

Abstract
Objective
We sought to determine whether interleukin (IL)-6 modulates myocardial infarction or the late phase of preconditioning (PC).
Methods
Wild-type and IL-6−/− mice underwent a 30-min coronary occlusion followed by 24 h of reperfusion with or without six cycles of coronary occlusion/reperfusion 24 h earlier. Myocardial IL-6 protein expression, activation of Janus kinase (JAK) 1 and JAK2, and signal transducers and activators of transcription (STAT) 1 and STAT3 after ischemic PC protocol were examined. The expression of the inducible nitric oxide synthase (iNOS) and cyclooxygenase (COX)-2 was determined 24 h after the PC ischemia.
Results
In preconditioned wild-type mice, infarct size was reduced from 60.5 ± 2.6% of the risk region to 33.5 ± 3.6%, indicating a late PC effect. In nonpreconditioned IL-6−/− mice, infarct size was similar to that observed in wild-type mice (59.9 ± 3.8%), indicating that the deletion of IL-6 has no effect on infarct size. However, in preconditioned IL-6−/− mice, infarct size was not reduced (65.1 ± 3.1%), indicating that the infarct-sparing effect was completely abrogated. Ischemic PC increased the expression of IL-6 in the cytoplasm of cardiomyocytes in the ischemic/reperfused zone. In IL-6−/− mice, the ischemic PC-induced activation of JAK1 and JAK2 and STAT1 and STAT3 was significantly reduced, and the increase in iNOS and COX-2 protein expression 24 h after the PC ischemia was markedly attenuated.
Conclusion
IL-6 does not modulate myocardial infarct size in naïve myocardium. However, following a PC stimulus, IL-6 is obligatorily required for the activation of the JAK–STAT pathway, the ensuing upregulation of iNOS and COX-2 (co-mediators of late PC), and the development of a cardioprotective phenotype.
Keywords: Interleukins, Preconditioning, Infarction, Signal transduction, Cyclooxygenase
1. Introduction
The late phase of ischemic preconditioning (PC) is the phenomenon whereby brief episodes of ischemia increase the tolerance of the myocardium to a subsequent ischemic insult 24–72 h later [1–3]. Intense research over the past decade has identified several molecules, including nitric oxide (NO) and reactive oxygen species (ROS), that serve as ‘triggers’ of this phenomenon [2,3]. Recent studies have revealed that the activation of the stress-responsive Janus kinase (JAK)-signal transducers and activators of transcription (STAT) pathway is essential for the development of late PC [4,5]. However, the complex signaling response elicited by ischemic PC remains incompletely understood [2,3]. In particular, nothing is known regarding the nature of the signaling molecule(s) that leads to the activation of the JAK–STAT pathway during the genesis of late PC.
Acting via gp130, the pleiotropic cytokine interleukin (IL)-6 is known to activate multiple signaling pathways, including the JAK–STAT, the Src family of PTKs, and phosphatidylinositol 3-kinase [6–9]. Although induction and release of myocardial IL-6 have been reported following ischemia/reperfusion injury and myocardial infarction [10–12], and cardiomyocytes themselves have been shown to produce IL-6 in response to hypoxia [13] and ischemia/reperfusion [12], very little is known about the importance of IL-6 signaling via gp130 and the JAK–STAT pathway in the setting of ischemia/reperfusion injury. Furthermore, it remains unknown whether IL-6 exerts a beneficial or a deleterious role on myocardial ischemia/reperfusion injury.
Recent evidence has revealed an essential role of the stress-responsive JAK–STAT pathway in the genesis of late PC [4,5]. Specifically, it has been shown that JAK1 and JAK2 are activated in the ischemic/reperfused myocardium 5 min after the ischemic PC stimulus, resulting in tyrosine phosphorylation and nuclear translocation of STAT1 and STAT3 [4]. Inhibition of JAK activation with AG-490 resulted in abrogation of the induction of iNOS and the protective effects of late PC [4]. Furthermore, activation of the JAK–STAT pathway has been implicated as the central mechanism of cyclooxygenase (COX)-2-induction 24 h after ischemic PC [5]. Although the IL-6 family of cytokines is known to activate the JAK–STAT pathway via gp130 [7,9], the role of IL-6 in the development of late PC has not been investigated, and the identity of the signals that cause JAK–STAT activation during the development of late PC remains unknown.
The present study was undertaken to determine the role of IL-6 signaling in modulating ischemia/reperfusion injury in nonpreconditioned myocardium and in the development of late PC against myocardial infarction. Specifically, we used IL-6−/− mice to conclusively establish: (i) whether the presence of IL-6 increases or decreases infarct size when ischemia is followed by reperfusion; (ii) whether ischemic PC upregulates myocardial IL-6 expression; (iii) whether IL-6 is necessary for the development of the infarct-sparing effects of late PC; and (iv) whether IL-6 signaling is required for JAK–STAT activation and induction of iNOS and COX-2 during late PC. A well-characterized murine model of late PC [14,15] was used to address these questions.
2. Methods
The investigation conforms with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996).
2.1. Animals
Wild-type and IL-6−/− mice (B6,129 strain, body weight 25–35 g, age 12–26 week) purchased from Jackson Laboratory were used for this study. The overall experimental design is summarized in Fig. 1.
Fig. 1.

Experimental protocol. Twenty groups of mice were used. On day 2, mice in groups I–IV underwent a 30-min coronary occlusion followed by 24 h of reperfusion. On day 1, mice in group II (n = 9, late PC group) and group IV (n = 9, IL-6−/− late PC group) underwent a sequence of six 4-min coronary occlusion (O)/4-min reperfusion (R) cycles, while mice in group I (n = 17, control group) and group III (n = 9, IL-6−/− control group) did not undergo any intervention. Mice in group V (n = 6) underwent 1 h of open-chest state (n = 3) or six cycles of 4-min coronary occlusion/4-min reperfusion (n = 3), and the hearts were harvested 30 min later for IL-6 immunolocalization. In groups VI–VIII (n = 6 in each group), wild-type mice underwent no intervention (group VI), 1 h of open-chest state (group VII), or six cycles of 4-min coronary occlusion/4-min reperfusion (group VIII), and myocardial tissue samples were harvested 2 h later for the measurement of myocardial IL-6 levels by ELISA. In groups IX–XX (n = 6 in each group), wild-type (groups IX, X, XIII, XIV, XVII, and XVIII) and IL-6−/− (groups XII, XV, XVI, XIX, and XX) mice underwent six cycles of 4-min coronary occlusion/4-min reperfusion (groups X, XII, XIV, XVI, XVIII, and XX) or 1-h open-chest state (groups IX, XIII, XV, XVII, and XIX), and myocardial tissue samples were harvested 5 min [groups IX–XII (JAK activity assays)], 30 min [groups XIII–XVI (STAT activation assays)] and 24 h [groups XVII–XX (iNOS and COX-2 levels)] later for biochemical assays.
