

Abstract
Objective:
To test the hypothesis that higher neuronal density in brainstem aminergic nuclei contributes to neural reserve.
Methods:
Participants are 165 individuals from the Rush Memory and Aging Project, a longitudinal clinical–pathologic cohort study. They completed a mean of 5.8 years of annual evaluations that included a battery of 19 cognitive tests from which a previously established composite measure of global cognition was derived. Upon death, they had a brain autopsy and uniform neuropathologic examination that provided estimates of the density of aminergic neurons in the locus ceruleus, dorsal raphe nucleus, substantia nigra, and ventral tegmental area plus summary measures of neuronal neurofibrillary tangles and Lewy bodies from these nuclei and medial temporal lobe and neocortex.
Results:
Neuronal densities in each nucleus were approximately normally distributed. In separate analyses, higher neuronal density in each nucleus except the ventral tegmental area was associated with slower rate of cognitive decline, but when modeled together only locus ceruleus neuronal density was related to cognitive decline (estimate = 0.003, SE = 0.001, p < 0.001). Higher densities of tangles and Lewy bodies in these brainstem nuclei were associated with faster cognitive decline even after controlling for pathologic burden elsewhere in the brain. Locus ceruleus neuronal density, brainstem tangles, and brainstem Lewy bodies had independent associations with rate of cognitive decline. In addition, at higher levels of locus ceruleus neuronal density, the association of Lewy bodies with cognitive decline was diminished.
Conclusion:
Density of noradrenergic neurons in the locus ceruleus may be a structural component of neural reserve.
According to the neural reserve hypothesis, individuals differ in their capacity to tolerate neuropathologic lesions,1,2 but the biologic basis of this capacity is poorly understood. Indicators of brain size such as head circumference,3 intracranial volume,4 brain volume,5 and brain weight6 have been used to validate the concept of brain reserve capacity. Another approach has been to measure neurons (number,6 size7) or their components (synapses8,9) in key locations. This approach allows collection of neuronal and pathologic data from the same brain regions, facilitating examination of their conjoint correlations with cognition.
The present study examines the associations among neuronal density, neurodegenerative lesions, and change in cognitive function. We assessed neuronal density in brainstem aminergic nuclei (i.e., locus ceruleus, dorsal raphe nucleus, substantia nigra, and ventral tegmental area) because these nuclei support multiple cognitive processes, synthesize important monoamines that function as neurotransmitters and neuromodulators, are therapeutic targets for cognitive enhancement,10 and bear a disproportionate burden of age-related neurodegeneration.11,12 Participants from the Rush Memory and Aging Project had annual cognitive testing for a mean of 5.8 years, died, and underwent a neuropathologic examination that yielded neuronal counts for each brainstem nucleus plus immunohistochemical measures of neuronal neurofibrillary tangles and Lewy bodies in the brainstem nuclei and elsewhere in the brain. We tested the hypothesis that higher neuronal density in brainstem aminergic nuclei is a structural indicator of neural reserve that limits the impact of common neurodegenerative lesions on cognitive function.
METHODS
Participants.
Participants were from the Rush Memory and Aging Project, an ongoing longitudinal clinical–pathologic study that began in 1997.13 Eligibility required age >55, absence of a previous dementia diagnosis, and agreement to annual clinical evaluations and brain autopsy upon death. Older individuals were recruited from retirement communities, social service agencies, and subsidized housing facilities in the Chicago metropolitan region.
At the time of these analyses, 548 of 1,536 participants had died. A brain autopsy was done in 431 (79%) and a uniform neuropathologic examination had been completed on the first consecutive 417 individuals. From this group, 170 cases were selected to provide a wide range of cognitive function, motor function, and depressed affect proximate to death for clinical-pathologic studies, as previously described.14 They had a mean age at death of 88.6 years (SD 5.7), a mean of 14.7 years of education (SD 2.7), and 66.5% were women. Those chosen did not differ from the 247 not selected in age at baseline, age at death, education, or limbic/neocortical tangles or Lewy bodies. They had better cognitive function (baseline global cognitive score of −0.039 vs −0.351, t[409.4] = 4.75, p < 0.001; last global cognitive score of −0.568 vs −0.993, t[402.3] = 4.16, p < 0.001) and longer follow-up (5.7 vs 5.0 years, t[402.5] = 2.25, p = 0.025).
Standard protocol approvals, registrations, and patient consents.
Following a presentation about the project, interested persons met for further discussion with project staff who obtained written informed consent. The study was approved by the institutional review board of Rush University Medical Center.
Assessment of cognitive function.
Cognition was assessed annually with 19 tests administered in an approximately 45-minute session, including 7 measures of episodic memory, 3 measures of semantic memory, 3 measures of working memory, 4 measures of perceptual speed, and 2 measures of visuospatial ability. To reduce error, a composite measure of global cognition based on 19 tests was used in analyses. Raw test scores were converted to z scores, using the baseline mean and SD from all participants, and z scores were averaged to yield the composite. Further information on the individual tests and the composite measure of global cognition is published elsewhere.15–17
Neuropathologic examination.
