Low coefficients of absorption and high quantum yield are also favorable dye properties. Commonly used dyes for animal imaging are, among many others, Cy5, AF647, IR800, and AF750.
Since light does not penetrate tissue particularly well, the most successful non-invasive imaging experiments will involve superficial tissues (for example, subcutaneous tumors). Systems like the IVIS detect reflected light (epifluorescence) and are less efficacious at detecting signal deeper than ~0.5 cm. With the FMT system (Fluorescence Mediated Tomography), light shines through the mouse and fluorescent signal is detected on the other side (trans-illumination). For this reason, the FMT is considerably better at detecting signal in deeper tissues, and its tomographic abilities result in 3D images. The most successful FMT imaging experiments result from models with robust and concentrated protease activity that result in very bright signal.
The typical mouse diet contains alfalfa, which is fluorescent in the Cy5 channel. This can result in unwanted background signal in the intestine of the mouse that often obscures the probe signal. If the tissue of interest is in the abdominal cavity, it is recommended to feed the mice non-fluorescent (alfalfa-free) chow for 4–7 days prior to the start of the experiment.
Hair is also a major hindrance in imaging studies. If the model does not use hairless nude mice, it is recommended to remove the hair in the imaging region. If using the IVIS, the hair only needs to be removed on the side of imaging. For FMT imaging, however, the hair needs to be removed on the front and back of the mouse to allow light to pass completely through. Using isoflurane to anesthetize the mice will facilitate this process immensely. Thin the hair using a small beard trimmer, using care not to cut the mouse with the blade. Then use Nair hair removal lotion to dissolve the remainder of the hair. Immediately after application, use cotton buds and paper towels to begin removing the hair and lotion. Be quick about this step and make sure to completely wash off the Nair, since extended exposure can burn the mice. Nicks and burns from this step not only cause pain to the mice, but they also tend to be autofluorescent. Also be sure to keep the mice warm during and after hair removal. This procedure can be very stressful for the mice, so it is recommended to do it a day or two in advance to give the mice a chance to recover before the start of the experiment (not too long, or the hair will grow back).
When testing a new probe in a new model, there are many steps to optimize. Models that involve constitutive protease activity involve less optimization than those requiring induction. For example, most apoptosis models require activation of caspases. Different drugs stimulate apoptosis at different rates. If data on the kinetics of caspase activation is not available in the literature, it must be determined experimentally. Do a time course, harvesting tissue at regular time points after drug administration. Harvest tissue, homogenize, and analyze caspase activity biochemically by labeling the lysate with the ABP followed by SDS-PAGE (Basic Protocol 6). Use the time point where caspase activity peaks for the initial non-invasive imaging experiments. This will maximize the chance for a successful pilot experiment. Models involving at least a 3-fold induction of protease activity will also lead to better contrast for imaging. Once conditions for the ABP have been optimized (see below), it may also be possible to determine the kinetics of caspase activation by imaging alone. If probe availability is a limiting factor, this is not recommended, since 500+ tissue lysates could be labeled with the same amount as one mouse injection.
Once the peak in protease activity has been determined, administration of the probe must be optimized. Dosing and formulation are the first things to consider. The dose may vary widely by the probe’s potency and PK/PD properties. In the Bogyo Lab, we typically start with 20 nmol of probe per injection and then adjust accordingly. The probe should be dissolved in a volume of 100 µl (or no more than 200 µl per injection). Depending on the solubility of the probe, increasing amounts of DMSO may be titrated in along with PBS. No more than 65 µl of DMSO should be injected at a time, as toxicity becomes an issue.Use less when possible, but make sure the probe is completely in solution prior to injection. (Note: Caspase ABPs typically use around 10% DMSO.) Other non-aqueous formulations are also available and can be tailored to the chemical properties of the probe. ABPs are typically administered by injection into the lateral tail vein using an insulin syringe while the mouse is restrained.
-
Once the ABP has been injected into the mouse, optimal circulation time must be determined. This is especially important for non-quenched probes. In this case, fluorescent signal will initially be high throughout the entire mouse. Probe molecules that bind the active protease will be retained within the tissue, whereas the unreacted probe will filter out of the body over time. This will create contrast in the target tissue relative to the surrounding areas. The rate at which this occurs can vary drastically by probe, ranging from 30 minutes to 24 hours. Quenched probes, on the other hand, are intrinsically dark until they covalently bind to the enzyme. Upon injection, there will be very low signal throughout the mouse, and then protease-specific signal will begin to accumulate in the target tissue over time. Again, the kinetics of this process varies by probe.