2.2. Protocol of studies of myocardial infarction
The experimental preparation has been described in detail [14,15]. In groups I–IV, myocardial infarction was produced by a 30-min coronary occlusion followed by 24 h of reperfusion [14,15]. Mice were anesthesized with pentobarbital sodium (50 mg/kg i.p.), intubated, and ventilated using a small rodent ventilator. Body temperature, heart rate, and arterial pH were carefully maintained within the physiological range throughout the experiments. Using a sterile technique, the chest was opened through a midline sternotomy. An 8-0 nylon suture was passed with a tapered needle under the left anterior descending coronary artery 2–3 mm from the tip of the left auricle, and a nontraumatic balloon occluder was applied on the artery. Coronary occlusion was induced by inflating the balloon occluder. Group I (control group) underwent the 30-min occlusion with no prior PC or any other intervention. Group II (late PC group) underwent a sequence of six 4-min coronary occlusion/4-min reperfusion cycles on day 1 and a 30-min coronary occlusion on day 2. Mice in groups III and IV (IL-6−/− mice) were subjected to protocols identical to those in groups I and II, respectively (Fig. 1). Since we have already demonstrated that a 30-min coronary occlusion/reperfusion 24 h after a sham operation produces infarct size similar to that observed in mice that do not undergo any prior intervention [14], mice in groups I and III were not subjected to a sham operation on day 1. At the conclusion of the study, infarct size was calculated as a percentage of the region at risk [14,15].
2.3. Immunohistochemistry
Wild-type mice (group V; n = 6) underwent six cycles of 4-min coronary occlusion/4-min reperfusion (n = 3) or 1-h open-chest state (n = 3) (Fig. 1). Thirty minutes after the last reperfusion, mice were euthanized and hearts were perfusion-fixed (10% neutral-buffered formalin). Immunohistochemical localization of IL-6 [primary: goat polyclonal antimouse IL-6 antibody (sc-1265; Santa Cruz); secondary: FITC-conjugated donkey anti-goat IgG (705-095-147; Jackson ImmunoResearch)] and cardiac troponin T [primary: goat polyclonal antitroponin T primary antibody (sc-8121; Santa Cruz); secondary: Texas red-conjugated bovine anti-goat IgG (sc-2786; Santa Cruz)] was performed according to standard methods. Nuclei were visualized by 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI; Sigma). All photomicrographs were acquired using a Zeiss LSM 510 confocal microscope.
2.4. Measurement of myocardial IL-6 levels
Mice were assigned to three groups (Fig. 1). Mice in group VI did not undergo any intervention. Mice in group VIII underwent six cycles of 4-min coronary occlusion/4-min reperfusion, while group VII underwent 1 h of open-chest state. Two hours later, myocardial tissue samples from the ischemic/reperfused region (group VIII) or the corresponding area (groups VI and VII) were harvested and frozen in liquid nitrogen immediately. Myocardial tissue was homogenized and the supernatant was used for the detection of IL-6 using a sandwich ELISA assay (DY406; R&D Systems) according to the manufacturer’s instructions. Each sample was tested in duplicate.
2.5. Preparation of cytosolic and membranous proteins
Cytosolic and membranous proteins were prepared from frozen myocardial samples according to the previously published methods [4,16]. Protein content was determined by a Bio-Rad protein assay kit.
2.6. Myocardial JAK1 and JAK2 activity assays
Wild-type (groups IX and X) and IL-6−/− (groups XI and XII) mice underwent a sham operation (groups IX and XI) or six cycles of coronary occlusion/reperfusion (groups X and XII) (Fig. 1). All mice were euthanized 5 min after the sixth reperfusion, and myocardial samples from the ischemic/reperfused area or the left ventricular anterior wall were frozen immediately in liquid nitrogen. The tyrosine-phosphorylated forms of JAK1 and JAK2 were determined by immunoprecipitation (IP) with anti-pTyr antibodies followed by Western blotting with anti-JAK1 or anti-JAK2 antibodies [4].
2.7. Preparation of nuclear extracts
Nuclear extracts were prepared from frozen myocardial samples using previously published methods [4].
2.8. STAT1 and STAT3 activity assays
Wild-type (groups XIII and XIV) and IL-6−/− (groups XV and XVI) mice underwent 1 h of open-chest state (groups XIII and XV) or six cycles of coronary occlusion/reperfusion (groups XIV and XVI) and were euthanized 30 min after the sixth reperfusion (Fig. 1). Nuclear extracts were prepared from the ischemic/reperfused region of preconditioned mice and the corresponding myocardial area in sham-operated mice [4]. Tyrosine-phosphorylated forms of STAT1 and STAT3 in nuclear extracts were analyzed by Western blotting with anti-pTyr(701) STAT1 or anti-pTyr(705) STAT3 antibodies [4].
2.9. Myocardial iNOS and COX-2 protein expression
On day 1, wild-type (groups XVII and XVIII) and IL-6−/− (groups XIX and XX) mice underwent 1 h of open-chest state (groups XVII and XIX) or six cycles of coronary occlusion/reperfusion (groups XVIII and XX) (Fig. 1). Twenty-four hours later, myocardial tissue samples were harvested from the ischemic/reperfused zone or the sham-operated heart. iNOS and COX-2 protein levels were determined by standard Western immunoblotting techniques in cytosolic and membranous fractions, respectively [16,17].
2.10. Statistical analysis
Data are reported as mean ± S.E.M. Heart rate, risk region, and infarct size were analyzed with a one-way or a two-way repeated-measures (time and group) analysis of variance (ANOVA), as appropriate, followed by Student’s t tests for unpaired data with the Bonferroni correction. The relationship between infarct size and risk region size was compared among groups with an analysis of covariance (ANCOVA), with size of the risk region used as the covariate. Differences among groups with respect to tyrosine-phosphorylated JAK1, JAK2, STAT1, and STAT3 were analyzed using one-way ANOVA. If the ANOVA showed an overall difference, post-hoc contrasts were performed with Student’s t tests for unpaired data with the Bonferroni correction. P < 0.05 was considered statistically significant. All statistical analyses were performed using the SPSS (version 8.0) statistical software (SPSS, Chicago, IL).