Participants died a mean of 10.9 months after the last clinical evaluation (SD 11.7). The brain was removed a mean of 7.3 hours after death (SD 4.3). A standard protocol was used for tissue preservation, tissue sectioning, and quantification of pathologic findings by examiners blinded to all clinical data.18
To assess neuronal density in the aminergic nuclei of interest, consecutive transverse blocks of fixed tissue were taken at 4 brainstem levels illustrated in figure 1 as previously described.14 The first block included the substantia nigra and paranigral nucleus of the ventral tegmental area. The second block contained the rostral level of the dorsal raphe nucleus, the trochlear nucleus, and the decussation of the superior cerebellar peduncles. The third block included the caudal level of the dorsal raphe nucleus, rostral levels of the locus ceruleus nuclei, and mesencephalic nucleus of the trigeminal nerve. The fourth block contained the main body of the locus ceruleus. Tissue blocks were embedded in paraffin and 6-μm sections were stained with hematoxylin & eosin to survey the regions of interest, and only blocks with anatomically matching levels of the regions of interest were included in the study. Sections (20 μm) were used for immunohistochemistry with a monoclonal anti-tyrosine hydroxylase antibody (Immunostar, Hudson, WI; 1:750) to identify tyrosine-hydroxylase-immunoreactive neurons in the substantia nigra, ventral tegmental area, and locus ceruleus. An anti-tryptophan hydroxylase monoclonal antibody (Sigma Chemical Co., St. Louis, MO; 1:1,000) was used to identify tryptophan-hydroxylase-immunoreactive neurons in the dorsal raphe nucleus. The immunohistochemical preparations were used to outline the regions of interest with Stereo Investigator Program software (MBF Biosciences, Williston, VT) attached to an Olympus BX60 microscope with a motorized stage. Sections missing more than 50% of the dorsal raphe nucleus or locus ceruleus were excluded.
Figure 1. Diagram of the posterior aspect of the brainstem extending rostrally from the level of the superior colliculus to the caudal end of the fourth ventricle.
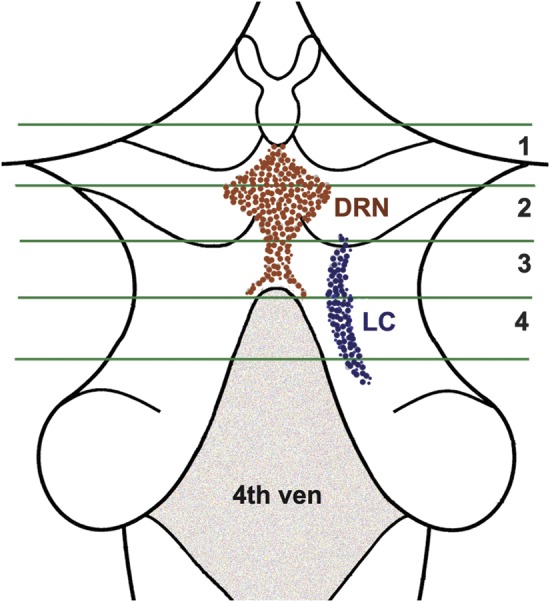
The green lines indicate the limits of the 4 consecutive blocks taken of the brainstem. The location of the dorsal raphe nucleus (DRN) and the right locus ceruleus (LC) are shown.
Neuronal density was measured unilaterally at one level of the substantia nigra and the ventral tegmental area and bilaterally at 2 levels of the dorsal raphe nucleus and locus ceruleus. Both unstained and immunostained neurons having nuclei were counted at a magnification of 400×. The density/mm2 of immunostained neurons was used in all analyses.
The density of τ-immunoreactive neurofibrillary tangles was assessed in each brainstem nucleus and in 8 limbic or neocortical regions (enthorhinal cortex, CA1/subiculum, anterior cingulate cortex, dorsal lateral prefrontal cortex, superior frontal cortex, inferior temporal cortex, inferior parietal cortex, primary visual cortex) using an anti-paired helical filaments-τ antibody clone AT8 (ThermoScientific, Rockford, IL: 1:2,000) and computer-assisted sampling.19 Composite measures of tangle density/mm2 in the brainstem nuclei (based on 4 sites) and limbic and neocortical regions (based on 8 sites) were constructed by converting raw scores for each site to standard scores and averaging standard scores across sites.
Lewy bodies were identified in each brainstem nucleus and 5 limbic and neocortical regions (entorhinal cortex, anterior cingulate cortex, midfrontal cortex, superior or middle temporal cortex, inferior parietal cortex) with a monoclonal phosphorylated antibody to α-synuclein (Wako Chemical USA Inc., Richmond, VA; 1:20,000).18 Lewy bodies were expressed as density/mm2 for the brainstem and present/absent for limbic/neocortical regions.
