

Abstract

Fluorogenic molecules are important tools for advanced biochemical and biological experiments. The extant collection of fluorogenic probes is incomplete, however, leaving regions of the electromagnetic spectrum unutilized. Here, we synthesize green-excited fluorescent and fluorogenic analogues of the classic fluorescein and rhodamine 110 fluorophores by replacement of the xanthene oxygen with a quaternary carbon. These anthracenyl “carbofluorescein” and “carborhodamine 110” fluorophores exhibit excellent fluorescent properties and can be masked with enzyme- and photolabile groups to prepare high-contrast fluorogenic molecules useful for live cell imaging experiments and super-resolution microscopy. Our divergent approach to these red-shifted dye scaffolds will enable the preparation of numerous novel fluorogenic probes with high biological utility.
Organic fluorophores are invaluable tools for many biological assays.1,2 A key advantage of small molecule dyes is the ability to tune and control the optical properties of the fluorophore using chemistry. In particular, many dyes can be “masked” by attachment of a blocking moiety that suppresses fluorescence. Unmasking by a specific chemical event elicits a large increase in fluorescence intensity. Fluorogenic molecules find broad utility in biology,1−4 being used as reporters for enzyme activity,5−8 sensors for analytes,9 and agents for sophisticated imaging experiments10−13 including super-resolution microscopy.14 Although the current collection of small molecule fluorophores easily spans the ultraviolet, visible, and near-infrared spectrum,2 only a subset of these dyes can be easily masked. Fluorogenic molecules excited in the ultraviolet (coumarins),5,8 blue (xanthenes),7,8,10,12,14 orange (resorufins),6,9,11 and red (acridinones)13 are well established. However, there is no general scaffold for activatable molecules excited with green light (∼545 nm), leaving this spectral region underutilized in experiments involving fluorogenic compounds.
An attractive solution to this problem is to extend the wavelength of activatable derivatives of the blue-excited xanthene fluorophores such as fluorescein (Fl, 1, Figure 1a) and rhodamine 110 (Rh110, 2). These dyes find extensive use in biological research owing to their high absorptivity, excellent fluorescence properties, modularity, and ever-improving synthetic chemistry.2,14−16 The xanthene dyes are particularly useful scaffolds for high contrast fluorogenic molecules due to an equilibrium between an open, highly absorbing, fluorescent quinoid form and a closed, colorless, nonfluorescent lactone. This open–closed equilibrium can be controlled by attachment of a broad collection of blocking groups on the oxygens of fluoresceins (O-alkylation/acylation) or the aniline nitrogens of rhodamines (N-acylation), which shifts the equilibrium significantly to the nonfluorescent lactone form. Removal of one or both labile moieties recapitulates the fluorescent form, yielding a large increase in fluorescence.12
Figure 1.

(a) Chemical structures of xanthene dyes and carbon-containing isologues. (b) Existing synthetic strategy to carborhodamine dyes. (c) Divergent synthesis of carborhodamines through carbofluorescein intermediates.
Nonetheless, most structural adjustments that increase the absorption (λmax) and emission (λem) wavelengths of the xanthene dyes also compromise their function as fluorogenic molecules. Halogenation of fluoresceins extends λmax and λem by up to 70 nm but also severely diminishes fluorescence quantum yield.17 Alkylation of the rhodamine nitrogens elicits a pronounced bathochromic shift in λmax and λem,15 but this structural modification complicates14 or precludes the installation of blocking groups. For example, tetramethylrhodamine (3, Figure 1a) is a widely used dye in biological imaging2,15 but cannot be masked using N-acylation strategies.
An alternative method to shift the absorption and emission wavelengths of rhodamine and fluorescein dyes is to modify the xanthene core. Substitution of the xanthene oxygen with other atoms (e.g., Se,18 C,19−21 Si,22−25 Te (26)) yields dyes with red-shifted spectra. In particular, substitution with a quaternary carbon elicits a 50-nm bathochromic shift,19 suggesting that “carbofluorescein” (CFl, 4, Figure 1a) and “carborhodamine 110” (CRh110, 5) could serve as valuable scaffolds for green-excited fluorogenic compounds. However, despite their deceptively simple structures, investigations of such modified fluoresceins and rhodamines have been severely limited by inefficient synthetic organic chemistry. The classic route to xanthene dyes, the condensation of a phthalic anhydride with a resorcinol or aminophenol,16 is not amenable to the synthesis of these anthracenyl scaffolds. Figure 1b shows the existing strategy to prepare carborhodamines, involving addition of a simple aryllithium species to a highly substituted diaminoanthrone. This convergent synthesis has been used to prepare only two N-alkylated carborhodamine scaffolds as red fluorescent labels,19,20 with a singular example of a masked derivative.21 Synthesis of less functionalized carborhodamines using this route would require robust, multistep protection–deprotection strategies; such considerations have stymied the preparation of fluorophores such as compound 5 and related fluorogenic N-acyl derivatives.
Figure 1c shows an alternative strategy to these xanthene dye isologues starting from derivatives of carbofluorescein (4), itself a novel fluorescent dye. Efficient conversion of these fluorescein isologues to rhodamine-like dyes could be accomplished using Buchwald–Hartwig cross-coupling.16,27 This divergent approach allows direct access to not only fluorescent N-alkyl derivatives but also fluorogenic N-acyl dyes and N-aryl carborhodamines. In this report we synthesize carbofluorescein (4), carborhodamine 110 (5), and an array of fluorescent and fluorogenic derivatives. We find these dyes exhibit interesting chemical and optical properties, making them excellent scaffolds for high contrast, green-absorbing fluorogenic derivatives useful for live cell fluorescence imaging and super-resolution microscopy.