3. Results
A total of 132 mice were used (44 mice for the infarct size studies and 88 mice for the biochemical and histologic analyses). Nineteen mice were excluded for the reasons specified in Table 1.
Table 1.
Reasons for excluding mice from the study
| Infarct studies
|
Immunohistochemistry
|
Biochemical analyses
|
Total | ||||
|---|---|---|---|---|---|---|---|
| Group I | Group II | Group III | Group IV | Group V | Groups VI–XX | ||
| Death | 0 | 4 | 0 | 3 | 0 | 2 | 9 |
| Technical problems | 1 | 2 | 0 | 0 | 0 | 4 | 7 |
| Poor postmortem staining | 2 | 1 | 0 | 0 | 0 | 0 | 3 |
| Mice excluded | 3 | 7 | 0 | 3 | 0 | 6 | 19 |
| Mice instrumented | 20 | 16 | 9 | 12 | 6 | 88 | 151 |
| Mice included in study | 17 | 9 | 9 | 9 | 6 | 82 | 132 |
| Mice included in study (%) | 85 | 56 | 100 | 75 | 100 | 93 | 87 |
3.1. Studies of myocardial infarction
3.1.1. Hemodynamic variables
Heart rate and the core body temperature did not differ significantly among the four groups 5 min prior to or at any time during the 30-min coronary occlusion or the ensuing reperfusion (Table 2).
Table 2.
Core body temperature and heart rate on the day of the 30-min coronary occlusion
| Preocclusion | Occlusion
|
Reperfusion
|
|||
|---|---|---|---|---|---|
| 5 min | 30 min | 5 min | 15 min | ||
| Temperature (°C) | |||||
| Group I | 36.8 ± 0.1 | 37.1 ± 0.1 | 37.2 ± 0.1 | 37.0 ± 0.1 | 37.0 ± 0.1 |
| Group II | 36.9 ± 0.1 | 37.3 ± 0.1 | 37.0 ± 0.1 | 37.3 ± 0.1 | 37.0 ± 0.1 |
| Group III | 36.9 ± 0.1 | 37.1 ± 0.0 | 37.0 ± 0.0 | 37.1 ± 0.1 | 37.1 ± 0.1 |
| Group IV | 36.7 ± 0.1 | 37.1 ± 0.1 | 37.0 ± 0.1 | 36.9 ± 0.0 | 37.0 ± 0.1 |
| Heart rate (beats/min) | |||||
| Group I | 544 ± 12 | 570 ± 14 | 588 ± 22 | 584 ± 23 | 600 ± 24 |
| Group II | 551 ± 14 | 566 ± 16 | 565 ± 17 | 572 ± 17 | 576 ± 18 |
| Group III | 511 ± 17 | 564 ± 7 | 555 ± 17 | 563 ± 14 | 597 ± 21 |
| Group IV | 566 ± 17 | 620 ± 21 | 615 ± 23 | 615 ± 24 | 628 ± 22 |
In groups I–IV, mice underwent a 30-min coronary occlusion followed by 24 h of reperfusion. Heart rate was measured 5 min before coronary occlusion (preocclusion), at 5 and 30 min into the 30-min coronary occlusion, and at 5 and 15 min after reperfusion. The experimental protocols are specified in the legend of Fig. 1. Data are mean ± S.E.M.
3.1.2. Region at risk and infarct size
There were no significant differences among groups I, II, III, and IV with respect to left ventricular weight or weight of the region at risk (Table 3). In wild-type mice, the average infarct size was 44.6% smaller in group II than in control animals (group I; Fig. 2), indicating a late PC effect against myocardial infarction. In nonpreconditioned IL-6−/− mice (group III; Fig. 2), infarct size was similar to that observed in the nonpreconditioned wild-type mice (group I; Fig. 2), indicating that in the absence of PC, the ablation of IL-6 neither alleviated nor aggravated myocardial infarction. In preconditioned IL-6−/− mice (group IV; Fig. 2), however, infarct size was similar to that in the control group and significantly larger than that in preconditioned wild-type mice (group II; Fig. 2), indicating that the absence of IL-6 completely abrogated the protective effects of late PC. In groups I, III, and IV, the size of the infarction was positively and linearly related to the size of the region at risk (r = 0.83, 0.92, and 0.99 in groups I, III, and IV, respectively). As expected, the regression line was significantly shifted to the right in group II compared with group I ( P < 0.05 by ANCOVA) (Fig. 3), indicating that late PC reduced infarct size independent of the risk region size. In contrast, in group IV, the regression line was very similar to that in group I and significantly different ( P < 0.05 by ANCOVA) from that in group II, indicating that for any given size of the region at risk, the resulting infarction was larger in preconditioned IL-6−/− than in preconditioned control mice (Fig. 3). Regression equations are given in the legend to Fig. 3.
Table 3.
Size of left ventricle, risk region, and infarct
| Group | Body weight (g) | Heart weight (g) | Left ventricular weight (mg) | Heart weight/body weight (%) | Risk region weight (mg) | Infarct weight (mg) | Risk region (% of left ventricle) | Infarct (% of risk region) | Infarct (% of left ventricle) |
|---|---|---|---|---|---|---|---|---|---|
| I | 31.1 ± 0.8 | 151.8 ± 5.7 | 114.2 ± 4.8 | 0.49 ± 0.02 | 45.5 ± 3.2 | 27.3 ± 2.2 | 40.0 ± 2.7 | 60.5 ± 2.6 | 24.1 ± 1.8 |
| II | 29.6 ± 1.0 | 155.1 ± 5.8 | 112.2 ± 4.1 | 0.53 ± 0.02 | 42.2 ± 2.2 | 14.0 ± 1.5 | 38.2 ± 2.9 | 33.5 ± 3.6* | 12.7 ± 1.5* |
| III | 33.0 ± 1.0 | 166.9 ± 5.7 | 128.0 ± 5.4 | 0.51 ± 0.02 | 49.7 ± 5.1 | 30.5 ± 4.1 | 38.2 ± 2.8 | 59.9 ± 3.8 | 23.4 ± 2.7 |
| IV | 29.5 ± 1.7 | 145.4 ± 9.2 | 110.1 ± 7.9 | 0.49 ± 0.02 | 48.4 ± 5.2 | 32.5 ± 4.8 | 43.2 ± 2.1 | 65.1 ± 3.1 | 28.4 ± 2.4 |
Data are mean ± S.E.M.