Statistical analysis.
We used mixed-effects models to assess the relationship of each postmortem variable to baseline level of cognitive function and annual rate of cognitive change. Analyses also included terms to control for age at death, sex, and education. We modeled each brainstem nucleus separately and then together. Subsequent models examined pathologic measures from brainstem and limbic/neocortical sites separately and then together. A final series of analyses included measures of locus ceruleus neurons and pathology in the same model and then tested for an interaction between them.
RESULTS
Neuronal density and cognitive decline.
Neuronal data were missing for 9 to 13 individuals per nucleus due to missing or insufficient tissue. The distributions of aminergic neuronal density in the locus ceruleus (mean = 41.8, SD = 18.1, skewness = 0.4, n = 156), dorsal raphe nucleus (mean = 104.2, SD = 27.0, skewness = 0.6, n = 154), substantia nigra (mean = 30.5, SD = 11.6, skewness = 0.0, n = 152), and ventral tegmental area (mean = 83.8, SD = 45.3, skewness = 0.3, n = 152) were approximately normal. Neuronal densities in the nuclei were modestly correlated with one another (range 0.13–0.48) and not related to age at death or sex. Education had a marginal association with nigral neuronal density (r = −0.17, p = 0.038) but no association with other neuronal measures.
We estimated the relation of neuronal density to change in the composite measure of global cognition (baseline mean = −0.028, SD = 0.525, skewness = −0.9) in mixed-effects models. In the initial analysis (table 1, model A), global cognition declined a mean of 0.123 unit per year, and higher locus ceruleus neuronal density was associated with higher baseline level of cognition and slower cognitive decline. Figure 2A shows that in those with high density (90th percentile, green line), cognition declined at less than half the rate associated with average density (50th percentile, black line) and less than one third the rate associated with low density (10th percentile, red line). In subsequent analyses, higher neuronal density in the dorsal raphe nucleus (table 1, model B) and substantia nigra (table 1, model C), but not the ventral tegmental area (table 1, model D), was associated with slower cognitive decline.
Table 1.
Neuronal density in brainstem aminergic nuclei and change in global cognitiona
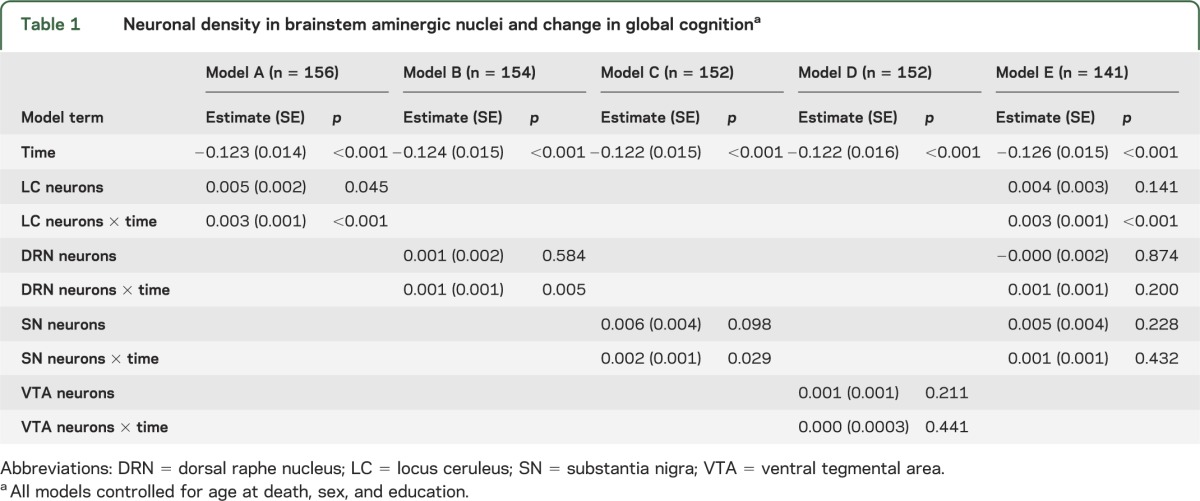
Figure 2. Cognitive decline.