Results and Discussion
Synthesis of Carbofluoresceins
We sought to synthesize carbofluorescein through the attachment of the bottom aryl ring through addition of an arylmetal species23,28,29 to an appropriately protected anthracenyl ketone derivative. To that end, a convenient, high-yielding synthesis of novel anthrone 12 was developed as shown in Scheme 1. Negishi cross-coupling30 of commercially available aryl bromide 6 with 4-methoxybenzylzinc chloride (7) afforded methyl benzylbenzoate 8 in nearly quantitative yield. Addition of methylmagnesium bromide to 8 provided the hydroxyisopropyl derivative 9 in 88% yield. Reaction of 9 with an excess of BBr3 resulted in demethylation and concomitant cyclization31 to efficiently form the necessary dihydroanthracene 10 in excellent yield (91%). DDQ-mediated oxidation installed the ketone moiety to yield 11 and was followed by protection with TBSCl to provide bis-protected anthrone 12 in multigram quantities.
Scheme 1. Synthesis of Carbofluoresceins.
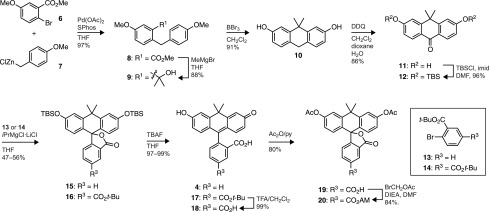
With ketone 12 in hand, we proceeded to install the bottom aryl group possessing the ortho-carboxyl group necessary for fluorogenicity. Previous attempts to assemble xanthene and xanthene-like dyes through this arylmetal–ketone approach have encountered difficulties in the metalation and subsequent addition of protected carboxyaryl halides. Aryllithium nucleophiles add efficiently to xanthones and isologous structures,20,23,25,28 but use of these highly reactive species requires highly stable carboxylic acid protecting groups. Two strategies have been used: tert- or sec-butyl esters23,25 and 4,4-dimethyl-2-oxazoline protected carboxyl groups.20 In initial experiments, we prepared carbofluorescein (4) via the lithiation of either 2-bromobenzoate esters or o-oxazolinylphenyl bromides and addition to anthrone 12. The ester protecting groups gave highly variable results, limiting the utility of this reaction sequence. Metalation and addition of o-oxazolinylphenyl bromides to anthrone 12 was more consistent, but only concentrated HCl at elevated temperature (100 °C) and extended reaction time (>48 h) could effect the desired hydrolysis of the carboxyl synthon in modest yield.
These synthetic difficulties led to the investigation of other halide/metal exchange conditions. Addition of aryl Grignard reagents to xanthones has also been used install pendant phenyl rings in xanthene dyes. The lower reactivity of aryl Grignard reagents is attractive, as these reagents tolerate a wider range of chemical functionality. In most cases, however, these syntheses use simple aryl groups;29,32 the successful addition of o-ester aryl Grignards to xanthones has not been described. We found that Knochel’s iPrMgCl·LiCl complex (the “Turbo Grignard”)33 allowed for mild Br/Mg exchange of tert-butyl ester 13 or 14 and subsequent addition to 12 in reasonable yield (56% and 47%, respectively, Scheme 1). In addition, gentle quenching of the reaction (NH4Cl) preserved the silyl ethers and directly provided the easily purified “lactone-locked” carbofluoresceins, presumably through an adventitious cyclization of the putative carbinol intermediate onto the adjacent tert-butyl ester. The use of diester 14 allowed for the introduction of a second ester as a functional handle, providing 5-carboxy-carbofluorescein derivative 16 as a single regioisomer bearing orthogonal protecting groups on the upper and lower portions of the molecule. Straightforward deprotection of 15 with TBAF gave the fluorescent carbofluorescein 4. A fluorogenic esterase substrate could be prepared by stepwise silyl ether and tert-butyl ester deprotection of 16, followed by esterification to access the acetoxymethyl (AM) ester of 5-carboxycarbofluorescein diacetate (20) in excellent overall yield.
Synthesis of Carborhodamines
In a previous communication, we detailed a general strategy for the conversion of fluoresceins to rhodamines via palladium-catalyzed C–N cross-coupling of fluorescein ditriflates.16 We envisioned using the same protocol to easily access carborhodamines from the novel carbofluoresceins. As illustrated in Table 1, carbofluoresceins 4 and 17 were converted to the corresponding ditriflates (21 and 22) through reaction with Tf2O. Cross-coupling of these triflates with tert-butyl carbamate cleanly afforded Boc-masked carborhodamines 23 (88%) and 24 (80%). Carborhodamine 110 (5) and 5-carboxycarborhodamine 110 (29) were then liberated in nearly quantitative yields via deprotection with TFA. More elaborate nitrogen nucleophiles were also tolerated; the photolabile o-nitroveratryl carbamate (NVOC-NH2) reacted with 22 to give NVOC-caged, tert-butyl-protected 5-carboxy-CRh110 (25, entry 3, 82%). A carborhodamine substrate bearing the esterase-labile “trimethyl lock” moiety12 (26) was similarly prepared through coupling of 21 with the trimethyl lock amide (entry 4). The scope of this process also encompassed amines (entries 5 and 6), as evidenced by the synthesis of presumed fluorescent dye 27 and N,N′-diarylcarborhodamine 28 in high yields.