Heart weight = total heart weight (ventricles and atria); LV = left ventricle.
P < 0.05 vs. groups I, III, and IV.
Fig. 2.
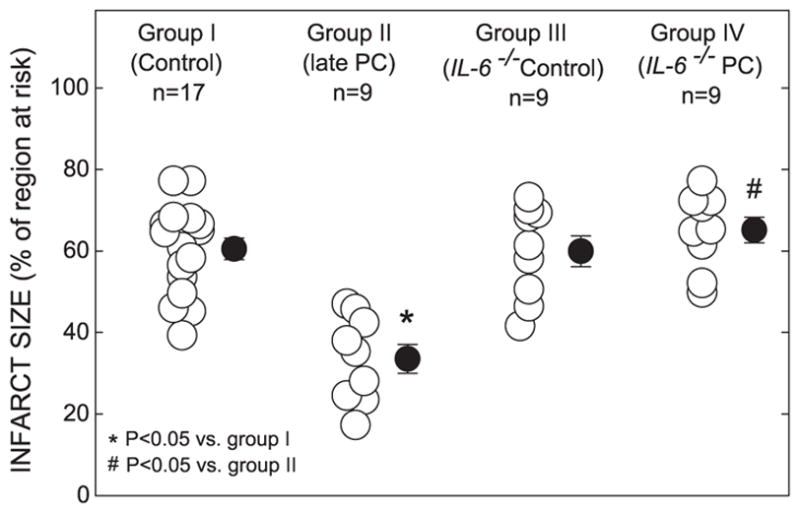
Myocardial infarct size in groups I (control group), II (PC group), III (IL-6−/− control group), and IV (IL-6−/− PC group). Infarct size is expressed as a percentage of the region at risk of infarction. (○) Individual mice. (●) Mean ± S.E.M. (n, number of mice).
Fig. 3.
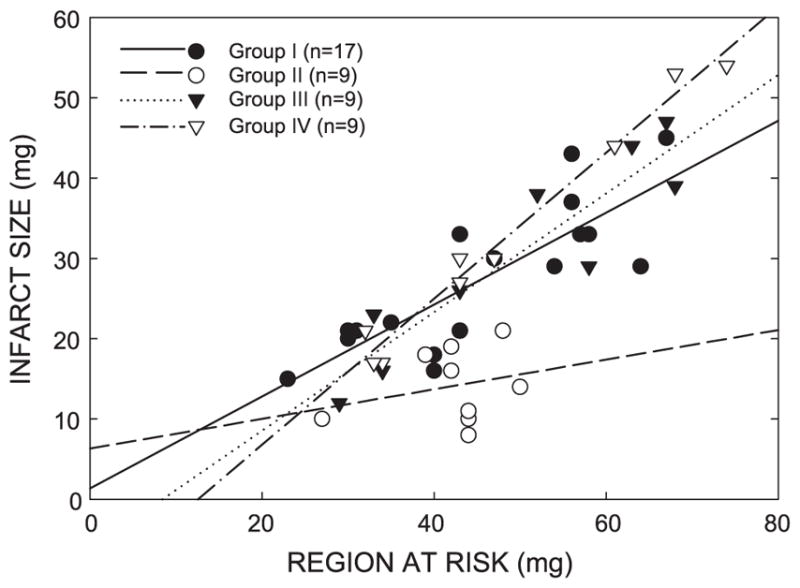
Relationship between size of the region at risk and size of myocardial infarction. Illustrated are both individual values and regression lines obtained by linear regression analysis for groups I (control group), II (PC group), III (IL-6−/− control group), and IV (IL-6−/− PC group). In groups I, III, and IV, infarct size was positively and linearly related to the risk region size. Linear regression equations were as follows: group I, y = 0.572x + 1.37 (r = 0.83); group III, y = 0.784x − 6.234 (r = 0.92); and group IV, y = 0.910x − 11.42 (r = 0.99). For any risk region size, infarct size was smaller in group II compared with groups I and IV. In contrast, for any risk region size, infarct size was similar in groups IV and III. These data demonstrate that late PC reduced infarct size independent of risk region size and that this effect was abrogated in mice lacking IL-6.
3.2. IL-6 expression in cardiomyocytes following ischemic PC
Minimal expression of IL-6 protein was detected in the hearts of sham-operated mice (Fig. 4, panel B). In preconditioned hearts, however, a marked upregulation of immunoreactive IL-6 expression was noted, mostly limited to the ischemic/reperfused region with minimal expression in the nonischemic zone (Fig. 4, panel A). Colocalization of IL-6 and troponin T signals by confocal microscopic examination confirmed the cytoplasmic distribution of intracellular IL-6 in cardiomyocytes (Fig. 4, panels C–E).
Fig. 4.
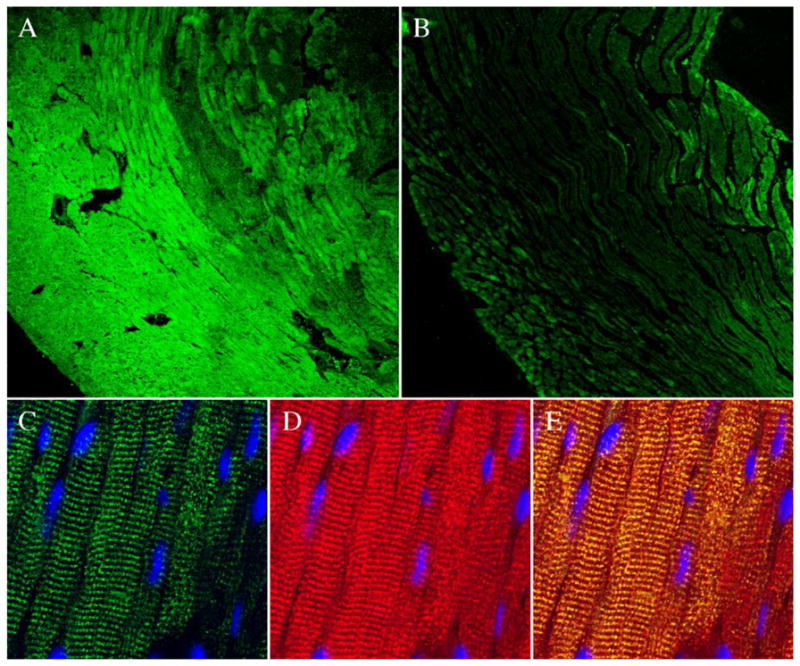
Immunolocalization of myocardial IL-6. Representative lower magnification images (100×, panels A and B) demonstrate increased expression of IL-6 in the ischemic/reperfused myocardium. Minimal IL-6 immunoreactivity (green fluorescence) was noted in the sham-operated hearts (panel B). In contrast, extensive IL-6 expression was noted in the ischemic/reperfused zone in the hearts of preconditioned mice 30 min after the preconditioning ischemia (panel A). Higher magnification images (630 ×, panels C–E) demonstrate the cytoplasmic distribution of IL-6 (panel C, green fluorescence) in cardiomyocytes. Cardiomyocytes are identified by the red fluorescence of troponin T (panel D). Colocalization of IL-6 and troponin T in yellow fluorescence (panel E) confirms the cytoplasmic distribution of intracellular IL-6 in cardiomyocytes. Nuclei are identified by DAPI (panel E, blue fluorescence).