The best way to determine the optimal time point is with an imaging time course. Immediately after injection, anesthetize the mouse with isoflurane and begin imaging at regular intervals. Shorter intervals will yield the most information about probe kinetics, but if the signal does not change drastically between images, the length of the interval can be increased. The mouse should be permitted to wake up and run around between imaging sessions.
Use the imaging software to quantify the signal intensity in the region of interest and also in an unrelated area as a comparative base. Choose the time point that yields the highest signal-to-noise ratio. When using models involving induction of protease activity (for example, models in which a drug is administered to induce caspase activity and apoptosis), a non-induced mouse serves as an excellent control since fold change between drug-treated and normal tissue can be observed and quantified. Once the probe kinetics have been determined, imaging need only be performed at the optimal time points in subsequent experiments. Furthermore, the kinetic data for each probe are often applicable to other models.
Lastly, it is generally a good idea to include a ‘no-probe’ mouse in each imaging experiment. This will help distinguish between probe fluorescence and autofluorescence.
Representative example: imaging anti-DR5 induced caspase activity with ABPs
This model has been optimized for imaging caspase activity using ABPs (Adams, et al., 2008; Edgington, et al., 2009 and 2012). A DR5 agonist antibody is used to induce apoptosis in subcutaneous COLO205 tumors implanted in nude mice. The caspase LE22 (Edgington et al., 2012) is then administered to visualize caspase activity.
Materials
Nude mice, 6 weeks old
COLO205 human colorectal tumor cells
Insulin syringes
Anti-DR5 monoclonal antibody (Genentech)
Anti-DR5 vehicle (see recipe)
LE22
DMSO
PBS
Isoflurane
IVIS
Inject 3 × 106 COLO205 cells in a total volume of 30 µl cold PBS into the skin of the backs of 6-week old nude mice. For initial imaging experiments, inject 5 control mice and 5 drug-treated mice plus 2–3 extra in case some of the tumors do not grow uniformly. Over the course of ~8 days, subcutaneous, xenografted tumors will grow. The preferred tumor size for imaging experiments is ~0.5 cm. Avoid letting tumors become too large.
Once tumors are established, choose a cohort of mice with evenly sized tumors. Because the mice are inherently nude, they do not need to be shaved. Inject 5 mice with anti-DR5 antibody (10 mg/kg in 100 µl vehicle; tail vein) to induce caspase activity and apoptosis. Inject 5 control mice with 100 µl of vehicle in parallel.
In this model, the caspase activity peaks at 12 hours. After 11 hours, inject 4 mice from each group with LE22 by tail vein (20 nmol in 10% DMSO/PBS). The remaining mouse from each group will serve as the ‘no-probe’ controls.
LE22 gives optimal contrast 1 hr after injection. At that time, anesthetize the mice with isoflurane and then image the mice using the IVIS according to the manufacturer’s instructions. LE22 contains a Cy5 fluorophore, so choose the appropriate excitation and emission wavelengths.
Quantify the fluorescence in the tumors using the IVIS software. Background fluorescence measured in the ‘no-probe’ control tumors may be subtracted from probe-treated tumors to eliminate the contribution of autofluorescence. The increase in fluorescence between vehicle-and anti-DR5-treated tumors will be ~3–4-fold.
Once imaging is complete, euthanize the mice and harvest the tumors for further analysis.
BASIC PROTOCOL 5
Ex vivo imaging with ABPs
Upon completion of a non-invasive imaging experiment, it is useful to follow up with ex vivo imaging of the tissues of interest. Not only does this produce an additional, quantifiable piece of data, it can also aid in trouble-shooting of not-so-successful imaging experiments. It is often much easier to see contrast when tissues are isolated from the numerous sources of confounding background signal found in vivo. This is especially true for deeper tissues which may be difficult to image non-invasively.
Materials
Dissection tools
PBS
Fluorescent imager
Humanely kill the mouse by IACUC-approved protocols. Note: prior to killing, the mouse may be intracardially perfused to remove blood from the tissues, but this step is not always necessary.
Isolate tissues of interest and rinse in cold PBS. (All tissues may be analyzed to gain information regarding the biodistribution of probe binding.) Tissues should be stored in a petri dish on ice once removed. Depending on additional planned analyses, ex vivo imaging may be performed immediately or tissue may be snap frozen in liquid nitrogen and stored at −80°C for later use.
Line tissues up and image according to the manufacturer’s instructions. If using the IVIS, be sure to adjust the imaging depth to obtain the correct focal plane. The tissues may also be placed on a piece of black paper to minimize scattering of light (except on the FMT, which requires light to shine through the tissue).
Quantify the tissue fluorescence using the imaging software.
Weigh the tissues. Since the isolated tissues may be of varying size, it is common practice to compare the data across samples by normalizing fluorescent signal by weight.