Table 1. Conversion of Carbofluoresceins to Carborhodaminesa.
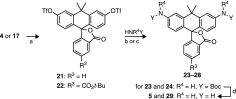
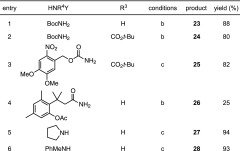
Reagents and conditions: (a) Tf2O,py,CH2Cl2, 0 °C to rt, 76–83%; (b)HNR4Y, Pd2dba3, Xantphos, Cs2CO3, dioxane, 80–100 °C; (c)HNR4Y, Pd2dba3, XPhos, Cs2CO3, dioxane, 80–100 °C; TFA/CH2Cl2, rt, 86–98%.
Properties of Carbofluoresceins and Carborhodamines
We then evaluated the optical properties of these novel dyes, comparing them to their xanthene-based fluorescein (1), rhodamine 110 (2), and tetramethylrhodamine (3) isologues. The results are shown in Figure 2a, and representative spectra are given in Supplementary Figure S1. The carbofluorescein (4) showed a significant (53-nm) bathochromic shift in λmax relative to fluorescein (1) with a higher extinction coefficient (ε = 108 000 M–1 cm–1) but lower quantum yield (Φ = 0.62). Both the λmax and λem values are similar to those of tetramethylrhodamine, and carbofluorescein exhibits a greater relative brightness (ε × Φ) compared to that of 3. We also determined the phenolic pKa value of 4 as shown in Figure 2b. CFl exhibits a pKa value of 7.4, close to the physiological pH and higher than that of fluorescein (pKa = 6.4).34 Another key difference between fluorescein and carbofluorescein is the optical properties of the protonated species. Although fluorescein retains significant absorption and fluorescence upon protonation, carbofluorescein is colorless and nonfluorescent at low pH (Supplementary Figure S2a,b). We note protonation of 4 is cooperative (Hill coefficient = 1.33, Figure 2b), suggesting the carbofluorescein monoanion can favorably adopt the lactone form, which can then undergo further protonation (see Supplementary Figure S2c for an equilibrium diagram). This attribute is similar to the properties of the isologous colormetric pH indicator phenolphthalein.35 Thus, in addition to excellent fluorescence properties, carbofluorescein could prove a useful scaffold for red-shifted, high contrast fluorogenic sensors for pH34 or other analytes.36
Figure 2.
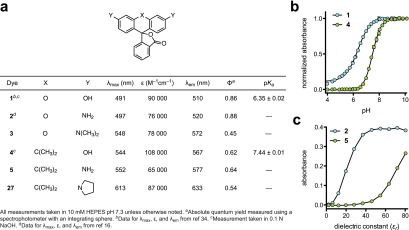
Properties of fluorescent dyes. (a) Spectral properties of dyes. (b) Normalized absorbance at λmax versus pH for fluorescein (1) and carbofluorescein (4). Error bars show standard error (SE; n = 2). Determined values (±SE): compound 1, pKa = 6.35 ± 0.02, Hill coefficient = 0.93 ± 0.02; compound 4, pKa = 7.44 ± 0.01, Hill coefficient = 1.33 ± 0.03. (c) Absorption at λmax versus dielectric constant for rhodamine 110 (2) and carborhodamine 110 (5). Error bars show SE (n = 2).
Like carbofluorescein, the carborhodamine 110 fluorophore (5) exhibited a 45-nm bathochromic shift relative to the parent rhodamine 2 with λmax/λem = 552/577 nm. These spectral properties are similar to those of tetramethylrhodamine (3), and the relative brightness (ε × Φ) of these dyes is comparable. Carborhodamine bears attachment points for blocking groups, however, allowing synthesis of fluorogenic N,N′-diacyl dyes 25 and 26, which are locked in the colorless, nonfluorescent form. As with the rhodamine dyes, N-alkylation elicits a further bathochromic shift. The tetraalkyl pyrrolidylcarborhodamine 27 exhibits λmax = 613 nm and λem = 633 nm with high ε and Φ values.
We also examined the open–closed equilibrium of both the rhodamine and carborhodamine system. In polar media, such as water, the open zwitterionic form should predominate. In lipophilic environments, however, the closed lactone form should be favored.37 We investigated the relative propensity of these dyes to adopt the closed lactone form by dissolving carborhodamine 5 and the corresponding rhodamine isologue 2 in different ratios of dioxane/water and measuring changes in absorption.38 Dioxane and water are fully miscible but have widely different solvent polarities; the dielectric constant (εr) value is 80.4 for water and 2.24 for dioxane.39 As shown in Figure 2c, rhodamine 110 (2) fully adopts the colorless lactone form only in 100% dioxane. Conversely, carborhodamine became colorless even in solutions of moderate polarity, with 40% dioxane (εr = 46.0) sufficient to fully shift the equilibrium to the closed form. We also found the N,N′-diaryl-carborhodamine dye 28 to be colorless in aqueous solution, contrasting the isologous N,N′-dimethyldiphenylrhodamine, which exhibits significant visible absorption in water (λmax = 549 nm, ε = 16 000 M–1 cm–1; Supplementary Figure S3).16 These data show the open–closed equilibrium of the carborhodamine scaffold is significantly shifted toward the closed lactone form relative to the rhodamine dyes.