3.3. Myocardial IL-6 protein content
Two hours after the ischemic PC protocol, IL-6 levels in the ischemic/reperfused myocardium were significantly upregulated in preconditioned wild-type mice (52.6 ± 3.2 pg/mg) compared with those in control (8.2 ± 3.5 pg/mg) and sham-operated (16.7 ± 2.6 pg/mg) wild-type mice.
3.4. Activation of JAK1 and JAK2
As shown in Fig. 5, the levels of tyrosine-phosphorylated JAK1 (panel A) and JAK2 (panel B) were similar in control wild-type and IL-6−/− mice. Six cycles of coronary occlusion/reperfusion induced a marked increase in the tyrosine-phosphorylated form of JAK1 and JAK2 in wild-type mice, indicating that both JAK1 and JAK2 are activated by the PC ischemia. However, in preconditioned IL-6−/− mice, the level of tyrosine phosphorylated JAK1 was not different from that in control mice, indicating that ischemic PC failed to activate JAK1 in the absence of IL-6. Activation of JAK2 following the PC protocol was also markedly attenuated in IL-6−/− mice. These results indicate that IL-6 signaling is critical for the ischemic PC-induced activation of JAK1 and is also responsible, at least in part, for the activation of JAK2.
Fig. 5.

Tyrosine phosphorylation of JAK1 and JAK2 by ischemic PC in wild-type and IL-6−/− mice. Myocardial samples were obtained from mice that underwent a sham operation (control) and from the ischemic/reperfused region of preconditioned (PC) wild-type and IL-6−/− mice. All mice were euthanized 5 min after the sixth reperfusion. Upper panels: Western blots showing that the immunoreactivity of the tyrosine-phosphorylated forms of JAK1 (panel A) and JAK2 (panel B) increased markedly 5 min after ischemic PC in wild-type mice, and that this increase was ablated in preconditioned IL-6−/− mice. Lower panels: Densitometric analysis of tyrosine-phosphorylated JAK1 and JAK2 signals. Data are mean ± S.E.M.
3.5. Activation of STAT1 and STAT3
In wild-type mice, six cycles of coronary occlusion and reperfusion resulted in a robust 3.8-fold increase in nuclear pTyr-STAT1 levels 30 min later (100 ± 19.5% in group XIII vs. 381 ± 52.7% in group XIV, P < 0.05; Fig. 6, panel A). In contrast, in IL-6−/−, pTyr-STAT1 levels 30 min after the PC ischemia were significantly lower than those observed in wild-type mice following ischemic PC (188 ± 24.0% in group XVI vs. 381 ± 52.7% in group XIV, P < 0.05; Fig. 6, panel A). Similarly, in wild-type mice, a 3.7-fold increase in nuclear pTyr-STAT3 level was noted 30 min after the ischemic PC stimulus (100 ± 15.7% in group XIII vs. 372 ± 36.5% in group XIV, P < 0.05; Fig. 6, panel B). In IL-6−/− mice, however, the increase in nuclear pTyr-STAT3 levels 30 min after the PC ischemia was significantly less than in wild-type mice (211 ± 11.8% in group XVI vs. 372 ± 36.5% in group XIV, P < 0.05; Fig. 6, panel B). These data indicate that IL-6 signaling plays a crucial role in the ischemic PC-induced tyrosine phosphorylation and subsequent nuclear translocation of activated STAT1 and STAT3. In conjunction with the previous data indicating that tyrosine phosphorylation of JAK1 and JAK2 is dependent on IL-6, these observations demonstrate that IL-6 is necessary for the activation of the myocardial JAK–STAT axis during the development of late PC.
Fig. 6.

Tyrosine phosphorylation of STAT1 and STAT3 by ischemic PC in wild-type and IL-6−/− mice. Nuclear extracts were prepared from mice that underwent a sham operation (control), and from the ischemic/reperfused region of preconditioned (PC) wild-type and IL-6−/− mice that were euthanized 30 min after the sixth reperfusion. Upper panels: Western blots showing that the immunoreactivity of tyrosine-phosphorylated STAT1 (panel A) and STAT3 (panel B) in nuclear extracts increased markedly 30 min after ischemic PC in wild-type mice, and that this increase was significantly attenuated in preconditioned IL-6−/− mice. Lower panels: Densitometric analysis of tyrosine-phosphorylated STAT1 and STAT3 in nuclear extracts at 30 min after PC. Data are mean ± S.E.M.
3.6. Myocardial iNOS and COX-2 protein expression
As expected [17], six cycles of coronary occlusion/reperfusion resulted, 24 h later, in a nearly twofold increase in myocardial iNOS protein content in preconditioned wild-type mice compared with sham-operated wild-type mice (199 ± 27.0% in group XVIII vs. 100 ± 10.1% in group XVII, P < 0.05; Fig. 7). This delayed increase in myocardial iNOS following ischemic PC, however, was abolished in preconditioned IL-6−/− mice (125 ± 8.6% in group XX vs. 100 ± 10.1% in group XVII, P = NS; Fig. 7). Myocardial COX-2 protein content 24 h after a sham operation was similar in wild-type and IL-6−/− mice (100 ± 12.3% in group XVII vs. 102 ± 15.0% in group XIX, P = NS; Fig. 8). The marked increase in myocardial COX-2 protein observed 24 h after ischemic PC in wild-type mice (374 ± 22.1% in group XVIII vs. 100 ± 12.3% in group XVII, P < 0.01; Fig. 8) was markedly blunted in preconditioned IL-6−/− mice (170 ± 19.6% in group XX vs. 374 ± 22.1% in group XVIII, P < 0.05; Fig. 8).