BASIC PROTOCOL 6
Biochemical analysis of probe-labeled tissues
One of the great advantages of activity-based probes is their tractability. Because of their covalent nature, fluorescent signal produced in vivo can be correlated with biochemical data demonstrating the actual labeling of protease targets. The tissues are homogenized and analyzed by SDS-PAGE allowing direct quantification of protease levels. In a successful imaging experiment, the levels should correlate with the fluorescent signal in the tissues. Biochemical analysis also reveals any labeling of off-target proteases that may occur in vivo.
Materials
Probe-labeled tissues with weights recorded
Muscle lysis buffer (see recipe)
Sonicator with tip for eppendorf tubes
4x sample buffer
After imaging the tissues ex vivo in Basic Protocol 5, they may be frozen and stored at −80°C or homogenized directly.
Add muscle lysis buffer to the tissue in an eppendorf tube. In general, add 10 µl of buffer for each 1 mg of tissue. If the tissue is larger than 60 mg, it may be better to do the lysis in a 15-mL conical tube. Alternatively, the tissue may be cut and a smaller portion used. This is practical when the protease activity is uniform throughout the tissue; however, if pockets of activity are expected, it is difficult to determine which fraction to analyze. In these cases, it is safer to homogenize the whole tissue and obtain an average signal throughout.
On ice, sonicate the tissue in short bursts until the tissue is completely homogenized. (Alternative homogenization methods are also suitable: glass douncer, BeadBeater, etc.)
Spin the homogenates at high speed in a tabletop centrifuge for 15 minutes. Transfer supernatant and quantify protein according to Basic Protocol 1, Step 9. Dilute samples in 4x sample buffer, boil, and analyze by SDS-PAGE according to Supporting Protocol 1.
Tissue homogenates may also be labeled with ABPs ex vivo. Follow the same procedure for preparing lysates, except instead of adding sample buffer in step 4, add probe in the same manner described in Basic Protocol 1, Steps 10–13. When labeling tissue lysates ex vivo, the lysis buffer may need to be adjusted to ensure activity of the target protease. For caspases, muscle lysis buffer works well. For cathepsins or legumain, a low pH buffer is required (see recipe).
To gain information regarding the percentage of active proteases that get labeled in vivo, it is also possible to re-label tissues ex vivo. For this experiment, lyse tissues in appropriate activity buffer for the target protease, and use an activity-based probe possessing a fluorophore that emits at a different wavelength than the in vivo administered probe.
BASIC PROTOCOL 7
Histological analysis of probe-labeled tissues
Activity-based probes may also be used to examine the distribution of protease activity at a microscopic level. For this protocol, frozen sections are required. Fluorescent probe signal cannot be detected after paraffin embedding.
Materials
Probe-labeled tissues
4% paraformaldehyde in PBS
30% sucrose in PBS
Tissue-Tek® OCT™ Compound
Tissue molds
For this protocol, use freshly isolated tissue. Signal is improved if the mouse is intracardially perfused with PBS prior to tissue harvest. It is strongly encouraged to use tissue from a mouse that did not receive a probe injection in parallel as a control. This will aid in discerning tissue autofluorescence from true probe signal.
Tissue fixation protocols vary widely and may affect fluorescent signal. We suggest 4–6 hours in 4% paraformaldehyde/PBS followed by overnight incubation in 30% sucrose/PBS. All incubation steps should be performed at 4°C.
Dab the tissue on a paper towel to remove excess moisture and place in a tissue mold that has been pre-filled with Tissue-Tek® OCT™ Compound. Avoid bubbles. Allow the OCT compound to permeate the tissue for 30 minutes at room temperature, and then freeze on a block of dry ice until compound becomes solid. Store at −80°C until sections can be cut. Eight to ten micron sections are generally sufficient, but this depends largely on the tissue/model in use.
Sections may then be visualized directly by fluorescence microscopy after a 10-minute hydration in PBS. Alternatively, they may be stained according to standard immunofluorescence protocols to correlate probe signals with other markers. Ensure that secondary antibodies are chosen such that they do not interfere with the fluorescent signal of the probe.
BASIC PROTOCOL 8
Flow cytometry of probe-labeled tissues
Activity-based probes are also highly amenable to flow cytometry assays. This application can be useful for determining the percentage of cells that possess protease activity within a tissue and can also correlate protease activity with cell type-specific markers.
Materials
Probe-labeled tissues
Razor blades
Digestion buffer (see recipe)
40 µm nylon mesh filters
ACK Lysing Buffer (Invitrogen)
FACS buffer (see recipe)
Use freshly isolated tissue with this protocol. As with histology, results are improved by intracardially perfusing the mice with PBS prior to tissue harvest. A ‘no-probe’ mouse is required to properly compensate the FACS machine. This will distinguish between cellular autofluorescence and fluorescent signal from the ABP.