Live Cell Imaging
We then tested if established masking strategies for fluorescein and rhodamine dyes could be applied to CFl and CRh110 and used in a cellular context. Fluorescein diacetate and its derivatives are widely used as a general cellular stain for microscopy and to determine cell viability.1,10 As shown in Figure 3a, the red-shifted carbofluorescein diacetate 20 also serves as a substrate for cellular esterases and gave bright, largely diffuse labeling throughout the cell, including the nucleus. We note the relatively high pKa and large contrast of carbofluorescein likely limits observable staining to cellular regions with pH > 6.5. Likewise, the carborhodamine-based trimethyl lock12 fluorogenic esterase substrate 26 shows reactivity with cellular esterases. Substrate 26 is cleaved more slowly than the carbofluorescein diacetate inside cells, however, and localizes in subcellular regions such as the mitochondria (Figure 3b, Supplementary Figure S5a). This result is consistent with the isologous rhodamine 110-based compound,7,16 which exhibited a similar unmasking rate in our experiments and also yields a reticulated staining pattern (Supplementary Figure S5b).
Figure 3.

Confocal microscopy of live, unwashed HeLa cells incubated with esterase substrates 20 or 26 and counterstained with Hoechst 33342; scale bars = 10 μm. (a) Compound 20, 1 h incubation. (b) Compound 26, 24 h incubation.
Super-Resolution Cellular Imaging
We then investigated derivatives of this dye class as labels for photoactivated localization microscopy (PALM).14,40 This super-resolution microscopy technique uses stochastic sequential triggering and localization of photoactivatable labels to enable imaging below the diffraction limit of light. Several types of photoactivatable fluorophores have emerged as useful labels for PALM, including photoactivatable fluorescent proteins40,41 and fluorogenic small molecules bearing photolabile “cages”.14,40,42−44 In addition, many conventional fluorophores can be caged by illumination in an oxygen-free “redox buffer” containing millimolar amounts of thiol, thereby shelving the molecules in a nonfluorescent, photoswitchable state.45−49
In all cases, effective labels for this super-resolution microscopy technique must meet several stringent criteria, including photon yields sufficient to enable precise molecular localization, high on:off contrast to reduce background and the probability of overlapping emitters in densely labeled biological structures, and an attachment strategy for the structure of interest.50 The caged rhodamine dyes comprise a promising class of photoactivatable labels for super-resolution microscopy. Photoactivatable rhodamines exhibit high contrast due to the open–closed equilibrium, utilize cages with well-known photochemistry, demonstrate excellent photon yields, and can employ established bioconjugation strategies. We and others recently showed NVOC-rhodamine 110 derivatives to be attractive labels for PALM.14,44 We therefore evaluated derivatives of the caged carborhodamine 110 25 as a label for super-resolution microscopy.
We first derivatized caged dye 25 is shown in Figure 4a. Removal of the tert-butyl ester with TFA afforded 5-carboxy derivative 30. This acid was converted to the NHS ester 31 by treatment with TSTU. Attachment of phalloidin via a short poly(ethylene glycol) (PEG) linker gave phallodin–PEG8–carborhodamine conjugate 32. As contrast is a key determinant of image quality, we determined the on:off contrast ratio of NVOC2-rhodamine 11014 and NVOC2-carborhodamine 30 by measuring the fluorescence of freshly purified caged dye before and after photoactivation. The NVOC2-rhodamine 110 shows low but significant background fluorescence with an on:off contrast ratio of 7000 (Supplementary Figure S4), which is consistent with other diacylrhodamine substrates.12 The carborhodamine system displays a contrast ratio of 6.5 × 104 (Supplementary Figure S4), significantly greater than the contrast ratio for caged rhodamine 110 and other photoactivatable dyes used for PALM (≤1500).43,44 The improved contrast value for carborhodamine 30 likely stems from the altered open–closed equilibrium observed with this fluorophore scaffold (Figure 2c) and ensures that PALM imaging can be performed on even densely labeled structures (>1 label/nm2).
Figure 4.

PALM imaging of F-actin in fixed mouse embryonic fibroblasts. (a) Synthesis of caged carborhodamine 110–phalloidin conjugate 32. Reagents and conditions: (a) TFA/CH2Cl2, rt, 98%. (b) TSTU, DIEA, DMF, 74%. (c) i. H2N-PEG8-CO2H, DIEA, DMF; ii. TSTU, DIEA, DMF, then phalloidin-NH2, 69%, two steps. (b, c) PALM images for labeling with CRh110–PEG8–phallodin conjugate 32. (d, e) Labeling with mEos2–actin. (f, g) Labeling with AF647–phalloidin. Images are rendered as a heat map of localization probability/nm2 with scale in lower right. For clarity, PALM images were plotted at a minimum localization precision of 22 nm (b, d, and f) and 11 nm (c, e, and g). Scale bars (lower left): 5 μm (b, d, and f) or 1 μm (c, e, and g).