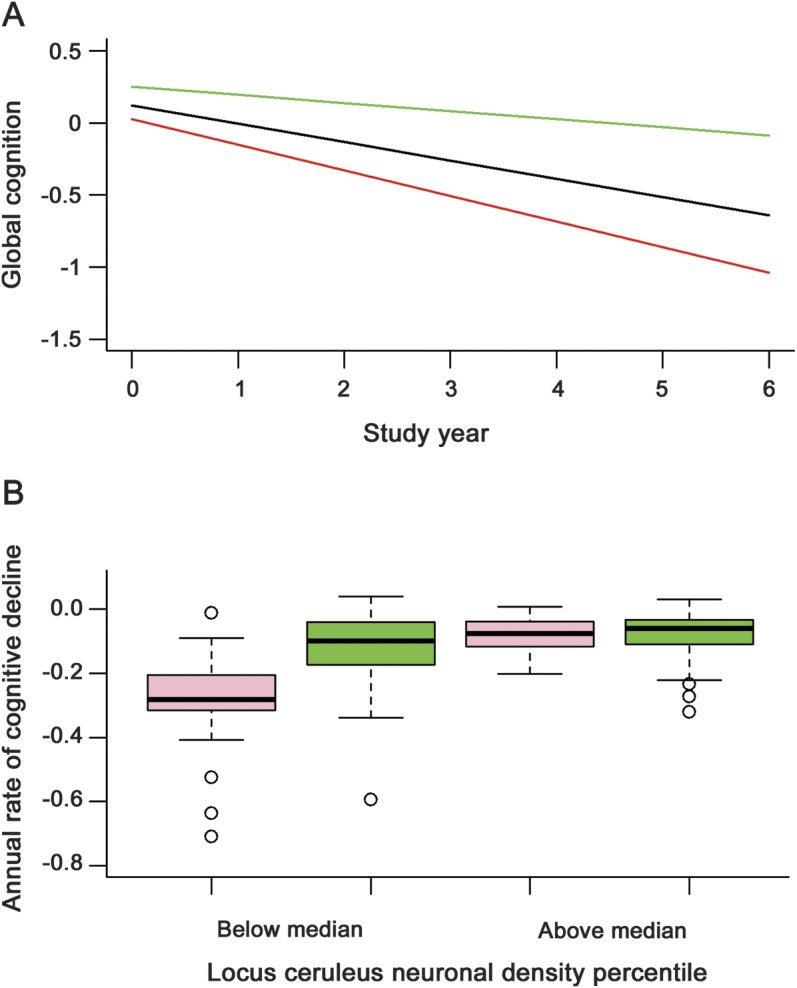
(A) Predicted 6-year paths of cognitive decline with low (10th percentile, red line), medium (50th percentile, black line), or high (90th percentile, green line) levels of locus ceruleus neuronal density adjusted for age at death, sex, and education. (B) Distribution of annual rate of cognitive decline at lower (below median) or higher (above median) levels of locus ceruleus neuronal density in those with (pink) or without (green) brainstem Lewy bodies adjusted for age at death, sex, and education.
To determine whether the neuronal density measures had independent associations with cognition, we analyzed the locus ceruleus, dorsal raphe nucleus, and substantia nigra together. Only locus ceruleus neuronal density was related to cognitive decline (table 1, model E).
Neuropathology and cognitive decline.
The densities of τ-immunoreactive tangles and α-synuclein-immunoreactive Lewy bodies in each nucleus were skewed. Because the tangle and Lewy body measures loaded on separate factors in a factor analysis, we averaged values in the 4 nuclei to yield composite brainstem measures of the density of tangles (mean = 1.99, SD = 1.78, skewness = 1.9, n = 165) and Lewy bodies (mean = 0.16, SD = 0.45, skewness = 3.0, n = 165). Higher tangle density was related to older age (r = 0.24, p = 0.002) but not to education or sex. Lewy body density was not related to age, education, or sex. Brainstem tangles and Lewy bodies were not related to one another (r = 0.10, p = 0.202) but were strongly related to composite limbic/neocortical measures of the same pathology (tangles: r = 0.70, p < 0.001; Lewy bodies: r = 0.80, p < 0.001).
In mixed-effects models, higher levels of tangles and Lewy bodies were each related to more rapid cognitive decline whether measured in the brainstem (table 2, model A) or limbic/neocortical (table 2, model B) regions. When all pathologic measures were simultaneously analyzed, brainstem tangles and Lewy bodies were still associated with cognitive decline but the limbic/neocortical measures were not (table 2, model C).
Table 2.
Relation of neuropathology to change in global cognitiona
Neuronal density, neuropathology, and cognitive decline.
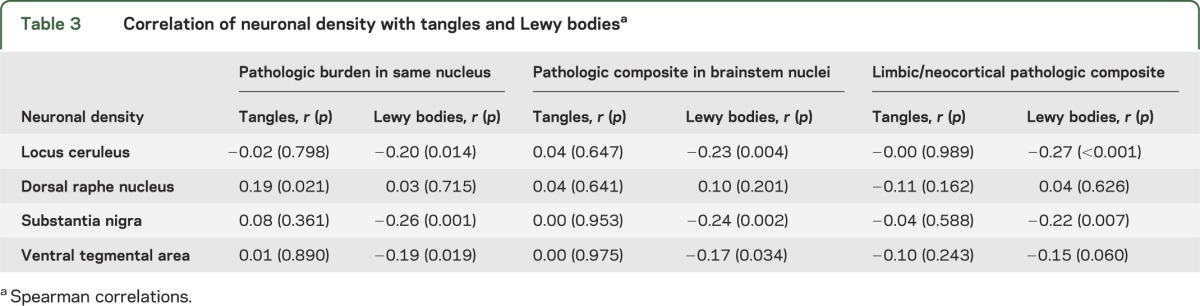
Table 3 shows the crude correlations of neuronal density with pathologic burden. Neuronal density had little association with tangles but was related to Lewy bodies. To determine whether neuronal density's association with cognitive decline was independent of neuropathologic burden, we simultaneously analyzed locus ceruleus neurons, brainstem tangles, and brainstem Lewy bodies (n = 156). With the effects of brainstem tangles (estimate = −0.023, SE = 0.007, p < 0.001) and Lewy bodies (estimate = −0.088, SE = 0.025, p < 0.001) controlled, locus ceruleus neurons were still related to cognitive decline (estimate = 0.002, SE = 0.001, p < 0.001), and the association persisted after also controlling for limbic/neocortical pathology (estimate = 0.002, SE = 0.001, p < 0.001, n = 156).