Protocols for making single cell suspensions vary by tissue. In general, mince the tissue using a razor blade until the pieces are 1–2 mm3 in size. Then add 10 mL digestion buffer and incubate at 37°C for approximately two hours, pipetting frequently.
Once in single cell suspension, filter cells with a 40 µm nylon mesh filter. Lyse red blood cells using ACK Lysing Buffer according to protocol. Then resuspend cells in FACS buffer at a density of 10 × 106 cells/mL.
The cells are then ready for flow cytometry analysis. Additional markers may be analyzed and staining is performed according to standard protocols.
REAGENTS AND SOLUTIONS
Hypotonic Lysis Buffer
50 mM PIPES pH 7.4
10 mM KCl
5 mM KCl2
2 mM EDTA
1% NP-40
4 mM DTT-add at the time of use
Store up to one year at 4°C
Low pH buffer for legumain or cathepsins
50 mM citrate, pH 5
0.5% CHAPS
1% NP-40
4 mM DTT-add at the time of use
Store up to one year at 4°C
4x Sample Buffer
40% Glycerol (v,v)
200 mM Tris/HCl pH 6.8
8% SDS (w,v)
0.04% bromophenol blue (w,v)
5% beta-mercaptoethanol (v,v)
Store in aliquots at −20°C, store at RT for 1–2 weeks
IP Buffer
1x PBS, pH 7.4
0.5% NP-40 (v,v)
1 mM EDTA
Store at RT for one year
Anti-DR5 Vehicle
10 mM histidine
0.8% sucrose (w,v)
0.02% Tween-20 (v,v)
pH 6
Store in aliquots at −20°C for one year
Muscle Lysis Buffer
1x PBS, pH 7.4
1% Triton X-100 (v,v)
0.1% SDS (w,v)
0.5% sodium deoxycholate (w,v)
Store for one year at 4°C
Digestion Buffer
Advanced DMEM/F12 (Invitrogen)
2 mM Glutamax (Invitrogen)
120 µg/ml penicillin
100 µg/ml streptomycin
0.25 µg/ml amphotericin-B
200 U/ml Collagenase type III (Worthington, Lakewood, NJ)
100 U/ml DNase I (Worthington, Lakewook, NJ)
Prepare fresh for each experiment
FACS Buffer
HBBS (Invitrogen)
120 µg/ml penicillin
100 µg/ml streptomycin
0.25 µg/ml amphotericin-B
2% heat inactivated fetal calf serum
COMMENTARY
Background information
In the last decade, the field of activity-based proteomics has expanded immensely and it continues to do so. The concept of activity-based probes has been a major breakthrough in studying the function of proteases in both normal physiology and disease(Blum, 2008; Deu et al., 2012; Edgington et al., 2011; Paulick and Bogyo, 2008; Serim et al., 2012). ABPs typically consist of three major components. The most critical moiety is a reactive electrophile that confers selectivity to a particular class of proteases. This element, often called a warhead, is susceptible to nucleophilic attack by the catalytic residue of the active enzyme, resulting in covalent, irreversible modification. An overview of electrophiles used for different proteases can be found in Serim et al., 2012. The second component is a peptide recognition element designed to confer selectivity towards a particular protease. This element is often peptidic in nature and is designed based on the natural protein substrate. The sequence can be engineered to increase potency towards the protease of interest and also to decrease affinity towards unwanted off-targets. Lastly, an ABP contains a tag that allows detection. For in vivo imaging applications, the tag is usually a fluorophore; however, ABPs with radiolabels have recently been reported for use with PET imaging modalities (Ren et al., 2011).
Some ABPs also contain a fourth component: a quenching group that absorbs photons emitted by the fluorophore until it is released by proteolytic attack of the warhead (Blum et al., 2005; Blum et al., 2007; Verdoes et al., 2012). Quenched probes are intrinsically dark until they bind the active protease; therefore, fluorescent signal specifically indicates activity.
One of the major advantages of ABPs is that they covalently and irreversibly modify the active protease (Edgington et al., 2011). This means that the fluorescent tag remains at the active site of the enzyme and does not diffuse away, allowing for precise studies of the localization of activity through imaging. Moreover, the modification survives the denaturing conditions of SDS-PAGE, which allows for a direct biochemical readout of protease binding. The fluorescent signal detected by imaging can directly be assigned to the proteases to which the probe binds. This is especially critical for identifying off-targets of probes for which absolute specificity has not been achieved.