On the basis of this promising in vitro result, NVOC2-carborhodamine 110 was used in a cellular PALM experiment. The phalloidin-containing affinity reagent 32 allowed ultrahigh resolution imaging of cellular actin in mouse embryonic fibroblasts as shown in Figure 4b,c. The carborhodamine conjugate exhibited a median photon yield of 2800 per activation event (Supplementary Figure S6a) and high labeling density, allowing the super-resolution imaging of both large stress fibers and finer actin filaments (Figure 4b,c) with a median localization precision of 8 nm (Supplementary Figure S6b). The probable emitting species in this experiment is a mixture of partially and fully uncaged carborhodamine as the monocaged dye exhibits modest fluorescence (Supplementary Figure S4). We compared this result with PALM experiments using the photoactivatable fluorescent protein mEos2–actin (41) (Figure 4d,e). The protein-based mEos2 label exhibited a median photon yield of 990 (Supplementary Figure S6c), providing an image with lower median localization precision (11 nm, Supplementary Figure S6d). We also calculated the total number of localizations per cell to estimate average labeling density. The carborhodamine label displayed similar localizations (842,372/cell) to the genetically encoded mEos2 (1,011,424/cell), showing comparable labeling of cellular actin with these two fluorophores.
We also performed imaging using a commercial phalloidin–AlexaFluor 647 (AF647) conjugate under redox buffer conditions (Figure 4f,g), allowing the first direct comparison of PALM imaging using a redox-switched conventional fluorophore or a caged dye. AF647 has been shown to exhibit the best switching properties for super-resolution microscopy.48 A key difference in these imaging modalities is the switching mechanism of the two labels. The photoactivation of AlexaFluor 647 in a redox buffer is a dynamic and reversible process where fluorescent molecules are first shelved in a dark excited state by illumination with excitation light.48 Return to the fluorescent ground state can be elicited with blue light but also occurs spontaneously; the extemporaneous activation rate depends on the specific fluorophore structure and buffer environment. At equilibrium, approximately 0.1% of AlexaFluor 647 molecules are in the fluorescent state under imaging conditions,48 limiting the effective on:off contrast ratio of this label to ∼1000. Moreover, excitation can lead to either fluorescence emission or reshelving of the molecule in the nonfluorescent state. Therefore, fluorophores in this imaging regime exhibit increased blinking and a lower photon yield. The AF647-phalloidin probe yielded 930 photons per activation event (Supplementary Figure S6e), resulting in an inferior localization precision value (17 nm, Supplementary Figure S6f).
In the carborhodamine case, however, photoactivation is not imposed by the environment but is built into the chemical structure. Because all molecules begin in a chemically caged state, the population density of activated molecules can be controlled exclusively with the blue activation light (405 nm). Excitation of uncaged carborhodamine labels leads primarily to fluorescence, giving a larger photon yield per activation event and a higher resolution image (Figure 4). Moreover, we note the imaging in Figure 4b,c was performed in atmosphere-saturated phosphate buffered saline without the addition of oxygen scavengers or other antibleaching agents. Although such reagents do improve photon yield (data not shown), the excellent characteristics of the carborhodamines is already sufficient for high resolution PALM imaging. The superior photophysics and flexibility in chemical environment greatly simplifies implementation of super-resolution microscopy and will enable compatibility with other imaging modalities and disparate biological samples. Overall, these results establish the N,N′-diacyl-carborhodamine system as a promising new platform for PALM labels. The dye displays an excellent photon yield and extremely large contrast ratio, enabling high resolution, high density microscopy without regard to chemical environment.
Conclusion
Fluorogenic compounds find wide use across many areas of biology. This high utility warrants the development of improved synthetic methodology and investigation of novel dye scaffolds. In this report we describe a divergent synthetic approach toward red-shifted isologues of the eminently useful fluoresceins and rhodamines. A key step in our synthesis was the unprecedented addition of ester-containing aryl Grignards to a novel anthrone to yield carbofluoresceins. We note this chemistry could also enable the efficient synthesis of other isomerically pure fluorescein and fluorescein-like dyes bearing the crucial o-carboxy substituent on the pendant phenyl ring. The carbofluoresceins can be taken on to diverse carborhodamine derivatives through a Pd-catalyzed cross-coupling with disparate N-alkyl, N-acyl, or N-aryl coupling partners.
Substitution of the xanthene oxygen with a quaternary carbon elicits a 50-nm bathochromic shift in absorption and emission wavelengths, yet the carbofluoresceins and carborhodamines retain many properties of the parent dyes, including high extinction coefficients and quantum yields (Figure 2a). In this work we demonstrate that carbofluoresceins and carborhodamines can be masked using established strategies to prepare red-shifted fluorogenic enzyme substrates and caged dyes. Attachment of other O- or N-linked functionalities (e.g., phosphatidyl, glycosyl, peptidyl) to this modular scaffold will provide numerous useful green-excited fluorogenic molecules to enable sophisticated multicolor biological assays.
Although carbofluoresceins and carborhodamines exhibit properties similar to those of the parent xanthene dyes, there are some notable differences, including the higher phenolic pKa of carbofluorescein (Figure 2b) and the higher propensity of the modified dyes to adopt a nonfluorescent lactone form in aqueous solution (Figure 2b,c). The pH sensitivity of fluorescein is exploited for numerous cellular experiments and modulated through structural changes;34 carbofluorescein could be used or modified in similar ways. The shift in open–closed equilibrium is advantageous for developing fluorogenic molecules with unexpectedly large on:off contrast ratios, which could enable a new generation of fluorogenic sensors3 and labels12 with extremely low background. We show photoactivatable carborhodamines to be a promising scaffold for super-resolution microscopy labels, based on the desirable combination of high contrast and high photon yields. Our facile synthesis of these modular anthracenyl isologs of rhodamine will permit the further fine-tuning of dye properties, photolabile groups, and attachment strategies to allow sophisticated cellular imaging experiments below the diffraction limit.