Table 3.
Correlation of neuronal density with tangles and Lewy bodiesa
To assess whether neuronal density modified the relation of pathology to cognition, we tested for an interaction between locus ceruleus neuronal density and pathology. There was no interaction with brainstem tangles (estimate for locus ceruleus neurons × time × brainstem tangles = 0.0003, SE = 0.0004, p = 0.454) but there was with brainstem Lewy bodies (estimate for locus neurons × time × brainstem Lewy bodies = 0.005, SE = 0.002, p < 0.001). The association of Lewy bodies with cognitive decline was only present at lower levels of neuronal density (figure 2B).
DISCUSSION
We assessed cognitive function annually for a mean of 5.8 years in more than 150 older persons who subsequently died and underwent neuropathologic examination. Higher density of noradrenergic neurons in the locus ceruleus was associated with reduced cognitive decline even after accounting for common neurodegenerative lesions in these nuclei and elsewhere in the brain.
Several studies have quantified aminergic nuclei neurons in persons with and without Alzheimer disease (AD). In a meta-analysis of this research, neuronal density in the locus ceruleus, dorsal raphe nucleus, and substantia nigra was reduced in AD.20 However, there has been little research on the relation of locus ceruleus noradrenergic neuronal density to cognitive functioning. Among persons with dementia, having few locus ceruleus neurons has been associated with lower level of cognition in some studies21,22 but not others.23 We are not aware of previous studies of locus ceruleus neurons and change in cognition over time. Thus, the present results extend previous research by showing that neuronal density in these brainstem nuclei is related to the primary clinical manifestation of AD, accelerated cognitive decline, and that it is noradrenergic neurons in the locus ceruleus that are primarily responsible for the association.
The most parsimonious explanation of the association between locus ceruleus neuronal density and cognitive decline is that some neuropathologic condition is affecting both. We found no evidence that either tangles or Lewy bodies are responsible for the association. Previous research has not suggested an association between Lewy bodies and nigral neuronal loss in Parkinson disease,24,25 but studies of individuals with and without dementia have found tangles to be associated with lower neuronal density in the dorsal raphe nucleus26 and medial temporal lobe.27 Most prior studies have included a substantial proportion of persons with severe dementia,23,28 but only 6.7% of individuals in the present study had a Mini-Mental State Examination score <10 at the last evaluation before death. The absence of a tangle–neuron association, therefore, might indicate that substantial loss of neurons in brainstem aminergic nuclei mainly occurs late in the course of AD as reported for the hippocampus and entorhinal cortex.29,30
It is possible that the association of locus ceruleus neuronal density with cognitive decline is due to other neuropathologic processes that were not measured (e.g., TDP-43) or whose footprint is not currently known. However, a pathologic process affecting neurons in some individuals but not others would be expected to skew the distribution of neuronal density, but the distribution in the locus ceruleus was approximately normal.
An alternate explanation of the association of locus ceruleus neuronal density with cognitive decline is that the locus ceruleus represents a structural component of neural reserve that contributes to brain reserve capacity. Although we think the data are most consistent with this hypothesis, we recognize that some portion of what we are attributing to neural reserve may actually be due to pathologic processes. In prior research, brain reserve capacity has been operationalized by indicators of neuronal density. In an early application of this approach, persons who died with pathologic AD but without dementia had a higher density of large neurons in the parietal cortex compared to persons with pathologic AD plus dementia though results in 2 other cortical regions were not significant.6 Similar findings have been reported for the entorhinal cortex and hippocampus.29,30 The present results are consistent with this research and extend it in important ways. First, these data suggest that brain reserve capacity may also depend on brainstem neuronal populations. Second, we assessed rate of cognitive decline rather than cross-sectional outcomes such as diagnosis or level of cognitive function. Third, the present study examined whether neuronal density added to or modified the association of pathology with cognition and did so for multiple types of pathology. Because neuronal density and pathology (i.e., tangles and Lewy bodies) each had independent associations with cognitive decline, higher neuronal density had the effect of subtracting from the effect of pathology on decline. Besides this additive effect, there was a multiplicative effect for Lewy bodies such that the correlation of Lewy bodies with cognitive decline was attenuated at higher neuronal density levels.