Fig. 7.

Myocardial iNOS expression 24 h after ischemic PC in wild-type and IL-6−/− mice. Myocardial samples were obtained from mice that underwent a sham operation (control) and from the ischemic/reperfused region of preconditioned (PC) wild-type and IL-6−/− mice. Upper panels: Western blot showing that the immunoreactivity of iNOS in the cytosolic fraction increased 24 h after ischemic PC, and that this increase was ablated in the absence of IL-6. Lower panels: Densitometric analysis of immunoreactive iNOS. Data are mean ± S.E.M.
Fig. 8.

Myocardial COX-2 expression 24 h after ischemic PC in wild-type and IL-6−/− mice. Myocardial samples were obtained from mice that underwent a sham operation (control) and from the ischemic/reperfused region of preconditioned (PC) wild-type and IL-6−/− mice. Upper panel: Western blots showing a marked increase in COX-2 immunoreactivity in the membranous fraction in the ischemic/reperfused region 24 h after ischemic PC in wild-type mice. Lower panel: Densitometric analysis of COX-2 signals in the membranous fraction. In all samples, the densitometric measurements of COX-2 immunoreactivity were expressed as a percentage of the average value measured in the anterior left ventricular wall of sham-operated wild-type mice. Data are mean ± S.E.M.
4. Discussion
Recent studies have revealed that IL-6 is produced in cardiomyocytes [12,13] and that the expression of IL-6, IL-6Rα, and gp130 is upregulated following myocardial ischemia/reperfusion [11]. However, the functional consequences of IL-6 signaling during myocardial infarction remain unknown. Furthermore, although IL-6 is known to activate the JAK–STAT pathway [7,9], which is an essential component of late PC signaling [4], the role of IL-6 in the development of late PC has not been explored. We addressed these unresolved issues in an in vivo murine model of myocardial ischemia/reperfusion injury in which physiologic variables that may potentially affect infarct size, including core body temperature, heart rate, arterial pressure, and arterial pH, were carefully controlled throughout the experiments [14,15].
4.1. Salient findings
The present findings can be summarized as follows: (i) in the naïve (nonpreconditioned) state, the absence of IL-6 does not affect the size of myocardial infarction; (ii) ischemic PC increases the expression of IL-6 in the cytosol of cardiomyocytes; and (iii) ablation of IL-6 prevents the activation of the JAK–STAT pathway, the ensuing upregulation of iNOS and COX-2 (the mediators of late PC), and the development of late PC. To our knowledge, this is the first study to investigate the effect of IL-6 gene deletion on myocardial infarct size following ischemia/reperfusion. This is also the first study to demonstrate an obligatory role of IL-6 in the genesis of late PC and to identify IL-6 as a proximal element required for activation of the cardiac JAK–STAT axis and induction of iNOS and COX-2 in the heart.
4.2. IL-6 signaling in acute myocardial ischemia/reperfusion injury
Deletion of the IL-6 gene has recently been reported to have no effect on infarct size and left ventricular remodeling after a permanent coronary occlusion in mice [18]. This finding, however, does not address the issue of whether IL-6 modulates infarct size because after a permanent coronary occlusion, cardiac myocytes in the ischemic region will die regardless of cytokines. In the present study, infarct size following a 30-min coronary occlusion and 24 h of reperfusion was not different between IL-6−/− mice and wild-type controls, indicating that IL-6 does not modulate the extent of lethal ischemia/reperfusion injury in naïve myocardium. Our results, however, demonstrate that the protection against infarction afforded by late PC was completely abrogated in IL-6−/− mice, indicating that IL-6-dependent signaling is essential for the development of this adaptation. This is the first report to demonstrate the involvement of IL-6 in late PC. The use of a mouse model of IL-6 gene deletion enabled us to specifically and conclusively determine the significance of IL-6 signaling in the genesis of this response.
4.3. Activation of the JAK–STAT pathway by IL-6
After demonstrating that IL-6 is obligatorily required for late PC, we conducted a series of studies to gain insights into the precise role of IL-6 in the signaling cascade that begins with ischemia and culminates in upregulation of protective genes. We have previously demonstrated that the JAK–STAT pathway plays an essential role in the genesis of late PC, and that JAK1 and JAK2 are activated very early (within minutes) after the ischemic PC stimulus [4]. However, the mechanism(s) leading to JAK–STAT activation after ischemic PC is virtually unknown. We reasoned that examining the role of IL-6 in JAK activation would help to elucidate this issue while clarifying the position of IL-6 in the cascade of late PC. It is well established that the JAK–STAT pathway can be recruited by several members of the IL-6 family of cytokines, IFN-γ, and other interleukins [19]. Our finding that ischemic PC-induced phosphorylation of JAK1 and JAK2 (and the consequent tyrosine phosphorylation of STAT1 and STAT3) was markedly suppressed in IL-6−/− mice (Figs. 5 and 6) demonstrates that IL-6 plays an essential and nonredundant role in the activation of the JAK–STAT pathway following the PC stimulus. It also implies that IL-6 is proximal to these kinases/transcription factors.
One possibility is that IL-6 may be a link between NO and JAKs. NO, possibly generated by the shear stress-induced activation of endothelial NO synthase (eNOS) during ischemia/reperfusion, is a ‘trigger’ of the late PC signaling cascade [2]. Although the interactions between NO and the cytokine network in the heart are poorly understood, inhibitors of the NO pathway have been reported to reduce IL-6 production in various other tissues and cell lines. Specifically, inhibition of NO generation by NG-monomethyl-L-arginine (L-NMMA) has been shown to reduce IL-6 production in Thy1+ fibroblasts and human endothelial cell lines in vitro [20] and in mice foot pads in vivo [21]. Thus, it is possible that NO generated by eNOS in response to the PC ischemia upregulates IL-6 synthesis in the ischemic/reperfused myocardium followed by the activation of the JAK–STAT pathway. Further studies will be necessary to determine the precise position of IL-6 in the signaling pathway that leads to late PC.