Methods
Chemical Synthesis
Detailed experimentals and characterization can be found in the Supporting Information. All reactions were performed under a nitrogen atmosphere in round-bottomed flasks or septum-capped crimp-top vials containing Teflon-coated magnetic stir bars. Reactions were monitored by thin layer chromatography (TLC) on precoated TLC glass plates or by LC–MS using a C18 column (ESI, positive ion mode; UV detection at 254 nm). Flash chromatography was performed on an automated purification system using prepacked silica gel columns. High-resolution mass spectrometry was performed by the Mass Spectrometry Center in the Department of Medicinal Chemistry at the University of Washington. NMR spectra were recorded on a 400 MHz spectrometer at the Janelia Farm Research Campus. 1H and 13C chemical shifts (δ) were referenced to TMS or residual solvent peaks, and 19F chemical shifts (δ) were referenced to CFCl3.
Optical Spectroscopy and Microscopy
Spectroscopy was performed using 1-cm path length quartz cuvettes. All measurements were taken at ambient temperature (22 ± 2 °C). Absorption spectra were recorded on a Cary Model 100 spectrometer (Varian), and fluorescence spectra were recorded on a Cary Eclipse fluorometer (Varian). Absolute quantum yields (Φ) were measured using a Quantaurus-QY spectrometer (Hamamatsu). Confocal microscopy was performed on live HeLa cells using a Zeiss LSM 510 META confocal microscope. PALM imaging was performed on a custom-built instrument based on an Olympus IX81 inverted wide field microscope. Photoactivatable 32 and mEos2 fluorophores were uncaged with a 405 nm laser and excited with a 561 nm laser. AlexaFluor 647-stained samples were incubated in a redox buffer consisting of 100 mM MEA, 50 μg/mL glucose oxidase, 40 μg/mL catalase, 10% w/v glucose, pH 8.5 and excited with a 642 nm laser. Full experimental details including PALM image analysis and plotting parameters are given in the Supporting Information.
Acknowledgments
We thank A. Arnold and H. White for cell culture and initial microscopy studies; G. Shtengel and H. Hess for microscopy assistance and supplies; and S. Sternson, D. Murphy, L. Wysocki, and P. Lee for contributive discussions. This work was supported by the Howard Hughes Medical Institute.
Supporting Information Available
Full details for synthesis and characterization of new materials, optical spectroscopy, cellular imaging, and supplementary figures. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Haugland R. P., Spence M. T. Z., Johnson I. D., and Basey A. (2005) The Handbook: A Guide to Fluorescent Probes and Labeling Technologies, 10th ed., Molecular Probes, Eugene, OR. [Google Scholar]
- Lavis L. D.; Raines R. T. (2008) Bright ideas for chemical biology. ACS Chem. Biol. 3, 142–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan J.; Dodani S. C.; Chang C. J. (2012) Reaction-based small-molecule fluorescent probes for chemoselective bioimaging. Nat. Chem. 4, 973–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm J. B.; Heckman L. M.; Lavis L. D. (2013) The chemistry of small-molecule fluorogenic probes. Prog. Mol. Biol. Transl. Sci. 113, 1–34. [DOI] [PubMed] [Google Scholar]
- Gee K. R.; Sun W.-C.; Bhalgat M. K.; Upson R. H.; Klaubert D. H.; Latham K. A.; Haugland R. P. (1999) Fluorogenic substrates based on fluorinated umbelliferones for continuous assays of phosphatases and β-galactosidases. Anal. Biochem. 273, 41–48. [DOI] [PubMed] [Google Scholar]
- Zhou M.; Diwu Z.; Panchuk-Voloshina N.; Haugland R. P. (1997) A stable nonfluorescent derivative of resorufin for the fluorometric determination of trace hydrogen peroxide: Applications in detecting the activity of phagocyte NADPH oxidase and other oxidases. Anal. Biochem. 253, 162–168. [DOI] [PubMed] [Google Scholar]
- Chandran S. S.; Dickson K. A.; Raines R. T. (2005) Latent fluorophore based on the trimethyl lock. J. Am. Chem. Soc. 127, 1652–1653. [DOI] [PubMed] [Google Scholar]
- Xie H.; Mire J.; Kong Y.; Chang M. H.; Hassounah H. A.; Thornton C. N.; Sacchettini J. C.; Cirillo J. D.; Rao J. (2012) Rapid point-of-care detection of the tuberculosis pathogen using a BlaC-specific fluorogenic probe. Nat. Chem. 4, 802–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller E. W.; Albers A. E.; Pralle A.; Isacoff E. Y.; Chang C. J. (2005) Boronate-based fluorescent probes for imaging cellular hydrogen peroxide. J. Am. Chem. Soc. 127, 16652–16659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotman B.; Papermaster B. W. (1966) Membrane properties of living mammalian cells as studied by enzymatic hydrolysis of fluorogenic esters. Proc. Natl. Acad. Sci. U.S.A. 55, 134–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theriot J. A.; Mitchison T. J. (1991) Actin microfilament dynamics in locomoting cells. Nature 352, 126–131. [DOI] [PubMed] [Google Scholar]