Although a comprehensive model of neural reserve would likely include multiple brain regions, the locus ceruleus has been hypothesized to be a critical component of neural reserve.31 Recent research has shown that the locus ceruleus norepinephrine neuromodulatory system interacts with multiple neural networks to regulate shifts in attention, behavioral adaptation, and memory consolidation and retrieval.32,33 In addition to directly supporting the efficiency of multiple cognitive networks, noradrenergic effects on neurotrophic factors, neurogenesis, synaptogenesis, and inflammation could also be contributing to neural reserve.31
Better understanding of key structural and functional components of neural reserve could suggest novel approaches to limiting cognitive decline. These approaches could have wide application because neural reserve is hypothesized to affect the clinical expression of multiple neuropathologic processes. The possible involvement of the locus ceruleus norepinephrine system in neural reserve is of particular interest. Dysfunction in this system is thought to contribute to cognitive impairment in several neuropsychiatric disorders (e.g., attention-deficit/hyperactivity disorder, depression). Pharmacologic research has shown that cognition can be enhanced in both affected and unaffected persons by α-2-adrenergic agonists (e.g., guanfacine34), β-adrenergic antagonists (e.g., propranolol35), and norepinephrine transport inhibitors (e.g., atomoxetine36). Further research on the role of the locus ceruleus norepinephrine system in cognitive aging is warranted.
Strengths and limitations of these data should be noted. Participation in clinical follow-up and brain autopsy was high, minimizing the likelihood that selective attrition biased results. The availability of longitudinal cognitive data proximate to death allowed us to assess the relation of neuronal density to rate of change while controlling for initial cognitive level. The results are based on a selected cohort and so their generalizability remains to be demonstrated.
Supplementary Material
ACKNOWLEDGMENT
The authors thank the Illinois residents who participated in the Rush Memory and Aging Project; Traci Colvin, MPH, and Karen Skish, MS, for study coordination; John Gibbons, MS, and Greg Klein, MS, for data management; and Wenqing Fan, MS, for statistical programming.
GLOSSARY
- AD
Alzheimer disease
Footnotes
AUTHOR CONTRIBUTIONS
Drafting/revising the manuscript for content: Dr. Wilson, Dr. Nag, Dr. Boyle, L.P. Hizel, Dr. Buchman, Dr. Schneider, Dr. Bennett. Study concept or design: Dr. Wilson, Dr. Nag, Dr. Boyle, L.P. Hizel, Dr. Buchman, Dr. Schneider, Dr. Bennett. Analysis or interpretation of the data: Dr. Wilson, Dr. Boyle, Dr. Yu, Dr. Buchman, Dr. Schneider, Dr. Bennett. Acquisition of data: Dr. Bennett, Dr. Nag, Dr. Schneider. Statistical analysis: Dr. Yu. Study supervision or coordination: Dr. Bennett. Obtaining funding: Dr. Boyle, Dr. Bennett.
STUDY FUNDING
Supported by the National Institute on Aging (R01AG17917, R01AG15819, R01AG33678, R01AG34374) and the Illinois Department of Public Health. The funding organizations had no role in the design or conduct of the study; collection, management, analysis, or interpretation of the data; or preparation, review, or approval of the manuscript.
DISCLOSURE
R. Wilson has served as a consultant for Pain Therapeutics, Inc. and receives research support from NIH (R01AG024871 [principal investigator], P30AG10161 [coinvestigator], R01AG11101 [coinvestigator], R01AG15819 [coinvestigator], U24AG026395 [coinvestigator], R01AG017917 [coinvestigator], R01AG009966 [coinvestigator], R01AG034374 [coinvestigator], RC2AG036547 [coinvestigator]). S. Nag receives support from NIH (P30AG010161 [coinvestigator], R01AG017917 [coinvestigator], R01AG033678 [coinvestigator]). P. Boyle receives research support from the NIH (R01AG034374 [principal investigator], R01AF034119 [coinvestigator], R01AG033678 [principal investigator]). L.P. Hizel receives research support from NIH (R01AG17917 [research assistant], P30AG10161 [research assistant]). L. Yu receives research support from NIH (R01AG033678 [coinvestigator], R01AG024871 [coinvestigator], R01AG024871 [coinvestigator], P30AG010161 [coinvestigator], R01AG015819], R01HL096944 [coinvestigator], R01AG036042 [coinvestigator], R01AG038651 [coinvestigator], R01AG040039 [coinvestigator], R01AG034374 [coinvestigator], U01AG032984 [coinvestigator]). A. Buchman receives support from NIH (R01AG24480 [principal investigator], R01AG034374 [coinvestigator], P30 AG010161 [coinvestigator], R01 AG017917 [coinvestigator], R01 AG022018, [coinvestigator], R01AG040039 [coinvestigator], P20MD006886 [coinvestigator], R01NS078009 [principal investigator]). J. Schneider receives research support from NIH (R01AG042210 [principal investigator], P30 AG010161 [coinvestigator], R01HL096944 [coinvestigator], R01AG039478 [coinvestigator], R01 AG017917 [coinvestigator], R01 AG015819 [coinvestigator], R01 AG022018 [coinvestigator], R01AG036042 [coinvestigator], R01AG040039 [coinvestigator], R01AG036836 [coinvestigator], R01AG034374 [coinvestigator], contract #23282007 [IDPH] [coinvestigator], R01 AG031553 [coinvestigator], P01 AG014449 [coinvestigator]). D. Bennett serves on the editorial board of Neurology®; has served as a consultant to Schering-Plough Corp., Medivation, Inc., and the Gerson Lehrman Group; and receives research support from Danone Inc., the NIH (R01AG017917 [principal investigator], R01AG015819 [principal investigator], R01AG036042 [principal investigator], RC2AG036547 [principal investigator], U01AG032984 [coprincipal investigator, leader of epidemiologic cohort studies], R01AG024480 [coinvestigator], R01AG024871 [coinvestigator], P01AG009466 [coinvestigator], U24AG026395 [coinvestigator], R01AG030142 [coinvestigator], P01AG014449 [coinvestigator], R01HL096944 [coinvestigator], R01AG033678 [coinvestigator], R01AG034374 [coinvestigator], R01AG032755 [coinvestigator], R01AG022018 [coinvestigator], R01AG034119 [coinvestigator], R01AG027040 [coinvestigator], R01AG026147 [coinvestigator], R01AG031553 [coinvestigator], and P30AG010161 [principal investigator–administrative core leader, Religious Orders Study core leader]), and the Illinois Department of Public Health. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Katzman R, Aronson M, Fuld P, et al. Development of dementing illnesses in an 80-year-old volunteer cohort. Ann Neurol 1989;25:317–324 [DOI] [PubMed] [Google Scholar]
- 2.Stern Y. What is cognitive reserve? Theory and research application of the reserve concept. J Int Neuropsychol Soc 2002;8:448–460 [PubMed] [Google Scholar]
- 3.Mortimer JA, Snowden DA, Markesbery WR. Head circumference, education and risk of dementia: findings from the Nun Study. J Clin Exp Neuropsychol 2003;25:671–679 [DOI] [PubMed] [Google Scholar]
- 4.Tate DF, Neeley ES, Norton MC, et al. Intracranial volume and dementia: some evidence in support of the cerebral reserve hypothesis. Brain Res 2011;1385:151–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Erten-Lyons D, Woltjer RL, Dodge H, et al. Factors associated with resistance to dementia despite high Alzheimer disease pathology. Neurology 2009;72:354–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Katzman R, Terry R, DeTeresa R, et al. Clinical, pathological, and neurochemical changes in dementia: a subgroup with preserved mental status and numerous neocortical plaques. Ann Neurol 1988;23:138–144 [DOI] [PubMed] [Google Scholar]
- 7.Riudavets MA, Iacono D, Resnick SM, et al. Resistance to Alzheimer’s pathology is associated with nuclear hypertrophy in neurons. Neurobiol Aging 2007;28:1484–1492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Head E, Corrada MM, Kahle-Wrobleski K, et al. Synaptic proteins, neuropathology and cognitive status in the oldest-old. Neurobiol Aging 2009;30:1125–1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Honer WG, Barr AM, Swada K, et al. Cognitive reserve, presynaptic proteins and dementia in the elderly. Transl Psychiatry 2012;2:e114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chamberlain SR, Muller U, Robbins TW, Sahakian BJ. Neuropharmacological modulation of cognition. Curr Opin Neurol 2006;19:607–612 [DOI] [PubMed] [Google Scholar]
- 11.Zweig R, Ross CA, Hedreen JC, et al. Neuropathology of aminergic nuclei in Alzheimer’s disease. Prog Clin Biol Res 1989;17:744–751 [PubMed] [Google Scholar]
- 12.Braak H, Del Tredici K, Rub U, et al. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 2003;24:197–211 [DOI] [PubMed] [Google Scholar]
- 13.Bennett DA, Schneider JA, Buchman AS, et al. Overview and findings from the rush memory and aging project. Curr Alzheimer Res 2012;9:646–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Buchman AS, Nag S, Shulman JM, et al. Locus ceruleus neuron density and parkinsonism in older adults without Parkinson’s disease. Mov Disord 2012;27:1625–1631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wilson RS, Barnes LL, Bennett DA. Assessment of lifetime participation in cognitively stimulating activities. J Clin Exp Neuropsychol 2003;25:634–642 [DOI] [PubMed] [Google Scholar]