While the covalent nature of activity-based probes is overall beneficial, one caveat is that probe binding also irreversibly inhibits enzyme activity (Edgington et al., 2011). Since proteases are often involved in complex signaling pathways, their inhibition may lead to disruption of biological processes. For most in vivo imaging experiments, the administered probe dose is so low that it only inhibits a small percentage of the total protease pool, thus it is unlikely to drastically alter the biology of the system. The use of ABPs is sufficient to obtain a snapshot of protease activity at a given time point; however, care should taken in interpreting results of experiments that monitor activity over time.
Another widely used class of probes for proteases is substrate-based probes (Edgington et al., 2011). Several adaptations of substrate-based probes exist, but the general concept is similar across all types. Like quenched ABPs, their spectral properties change upon proteolytic cleavage. Unlike APBs, however, they lack a reactive electrophile, therefore no covalent modification occurs. This means that enzyme activity is retained after substrate cleavage, which allows for signal amplification. On the other hand, the fluorescent signal is able to diffuse away from the site of cleavage, making localization studies less precise. For a more thorough discussion of the pros and cons of activity- and substrate-based probes for use in in vivo imaging studies, refer to (Blum et al., 2009; Edgington et al., 2011).
Critical Parameters and Troubleshooting
Perhaps the most critical parameter for a successful in vivo imaging experiment is timing. In order to detect protease activity, the ABP must make contact with the protease at the time that it is active. As outlined in the imaging section, achieving this may take several rounds of optimization. Information is needed regarding when the proteases are active and for how long. If the window of activity is short (as in some apoptosis models, for example, when the cells die and are quickly removed by macrophages), the timing of probe delivery will be even more important. In turn, the length of time it takes the probe to reach the target tissue is another critical factor. If using a non-quenched probe, it must also be determined how long it will take for the free probe to clear from the tissue. If little signal is observed in vivo, take time to follow up with ex vivo imaging and biochemical analysis. If no bands are observed by gel, the timing may need to be adjusted.
Another critical parameter is signal strength. In general, fairly robust protease activity is required to detect by non-invasive imaging methods. This is particularly true if the tissues are deep and the light must travel further and through other tissues. Models where caspases become activated in just a few cells, for example, are not good candidates for whole animal imaging experiments. If the precise location of these cells is known, it may be possible to see fluorescence in single cells, either by microscopy or flow cytometry. If protease activity is low, it may or may not be detected by SDS-PAGE. Typically, if the signal is strong enough to be detected by western blot (by cleaved caspase-3 staining, for example), then the threshold should be high enough for detection of ABP labeling.
Pharmacodynamic properties of the probe are also important factors that govern the effectiveness of imaging experiments. Ideally, the probe will circulate well and promptly reach the tissue of interest. Some probes are cleared by the kidney or liver before they ever reach the target tissue, and some are unstable in vivo and degrade immediately upon injection. Often, these properties are impossible to predict and require more thorough PK/PD studies to understand. Sometimes delivery or targeting strategies can be used to direct the probe to the tissue of interest (Mikhaylov et al., 2011, for example). If the probe works well in in vitro assays, it may be possible to glean some data by labeling tissue homogenates ex vivo.
Sometimes background fluorescence is so high that it obscures the true signal. Using higher wavelength, near-infrared probes, shaving the mice, and feeding non-fluorescent chow are all good practices to minimize autofluorescence. As previously mentioned, it is always a good idea to use a ‘no-probe’ mouse in parallel as a control to distinguish other areas of autofluorescence. Some probes become sequestered in the liver, gallbladder, kidneys and bladder as they clear from the body, leading to high background fluorescence in these tissues that may bleed into other areas. Imaging tissues within the peritoneum is challenging for this reason. Quenched probes are vastly preferred if available. Additionally, probes that lack absolute specificity for the protease of interest will also contribute additional background due to labeling of off-target proteases. Following up imaging studies with biochemical analysis is a must do.
Anticipated Results
These protocols should yield a reproducible and quantifiable readout of the levels of protease activity across numerous fluorescent-based applications both in vitro and in vivo, including the ability to visualize such activity by optical imaging of a live animal. Cellular localization of protease activity can also be determined using flow cytometry and histology protocols. Additionally, targets of ABPs can be confirmed using biochemical and immunoprecipitation assays.