- Lavis L. D.; Chao T.-Y.; Raines R. T. (2006) Fluorogenic label for biomolecular imaging. ACS Chem. Biol. 1, 252–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warther D.; Bolze F.; Leonard J.; Gug S.; Specht A.; Puliti D.; Sun X. H.; Kessler P.; Lutz Y.; Vonesch J. L.; Winsor B.; Nicoud J. F.; Goeldner M. (2010) Live-cell one- and two-photon uncaging of a far-red emitting acridinone fluorophore. J. Am. Chem. Soc. 132, 2585–2590. [DOI] [PubMed] [Google Scholar]
- Wysocki L. M.; Grimm J. B.; Tkachuk A. N.; Brown T. A.; Betzig E.; Lavis L. D. (2011) Facile and general synthesis of photoactivatable xanthene dyes. Angew. Chem., Int. Ed. 50, 11206–11209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beija M.; Afonso C. A. M.; Martinho J. M. G. (2009) Synthesis and applications of rhodamine derivatives as fluorescent probes. Chem. Soc. Rev. 38, 2410–2433. [DOI] [PubMed] [Google Scholar]
- Grimm J. B.; Lavis L. D. (2011) Synthesis of rhodamines from fluoresceins using Pd-catalyzed C–N cross-coupling. Org. Lett. 13, 6354–6357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming G. R.; Knight A. W. E.; Morris J. M.; Morrison R. J. S.; Robinson G. W. (1977) Picosecond fluorescence studies of xanthene dyes. J. Am. Chem. Soc. 99, 4306–4311. [Google Scholar]
- Ohulchanskyy T. Y.; Donnelly D. J.; Detty M. R.; Prasad P. N. (2004) Heteroatom substitution induced changes in excited-state photophysics and singlet oxygen generation in chalcogenoxanthylium dyes: Effect of sulfur and selenium substitutions. J. Phys. Chem. B 108, 8668–8672. [Google Scholar]
- Arden-Jacob J.; Frantzeskos J.; Kemnitzer N. U.; Zilles A.; Drexhage K. H. (2001) New fluorescent markers for the red region. Spectrochim. Acta, Part A 57, 2271–2283. [DOI] [PubMed] [Google Scholar]
- Kolmakov K.; Belov V. N.; Wurm C. A.; Harke B.; Leutenegger M.; Eggeling C.; Hell S. W. (2010) A versatile route to red-emitting carbopyronine dyes for optical microscopy and nanoscopy. Eur. J. Org. Chem. 2010, 3593–3610. [Google Scholar]
- Kolmakov K.; Wurm C.; Sednev M. V.; Bossi M. L.; Belov V. N.; Hell S. W. (2012) Masked red-emitting carbopyronine dyes with photosensitive 2-diazo-1-indanone caging group. Photochem. Photobiol. Sci. 11, 522–532. [DOI] [PubMed] [Google Scholar]
- Egawa T.; Koide Y.; Hanaoka K.; Komatsu T.; Terai T.; Nagano T. (2011) Development of a fluorescein analogue, TokyoMagenta, as a novel scaffold for fluorescence probes in red region. Chem. Commun. 47, 4162–4164. [DOI] [PubMed] [Google Scholar]
- Koide Y.; Urano Y.; Hanaoka K.; Terai T.; Nagano T. (2011) Evolution of Group 14 rhodamines as platforms for near-infrared fluorescence probes utilizing photoinduced electron transfer. ACS Chem. Biol. 6, 600–608. [DOI] [PubMed] [Google Scholar]
- Kushida Y.; Hanaoka K.; Komatsu T.; Terai T.; Ueno T.; Yoshida K.; Uchiyama M.; Nagano T. (2012) Red fluorescent scaffold for highly sensitive protease activity probes. Bioorg. Med. Chem. Lett. 22, 3908–3911. [DOI] [PubMed] [Google Scholar]
- Lukinavičius G.; Umezawa K.; Olivier N.; Honigmann A.; Yang G.; Plass T.; Mueller V.; Reymond L.; Corrêa I. R. Jr; Luo Z. G.; Schultz C.; Lemke E. A.; Heppenstall P.; Eggeling C.; Manley S.; Johnsson K. (2013) A near-infrared fluorophore for live-cell super-resolution microscopy of cellular proteins. Nat. Chem. 5, 132–139. [DOI] [PubMed] [Google Scholar]
- Calitree B.; Donnelly D. J.; Holt J. J.; Gannon M. K.; Nygren C. L.; Sukumaran D. K.; Autschbach J.; Detty M. R. (2007) Tellurium analogues of rosamine and rhodamine dyes: Synthesis, structure, 125Te NMR, and heteroatom contributions to excitation energies. Organometallics 26, 6248–6257. [Google Scholar]
- Jiang L., and Buchwald S. L. (2004) in Metal-Catalyzed Cross-Coupling Reactions (de Meijere A., and Diederich F., Eds.), p 699, Wiley-VCH, Weinheim. [Google Scholar]
- Minta A.; Kao J. P.; Tsien R. Y. (1989) Fluorescent indicators for cytosolic calcium based on rhodamine and fluorescein chromophores. J. Biol. Chem. 264, 8171–8178. [PubMed] [Google Scholar]
- Wu L.; Burgess K. (2008) Synthesis and spectroscopic properties of rosamines with cyclic amine substituents. J. Org. Chem. 73, 8711–8718. [DOI] [PubMed] [Google Scholar]