4.4. IL-6 signaling and the induction of myocardial iNOS and COX-2
Extensive evidence has shown that iNOS is an essential co-mediator of late PC [2,22,23]. Various PC stimuli upregulate iNOS [17,24,25], and both pharmacologic inhibition of iNOS [22,23] and genetic deletion of the iNOS gene [15] abrogate the beneficial effects of late PC. Inhibition of nuclear factor-κB (NF-κB) activation prevents iNOS upregulation following ischemic PC [26]. However, the promoter region of the iNOS gene harbors binding sites for multiple transcription factors, including interferon (IFN)-γ response elements (IREs), γ-IFN activation sites (GAS), and κB sites [27], and iNOS transcription is known to be controlled by several transcription factors that act synergistically [28,29]. The interaction of STAT1 with the GAS site has been shown to be necessary for the expression of iNOS after exposure to cytokines or LPS in murine macrophages [30]. IL-6-induced JAK2/STAT3 activation has been reported to increase iNOS expression and NO production in adult rat ventricular myocytes [31]. Our finding that absence of IL-6 signaling resulted in complete abrogation of iNOS induction (Fig. 7) indicates that IL-6-induced activation of STAT1 and/or STAT3 plays a critical role in the transcriptional activation of the iNOS gene after the PC ischemia. These data are the first to implicate IL-6 in the regulation of cardiomyocyte iNOS. In conjunction with our previous findings of an obligatory role of NF-κB in iNOS induction [26], the present results suggest a synergistic role of NF-κB and STAT1/STAT3 in the transcriptional upregulation of iNOS following ischemic PC, consistent with the concept that late PC requires the concerted activation of multiple molecular pathways [2]. A previous study has suggested a synergistic role of NF-κB activation and IRF-1 induction in LPS-induced iNOS induction in neonatal rat cardiomyocytes [32]. However, to our knowledge, this is the first evidence that the upregulation of cardiac iNOS requires the combinatorial action of STAT1/STAT3 and NF-κB.
COX-2 plays a nonredundant role as a co-mediator of late PC [16]. Recent evidence from our laboratory has identified an obligatory role of the JAK–STAT pathway in the upregulation of myocardial COX-2, 24 h after ischemic PC [5]. The present results are consistent with these findings, since deletion of IL-6 not only prevented the activation of the JAK–STAT pathway but also significantly attenuated the increase in COX-2 expression. As was the case for iNOS, this is the first demonstration of a causal role played by IL-6 in myocardial COX-2 induction.
5. Conclusions
We have demonstrated that IL-6 does not aggravate or ameliorate myocardial infarction in the naïve state. However, ischemic PC markedly upregulates IL-6 expression in cardiomyocytes, and IL-6 signaling is critical for ischemic PC-induced activation of the JAK–STAT pathway, induction of the mediators of protection, upregulation of iNOS and COX-2, and development of protection. We have also shown that IL-6 acts at a very early stage of the signaling pathway of late PC and that it is necessary for (and proximal to) JAK–STAT activation. These results add another element to the already complex cascade that underlies the adaptation of the heart to ischemic stress. The identification of IL-6 as an essential molecule upstream of the JAK–STAT pathway expands our understanding of the molecular mechanisms that underlie the phenomenon of late PC as well as myocardial IL-6 signaling during ischemia/reperfusion injury.
Acknowledgments
This study was supported, in part, by NIH grants R01 HL-72410 (B.D.), HL-65660 (Y.-T.X), HL-43151, HL-55757, HL-68088, HL-70897, and HL-76794 (R.B.); AHA Scientist Development grant 0130146N (B.D.); AHA Ohio Valley Affiliate grants 0265087B (Y.G.) and 0325372B (A.B.S.); the Medical Research Grant Program of the Jewish Hospital Foundation (Louisville, KY); and the Commonwealth of Kentucky Research Challenge Trust Fund. We gratefully acknowledge Sheron Lear for expert technical assistance.
References
- 1.Kuzuya T, Hoshida S, Yamashita N, et al. Delayed effects of sublethal ischemia on the acquisition of tolerance to ischemia. Circ Res. 1993;72:1293–9. doi: 10.1161/01.res.72.6.1293. [DOI] [PubMed] [Google Scholar]
- 2.Bolli R. The late phase of preconditioning. Circ Res. 2000;87:972–83. doi: 10.1161/01.res.87.11.972. [DOI] [PubMed] [Google Scholar]
- 3.Yellon DM, Downey JM. Preconditioning the myocardium: from cellular physiology to clinical cardiology. Physiol Rev. 2003;83:1113–51. doi: 10.1152/physrev.00009.2003. [DOI] [PubMed] [Google Scholar]
- 4.Xuan YT, Guo Y, Han H, Zhu Y, Bolli R. An essential role of the JAK–STAT pathway in ischemic preconditioning. Proc Natl Acad Sci U S A. 2001;98:9050–5. doi: 10.1073/pnas.161283798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xuan YT, Guo Y, Zhu Y, et al. Mechanism of cyclooxygenase-2 upregulation in late preconditioning. J Mol Cell Cardiol. 2003;35:525–37. doi: 10.1016/s0022-2828(03)00076-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hirano T. The biology of interleukin-6. Chem Immunol. 1992;51:153–80. [PubMed] [Google Scholar]
- 7.Heinrich PC, Behrmann I, Muller-Newen G, Schaper F, Graeve L. Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem J. 1998;334:297–314. doi: 10.1042/bj3340297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hirano T. Interleukin 6 and its receptor: ten years later. Int Rev Immunol. 1998;16:249–84. doi: 10.3109/08830189809042997. [DOI] [PubMed] [Google Scholar]
- 9.Hirano T, Nakajima K, Hibi M. Signaling mechanisms through gp130: a model of the cytokine system. Cytokine Growth Factor Rev. 1997;8:241–52. doi: 10.1016/s1359-6101(98)80005-1. [DOI] [PubMed] [Google Scholar]
- 10.Kukielka GL, Smith CW, Manning AM, Youker KA, Michael LH, Entman ML. Induction of interleukin-6 synthesis in the myocardium. Potential role in postreperfusion inflammatory injury. Circulation. 1995;92:1866–75. doi: 10.1161/01.cir.92.7.1866. [DOI] [PubMed] [Google Scholar]