Figure 2 provides data from representative experiments using a fluorescent ABP for caspases, LE22 (Edgington, et al., 2012). LE22 contains a Cy5 fluorophore, a Val-Glu-Ile-Asp peptide sequence, and an acyloxmethyl ketone warhead (Figure 2A). Figure 2B demonstrates the application of LE22 to label caspases in intact cells. COLO205 human colorectal cancer cells were treated with a DR-5 agonist antibody for 3.5 hours to induce caspase activity, followed by incubation with increasing amounts of LE22 for 30 minutes. Cells were then harvested according to the procedure described in Basic Protocol 2, and analyzed by SDS-PAGE (Supporting Protocol 1). Saturation of labeling occurs at 0.5 µM, indicating that this would be an optimal concentration to use for future in vitro experiments.
Figure 2.
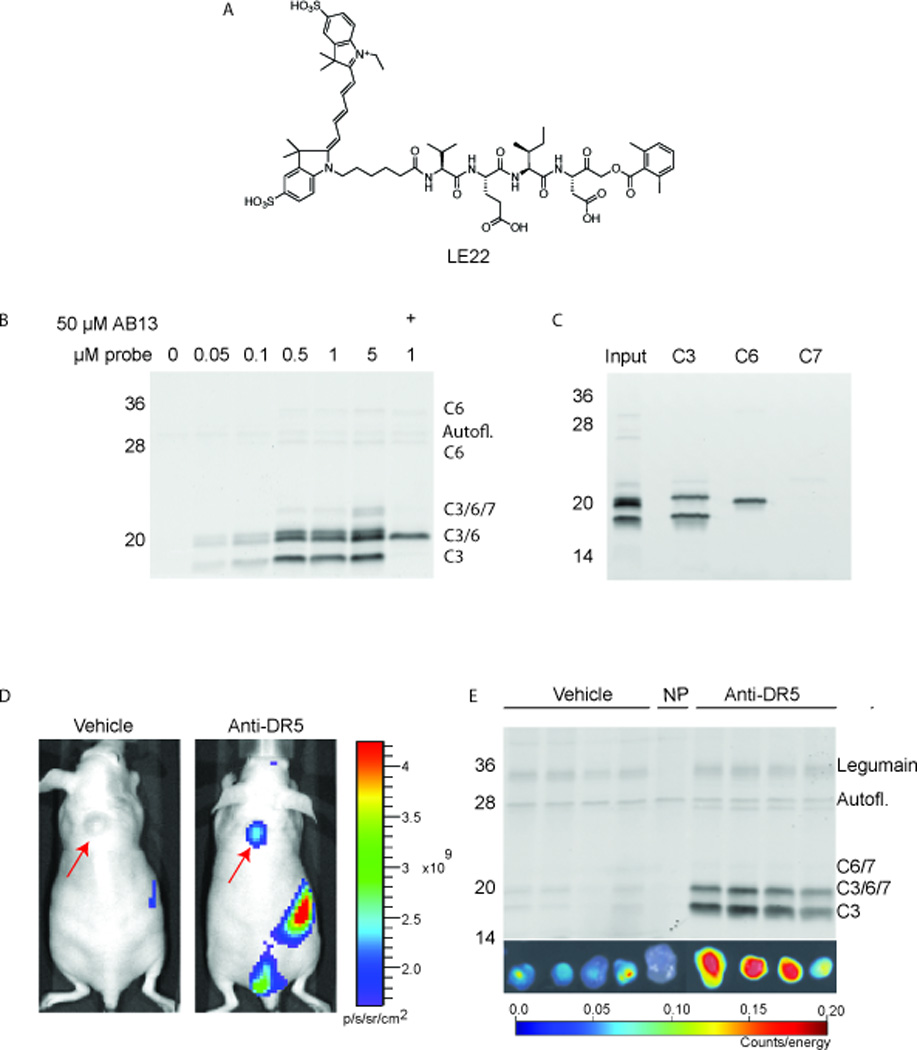
Representive example. (A) Structure of LE22, a fluorescent ABP that targets caspase-3,−6,−7 (C3, C6, C7, respectively). (B) Dose curve of LE22 labeling in apoptotic COLO205 cells. Cells were treated with anti-DR5 for 3.5 hours to activate caspases, followed by 30 minute labeling with the indicated dose of LE22. In the last lane, cells were pretreated with AB13, a caspase-3/−7 selective inhibitor, prior to LE22 labeling, allowing for selective labeling of caspase-6. The band appearing in the ‘no-probe’ control lane (0 µM LE22) is autofluorescent and is marked accordingly. (C) Immunoprecipitation experiment to confirm that species labeled by LE22 are indeed caspases-3, −6, and −7. (D) Non-invasive imaging of mice treated with vehicle only (left) or anti-DR5 (right), followed by administration of LE22. Fluorescent accumulates in the tumor only when caspases are activated by anti-DR5 treatment. (E) Ex vivo imaging of tumors imaged in (D) and subsequent biochemical analysis. Upon administration with anti-DR5, tumors exhibit a significant increase in LE22 fluorescence, corresponding to an increase in caspase labeling. A band appearing in the ‘no-probe’ control tumor (NP) is marked as autofluorescent.