- 16.Wilson RS, Barnes LL, Krueger KR, et al. Early and late life cognitive activity and cognitive systems in old age. J Int Neuropsychol Soc 2005;11:400–407 [PubMed] [Google Scholar]
- 17.Wilson RS, Scherr PA, Schneider JA, et al. Relation of cognitive activity to risk of developing Alzheimer disease. Neurology 2007;69:1911–1920 [DOI] [PubMed] [Google Scholar]
- 18.Schneider JA, Li JL, Li Y, et al. Substantia nigra tangles are related to gait impairment in older persons. Ann Neurol 2006;59:166–173 [DOI] [PubMed] [Google Scholar]
- 19.Wilson RS, Arnold SE, Schenider JA, et al. Chronic distress, age-related neuropathology, and late-life dementia. Psychosom Med 2007;89:47–53 [DOI] [PubMed] [Google Scholar]
- 20.Lyness SA, Zarow C, Chui HC. Neuron loss in key cholinergic and aminergic nuclei in Alzheimer disease: a meta-analysis. Neurobiol Aging 2003;24:1–23 [DOI] [PubMed] [Google Scholar]
- 21.Bondareff W, Mountjoy CQ, Roth M. Selective loss of neurones of origin of adrenergic projection to cerebral cortex (nucleus locus ceruleus) in senile dementia. Lancet 1981;1:783–784 [DOI] [PubMed] [Google Scholar]
- 22.Szot P, White SS, Greenup JL, et al. Compensatory changes in the noradrenergic nervous system in the locus ceruleus and hippocampus of postmortem subjects with Alzheimer’s disease and dementia with Lewy bodies. J Neurosci 2006;26:467–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Matthews KL, Chen CPLH, Esiri MM, et al. Noradrenergic changes, aggressive behavior, and cognition in patients with dementia. Biol Psychiatry 2002;51:407–416 [DOI] [PubMed] [Google Scholar]
- 24.Parkkinen L, O’Sullivan SS, Collins C, et al. Disentangling the relationship between Lewy bodies and nigral neuronal loss in Parkinson’s disease. J Park Dis 2011;1:277–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Greffard S, Verny M, Bonnet AM, et al. A stable proportion of Lewy body bearing neurons in the substantia nigra suggests a model in which the Lewy body causes neuronal death. Neurobiol Aging 2010, 31:99–103 [DOI] [PubMed] [Google Scholar]
- 26.Hendricksen M, Thomas AJ, Ferrier IN, et al. Neuropathological study of the dorsal raphe nuclei in late-life depression and Alzheimer’s disease with and without depression. Am J Psychiatry 2004;161:1092–1102 [DOI] [PubMed] [Google Scholar]
- 27.Von Guten A, Kövari E, Bussière T, et al. Cognitive impact of neuronal pathology in the entorhinal cortex and CA1 field in Alzheimer’s disease. Neurobiol Aging 2006;27:270–277 [DOI] [PubMed] [Google Scholar]
- 28.Chen CPLH, Eastwood SL, Hope T, McDonald B, Francis PT, Esiri MM. Immunocytochemical study of the dorsal and median raphe nuclei in patients with Alzheimer’s disease prospectively assessed for behavioral changes. Neuropathol Appl Neurobiol 2000;26:347–355 [DOI] [PubMed] [Google Scholar]
- 29.Price JL, Ko AI, Wade MJ, et al. Neuron number in the entorhinal cortex and CA1 in preclinical Alzheimer disease. Arch Neurol 2001;58:1395–1402 [DOI] [PubMed] [Google Scholar]
- 30.West MJ, Kawas CH, Steward WF, et al. Hippocampal neurons in pre-clinical Alzheimer’s disease. Neurobiol Aging 2004;25:1205–1212 [DOI] [PubMed] [Google Scholar]
- 31.Robertson IH. A noradrenergic theory of cognitive reserve: implications for Alzheimer’s disease. Neurobiol Aging 2013;34:298–308 [DOI] [PubMed] [Google Scholar]
- 32.Aston-Jones G, Cohen JD. Adaptive gain and the role of the locus ceruleus-norepinephrine system in optimal performance. J Comp Neurol 2005;493:99–110 [DOI] [PubMed] [Google Scholar]
- 33.Sara SJ. The locus ceruleus and noradrenergic modulation of cognition. Nat Rev Neurosci 2009;10:211–223 [DOI] [PubMed] [Google Scholar]
- 34.Jakala P, Riekkinen M, Sirvio J, et al. Guanfacine, but not clonidine, improves planning and working memory performance in humans. Neuropsychopharmacology 1999;20:460–470 [DOI] [PubMed] [Google Scholar]
- 35.Campbell HL, Tivarus ME, Hillier A, Beversdorf DQ. Increased task difficulty results in greater impact of noradrenergic modulation of cognitive flexibility. Pharmacol Biochem Behav 2008;88:222–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chamberlain SR, Del Campo N, Dowson J, et al. Atomoxetine improved response inhibition in adults with attention deficit disorder. Biol Psychiatry 2007;62:977–984 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.