- 11.Chandrasekar B, Mitchell DH, Colston JT, Freeman GL. Regulation of CCAAT/enhancer binding protein, interleukin-6, interleukin-6 receptor, and gp130 expression during myocardial ischemia/reperfusion. Circulation. 1999;99:427–33. doi: 10.1161/01.cir.99.3.427. [DOI] [PubMed] [Google Scholar]
- 12.Gwechenberger M, Mendoza LH, Youker KA, et al. Cardiac myocytes produce interleukin-6 in culture and in viable border zone of reperfused infarctions. Circulation. 1999;99:546–51. doi: 10.1161/01.cir.99.4.546. [DOI] [PubMed] [Google Scholar]
- 13.Yamauchi-Takihara K, Ihara Y, Ogata A, Yoshizaki K, Azuma J, Kishimoto T. Hypoxic stress induces cardiac myocyte-derived inter-leukin-6. Circulation. 1995;91:1520–4. doi: 10.1161/01.cir.91.5.1520. [DOI] [PubMed] [Google Scholar]
- 14.Guo Y, Wu WJ, Qiu Y, Tang XL, Yang Z, Bolli R. Demonstration of an early and a late phase of ischemic preconditioning in mice. Am J Physiol. 1998;275:H1375–87. doi: 10.1152/ajpheart.1998.275.4.H1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guo Y, Jones WK, Xuan YT, et al. The late phase of ischemic preconditioning is abrogated by targeted disruption of the inducible NO synthase gene. Proc Natl Acad Sci USA. 1999;96:11112–507. doi: 10.1073/pnas.96.20.11507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shinmura K, Tang XL, Wang Y, et al. Cyclooxygenase-2 mediates the cardioprotective effects of the late phase of ischemic preconditioning in conscious rabbits. Proc Natl Acad Sci USA. 2000;97:10197–202. doi: 10.1073/pnas.97.18.10197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xuan YT, Tang XL, Qiu Y, et al. Biphasic response of cardiac NO synthase isoforms to ischemic preconditioning in conscious rabbits. Am J Physiol, Heart Circ Physiol. 2000;279:H2360–71. doi: 10.1152/ajpheart.2000.279.5.H2360. [DOI] [PubMed] [Google Scholar]
- 18.Fuchs M, Hilfiker A, Kaminski K, et al. Role of interleukin-6 for LV remodeling and survival after experimental myocardial infarction. FASEB J. 2003;17:2118–20. doi: 10.1096/fj.03-0331fje. [DOI] [PubMed] [Google Scholar]
- 19.Imada K, Leonard WJ. The JAK – STAT pathway. Mol Immunol. 2000;37:1–11. doi: 10.1016/s0161-5890(00)00018-3. [DOI] [PubMed] [Google Scholar]
- 20.Willis RA, Nussler AK, Fries KM, Geller DA, Phipps RP. Induction of nitric oxide synthase in subsets of murine pulmonary fibroblasts: effect on fibroblast interleukin-6 production. Clin Immunol Immunopathol. 1994;71:231–9. doi: 10.1006/clin.1994.1077. [DOI] [PubMed] [Google Scholar]
- 21.Ianaro A, O’Donnell CA, Di Rosa M, Liew FY. A nitric oxide synthase inhibitor reduces inflammation, down-regulates inflammatory cytokines and enhances interleukin-10 production in carrageenin-induced oedema in mice. Immunology. 1994;82:370–5. [PMC free article] [PubMed] [Google Scholar]
- 22.Bolli R, Manchikalapudi S, Tang XL, et al. The protective effect of late preconditioning against myocardial stunning in conscious rabbits is mediated by nitric oxide synthase: evidence that nitric oxide acts both as a trigger and as a mediator of the late phase of ischemic preconditioning. Circ Res. 1997;81:1094–107. doi: 10.1161/01.res.81.6.1094. [DOI] [PubMed] [Google Scholar]
- 23.Takano H, Manchikalapudi S, Tang XL, et al. Nitric oxide synthase is the mediator of late preconditioning against myocardial infarction in conscious rabbits. Circulation. 1998;98:441–9. doi: 10.1161/01.cir.98.5.441. [DOI] [PubMed] [Google Scholar]
- 24.Guo Y, Xuan YT, Wu WJ, Li Q, Tang XL, Bolli R. Nitric oxide plays a dual role in exercise-induced late preconditioning. Circulation. 2001;104(Suppl II):II-60. [Google Scholar]
- 25.Zhao TC, Kukreja RC. Late preconditioning elicited by activation of adenosine A(3) receptor in heart: role of NF-kappa B, iNOS and mitochondrial K(ATP) channel. J Mol Cell Cardiol. 2002;34:263–77. doi: 10.1006/jmcc.2001.1510. [DOI] [PubMed] [Google Scholar]
- 26.Xuan YT, Tang XL, Banerjee S, et al. Nuclear factor-kappaB plays an essential role in the late phase of ischemic preconditioning in conscious rabbits. Circ Res. 1999;84:1095–109. doi: 10.1161/01.res.84.9.1095. [DOI] [PubMed] [Google Scholar]
- 27.Xie QW, Whisnant R, Nathan C. Promoter of the mouse gene encoding calcium-independent nitric oxide synthase confers inducibility by interferon gamma and bacterial lipopolysaccharide. J Exp Med. 1993;177:1779–84. doi: 10.1084/jem.177.6.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sakitani K, Nishizawa M, Inoue K, Masu Y, Okumura T, Ito S. Synergistic regulation of inducible nitric oxide synthase gene by CCAAT/enhancer-binding protein beta and nuclear factor-kappaB in hepatocytes. Genes Cells. 1998;3:321–30. doi: 10.1046/j.1365-2443.1998.00193.x. [DOI] [PubMed] [Google Scholar]
- 29.Saura M, Zaragoza C, Bao C, McMillan A, Lowenstein CJ. Interaction of interferon regulatory factor-1 and nuclear factor kappaB during activation of inducible nitric oxide synthase transcription. J Mol Biol. 1999;289:459–71. doi: 10.1006/jmbi.1999.2752. [DOI] [PubMed] [Google Scholar]
- 30.Gao J, Morrison DC, Parmely TJ, Russell SW, Murphy WJ. An interferon-gamma-activated site (GAS) is necessary for full expression of the mouse iNOS gene in response to interferon-gamma and lipopolysaccharide. J Biol Chem. 1997;272:1226–30. doi: 10.1074/jbc.272.2.1226. [DOI] [PubMed] [Google Scholar]
- 31.Yu X, Kennedy RH, Liu SJ. JAK2/STAT3, not ERK1/2, mediates interleukin-6-induced activation of inducible nitric-oxide synthase and decrease in contractility of adult ventricular myocytes. J Biol Chem. 2003;278:16304–9. doi: 10.1074/jbc.M212321200. [DOI] [PubMed] [Google Scholar]
- 32.Kinugawa K, Shimizu T, Yao A, Kohmoto O, Serizawa T, Takahashi T. Transcriptional regulation of inducible nitric oxide synthase in cultured neonatal rat cardiac myocytes. Circ Res. 1997;81:911–21. doi: 10.1161/01.res.81.6.911. [DOI] [PubMed] [Google Scholar]