Multiple species are labeled by LE22 including various maturation forms of caspase-3, −6, and 7. To confirm the identity of the labeled bands, two approaches were used. The first was to pretreat the cells with a caspase-3-/7 selective inhibitor, AB13, prior to the addition of LE22. The inhibitor blocked labeling of caspase-3/−7, and the remaining bands corresponded to caspase-6. To confirm this with more certainty, an immuprecipitation experiment was performed according to Basic Protocol 3 (Figure 2C). Indeed, all of the labeled species could be assigned to caspase-3, −6, or −7, with one exception. There is one band that appears in the lane in which no probe was added (0 µM, Figure 2B). This indicates and autofluorescent protein, and highlights the importance of including a ‘no-probe’ control.
Figure 2D shows data obtained using the procedure described in the representative example of Basic Protocol 4. Nude mice bearing COLO205 human colorectal xenograft tumors were treated with anti-DR5 to induce tumor-specific caspase activity/apoptosis. At the peak of caspase activation (~11 hours in this case), LE22 was administered, and mice were imaged by IVIS one hour later. Tumors on the mice treated with anti-DR5 exhibited significantly increased accumulation of fluorescence compared to the vehicle-treated control mice. This strongly suggests that the increase in fluorescence was caspase-dependent. When the tumors were removed and imaged ex vivo according to Basic Protocol 5, the same trend in fluorescence was observed (Figure 2E). To confirm the in vivo targets of LE22, a biochemical analysis was also performed according to Basic Protocol 6 (Figure 2E). Indeed, tumors treated with anti-DR5 showed a sharp increase in the labeling of caspase-3, −6, and −7 compared to the vehicle-treated control tumors, which corresponds to the in vivo and ex vivo imaging data. Moreover, the biochemical analysis reveals that, in addition to caspases, LE22 labels low levels of the off-target protease, legumain. This suggests that some of the fluorescence detected by imaging is due to legumain rather than caspases. Caution should therefore be taken in interpreting the data. In this case, legumain levels do not change upon treatment with anti-DR5, therefore the fold-change in fluorescence is entirely due to caspase labeling. LE22 would also be amenable to histology and flow cytometry as described in Basic Protocols 7 and 8; however, this data is not shown. A similar probe, AB50, was used for these applications and data can be found in Edgington et al., 2009.
Time Considerations
Sample preparation and probe labeling can be performed in <3 hours from the time of harvest. This does not include time to induce protease activity, as this varies widely by model. SDS-PAGE analysis also takes ~3 hours. The length of animal imaging experiments varies widely by model and the probe used and can range from 2–24 hours (sometimes more). Ex vivo imaging takes 15 minutes, or longer if more tissues are imaged. Tissue preparation for SDS-PAGE can be performed in 4 hours or less. Preparing tissues for histology takes 1.5 days, and FACS preparation will take ~ 5 hours.
ACKNOWLEDGEMENTS
This work was supported by NIH grant R01 EB005011.
LITERATURE CITED
- Adams C, Totpal K, Lawrence D, Marsters S, Pitti R, Yee S, Ross S, Deforge L, Koeppen H, Sagolla M, et al. Structural and functional analysis of the interaction between the agonistic monoclonal antibody Apomab and the proapoptotic receptor DR5. Cell DeathDiffer. 2008;15:751–761. doi: 10.1038/sj.cdd.4402306. [DOI] [PubMed] [Google Scholar]
- Blum G. Use of fluorescent imaging to investigate pathological protease activity. Curr Opin Drug Discov Devel. 2008;11:708–716. [PubMed] [Google Scholar]
- Blum G, Mullins SR, Keren K, Fonovic M, Jedeszko C, Rice MJ, Sloane BF, Bogyo M. Dynamic imaging of protease activity with fluorescently quenched activity-based probes. Nat Chem Biol. 2005;1:203–209. doi: 10.1038/nchembio728. [DOI] [PubMed] [Google Scholar]
- Blum G, von Degenfeld G, Merchant MJ, Blau HM, Bogyo M. Noninvasive optical imaging of cysteine protease activity using fluorescently quenched activity-based probes. Nat Chem Biol. 2007;3:668–677. doi: 10.1038/nchembio.2007.26. [DOI] [PubMed] [Google Scholar]