- Negishi E.-i., Zeng X., Tan Z., Qian M., Hu Q., and Huang Z. (2004) in Metal-Catalyzed Cross-Coupling Reactions (de Meijere A., and Diederich F., Eds.) 2nd ed., pp 815–889, Wiley-VCH, Weinheim. [Google Scholar]
- Gu L.; Yang B.; Liu F.; Bai Y. (2009) Chin. J. Chem. 27, 1199–1201. [Google Scholar]
- Urano Y.; Kamiya M.; Kanda K.; Ueno T.; Hirose K.; Nagano T. (2005) Evolution of fluorescein as a platform for finely tunable fluorescence probes. J. Am. Chem. Soc. 127, 4888–4894. [DOI] [PubMed] [Google Scholar]
- Krasovskiy A.; Knochel P. (2004) A LiCl-Mediated Br/Mg exchange reaction for the preparation of functionalized aryl-and heteroarylmagnesium compounds from organic bromides. Angew. Chem., Int. Ed. 43, 3333–3336. [DOI] [PubMed] [Google Scholar]
- Lavis L. D.; Rutkoski T. J.; Raines R. T. (2007) Tuning the pKa of fluorescein to optimize binding assays. Anal. Chem. 79, 6775–6782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger S. (1981) The pH dependence of phenolphthalein: a 13C NMR study. Tetrahedron 37, 1607–1611. [Google Scholar]
- Burdette S. C.; Walkup G. K.; Spingler B.; Tsien R. Y.; Lippard S. J. (2001) Fluorescent sensors for Zn2+ based on a fluorescein platform: Synthesis, properties and intracellular distribution. J. Am. Chem. Soc. 123, 7831–7841. [DOI] [PubMed] [Google Scholar]
- Ioffe I. S.; Otten V. F. (1965) Rhodamine dyes and related compounds. XIV. Mutual conversions of colorless and colored forms of rhodamine and rhodol. Zh. Obshch. Khim. 1, 343–346. [Google Scholar]
- Watkins R. W.; Lavis L. D.; Kung V. M.; Los G. V.; Raines R. T. (2009) Fluorogenic affinity label for the facile, rapid imaging of proteins in live cells. Org. Biomol. Chem. 7, 3969–3975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Critchfield F. E.; Gibson J. A. Jr; Hall J. L. (1953) Dielectric constant for the dioxane–water system from 20 to 35°. J. Am. Chem. Soc. 75, 1991–1992. [Google Scholar]
- Betzig E.; Patterson G. H.; Sougrat R.; Lindwasser O. W.; Olenych S.; Bonifacino J. S.; Davidson M. W.; Lippincott-Schwartz J.; Hess H. F. (2006) Imaging intracellular fluorescent proteins at nanometer resolution. Science 313, 1642–1645. [DOI] [PubMed] [Google Scholar]
- McKinney S. A.; Murphy C. S.; Hazelwood K. L.; Davidson M. W.; Looger L. L. (2009) A bright and photostable photoconvertible fluorescent protein. Nat. Methods 6, 131–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fölling J.; Belov V.; Kunetsky R.; Medda R.; Schönle A.; Egner A.; Eggeling C.; Bossi M.; Hell S. W. (2007) Photochromic rhodamines provide nanoscopy with optical sectioning. Angew. Chem., Int. Ed. 46, 6266–6270. [DOI] [PubMed] [Google Scholar]
- Lee H. D.; Lord S. J.; Iwanaga S.; Zhan K.; Xie H.; Williams J. C.; Wang H.; Bowman G. R.; Goley E. D.; Shapiro L.; Twieg R. J.; Rao J.; Moerner W. E. (2010) Superresolution imaging of targeted proteins in fixed and living cells using photoactivatable organic fluorophores. J. Am. Chem. Soc. 132, 1642–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banala S.; Maurel D.; Manley S.; Johnsson K. (2011) A caged, localizable rhodamine for superresolution microscopy. ACS Chem. Biol. 7, 289–293. [DOI] [PubMed] [Google Scholar]
- Heilemann M.; Margeat E.; Kasper R.; Sauer M.; Tinnefeld P. (2005) Carbocyanine dyes as efficient reversible single-molecule optical switch. J. Am. Chem. Soc. 127, 3801–3806. [DOI] [PubMed] [Google Scholar]
- Heilemann M.; van de Linde S.; Schüttpelz M.; Kasper R.; Seefeldt B.; Mukherjee A.; Tinnefeld P.; Sauer M. (2008) Subdiffraction-resolution fluorescence imaging with conventional fluorescent probes. Angew. Chem., Int. Ed. 47, 6172–6176. [DOI] [PubMed] [Google Scholar]
- Dempsey G. T.; Bates M.; Kowtoniuk W. E.; Liu D. R.; Tsien R. Y.; Zhuang X. (2009) Photoswitching mechanism of cyanine dyes. J. Am. Chem. Soc. 131, 18192–18193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempsey G. T.; Vaughan J. C.; Chen K. H.; Bates M.; Zhuang X. (2011) Evaluation of fluorophores for optimal performance in localization-based super-resolution imaging. Nat. Methods 8, 1027–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Linde S.; Löschberger A.; Klein T.; Heidbreder M.; Wolter S.; Heilemann M.; Sauer M. (2011) Direct stochastic optical reconstruction microscopy with standard fluorescent probes. Nat. Protoc. 6, 991–1009. [DOI] [PubMed] [Google Scholar]
- Ji N.; Shroff H.; Zhong H.; Betzig E. (2008) Advances in the speed and resolution of light microscopy. Curr. Opin. Neurobiol. 18, 605–616. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.