- Blum G, Weimer RM, Edgington LE, Adams W, Bogyo M. Comparative assessment of substrates and activity based probes as tools for non-invasive optical imaging of cysteine protease activity. PLoS One. 2009;4:e6374. doi: 10.1371/journal.pone.0006374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deu E, Verdoes M, Bogyo M. New approaches for dissecting protease functions to improve probe development and drug discovery. Nat Struct Mol Biol. 2012;19:9–16. doi: 10.1038/nsmb.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgington LE, Berger AB, Blum G, Albrow VE, Paulick MG, Lineberry N, Bogyo M. Noninvasive optical imaging of apoptosis by caspase-targeted activity-based probes. Nat Med. 2009;15:967–973. doi: 10.1038/nm.1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgington LE, van Raam BJ, Verdoes M, Wierschem C, Salvesen GS, Bogyo M. An optimized activity-based probe for the study of caspase-6 activation. Chem Biol. 2012;19:340–352. doi: 10.1016/j.chembiol.2011.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgington LE, Verdoes M, Bogyo M. Functional imaging of proteases: recent advances in the design and application of substrate-based and activity-based probes. Curr Opin Chem Biol. 2011;15:798–805. doi: 10.1016/j.cbpa.2011.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gocheva V, Wang HW, Gadea BB, Shree T, Hunter KE, Garfall AL, Berman T, Joyce JA. IL-4 induces cathepsin protease activity in tumor-associated macrophages to promote cancer growth and invasion. Genes Dev. 2010;24:241–255. doi: 10.1101/gad.1874010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyce JA, Baruch A, Chehade K, Meyer-Morse N, Giraudo E, Tsai FY, Greenbaum DC, Hager JH, Bogyo M, Hanahan D. Cathepsin cysteine proteases are effectors of invasive growth and angiogenesis during multistage tumorigenesis. Cancer Cell. 2004;5:443–453. doi: 10.1016/s1535-6108(04)00111-4. [DOI] [PubMed] [Google Scholar]
- Lee J, Bogyo M. Development of near-infrared fluorophore (NIRF)-labeled activity-based probes for in vivo imaging of legumain. ACS Chem Biol. 2010;5:233–243. doi: 10.1021/cb900232a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Hsu HC, Yang P, Wu Q, Li H, Edgington LE, Bogyo M, Kimberly RP, Mountz JD. Treatment of arthritis by macrophage depletion and immunomodulation: testing an apoptosis-mediated therapy in a humanized death receptor mouse model. Arthritis Rheum. 2012;64:1098–1109. doi: 10.1002/art.33423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Patricelli MP, Cravatt BF. Activity-based protein profiling: the serine hydrolases. Proc Natl Acad Sci U S A. 1999;96:14694–14699. doi: 10.1073/pnas.96.26.14694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikhaylov G, Mikac U, Magaeva AA, Itin VI, Naiden EP, Psakhye I, Babes L, Reinheckel T, Peters C, Zeiser R, et al. Ferri-liposomes as an MRI-visible drug-delivery system for targeting tumours and their microenvironment. Nat Nanotechnol. 2011;6:594–602. doi: 10.1038/nnano.2011.112. [DOI] [PubMed] [Google Scholar]
- Paulick MG, Bogyo M. Application of activity-based probes to the study of enzymes involved in cancer progression. Curr Opin Genet Dev. 2008;18:97–106. doi: 10.1016/j.gde.2007.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren G, Blum G, Verdoes M, Liu H, Syed S, Edgington LE, Gheysens O, Miao Z, Jiang H, Gambhir SS, et al. Non-invasive imaging of cysteine cathepsin activity in solid tumors using a 64Cu-labeled activity-based probe. PLoS One. 2011;6:e28029. doi: 10.1371/journal.pone.0028029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serim S, Haedke U, Verhelst SH. Activity-based probes for the study of proteases: recent advances and developments. ChemMedChem. 2012;7:1146–1159. doi: 10.1002/cmdc.201200057. [DOI] [PubMed] [Google Scholar]
- Verdoes M, Edgington LE, Scheeren FA, Leyva M, Blum G, Weiskopf K, Bachmann MH, Ellman JA, Bogyo M. A nonpeptidic cathepsin S activity-based probe for noninvasive optical imaging of tumor-associated macrophages. Chem Biol. 2012;19:619–628. doi: 10.1016/j.chembiol.2012.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdoes M, Florea BI, Menendez-Benito V, Maynard CJ, Witte MD, van der Linden WA, van den Nieuwendijk AM, Hofmann T, Berkers CR, van Leeuwen FW, et al. A fluorescent broad-spectrum proteasome inhibitor for labeling proteasomes in vitro and in vivo. Chem Biol. 2006;13:1217–1226. doi: 10.1016/j.chembiol.2006.09.013. [DOI] [PubMed] [Google Scholar]