

Abstract
Objective:
To investigate the frequency of Coats syndrome and its association with D4Z4 contraction size in patients with facioscapulohumeral muscular dystrophy type 1 (FSHD1).
Methods:
We searched a North American FSHD registry and the University of Rochester (UR) FSHD research database, reviewed the literature, and sent surveys to 14 FSHD referral centers in the United States and overseas to identify patients with genetically confirmed FSHD1 with a diagnosis of Coats syndrome.
Results:
Out of 357 genetically confirmed patients in a North American FSHD registry and 51 patients in the UR database, 3 patients had a self-reported history of Coats disease (0.8%; 95% confidence interval 0.2%–2.2%). In total, we identified 14 patients with FSHD with known genetic contraction size and Coats syndrome confirmed by ophthalmologic examination: 10 from our survey and 4 from the literature. The median age at diagnosis of Coats syndrome was 10 years (interquartile range 14 years). The median D4Z4 fragment size was 13 kilobases (kb) (interquartile range 1 kb). One patient was mosaic (55% 11 kb, and 45% 78 kb).
Conclusions:
Coats syndrome is a rare extramuscular complication of FSHD1 associated with large D4Z4 contractions. Closer surveillance for retinal complications is warranted in patients with D4Z4 fragments ≤15 kb.
Facioscapulohumeral muscular dystrophy type 1 (FSHD1) is caused by loss of a critical number of subtelomeric D4Z4 macrosatellite repeat units on chromosome 4q35. Whereas normal individuals have 11–100 repeats, individuals with FSHD have 1–10 repeats, which translates to D4Z4 EcoRI restriction fragment size between 10 and 38 kilobases (kb) by conventional genetic testing.1 Extramuscular manifestations include high-frequency hearing loss and retinal vascular tortuosity, which can progress to a treatable symptomatic condition called Coats syndrome.
The idiopathic form of this condition, Coats disease, is typically unilateral and predominately affects males with an age at onset in the first 2 decades of life. Retinal examination reveals telangiectasias and leakage that can lead to exudative retinal detachment and blindness.2 Treatment ranges from laser photocoagulation and cryotherapy in mild disease to vitreoretinal surgery or enucleation in advanced stages.2 Coats disease as seen in FSHD (Coats syndrome) can be bilateral, occur at any age, and affect males and females equally.
While more than half of patients with FSHD1 show peripheral retinal vascular abnormalities on fluorescein angiography, the prevalence of Coats syndrome is much lower, and the relationship to the D4Z4 contraction size is unknown.3,4 Understanding which patients are most at risk for Coats syndrome is an important clinical question with direct implications for screening and surveillance.
METHODS
We performed a cross-sectional database review and a deidentified patient survey of international FSHD1 neuromuscular referral centers. This study was considered exempt after institutional review board review.
Registry and database review.
We analyzed deidentified data from patients with genetically confirmed FSHD (D4Z4 fragment <38 kb) from the National Registry of FSHD Patients and Family Members (n = 357) and the University of Rochester (UR)-FSHD database (n = 51). Participants were self-selected, from North America, and over 18 years of age (UR-FSHD database), or any age (Registry). We used self-reported history of Coats disease for prevalence estimates. Because data are deidentified, participants may be double represented in the Registry and UR-FSHD database. We compared participants based on D4Z4 deletion size (±1 kb), sex, and age at diagnosis (±5 years). The denominator used for point prevalence estimates (n = 396 total, n = 24 D4Z4 ≤15 kb) was the sum of participants from the 2 databases minus possible overlapping subjects (n = 12).
Survey.
We sent a one-page deidentified survey to 14 neuromuscular FSHD referral centers in the United States and overseas. In addition, we solicited cases at international meetings, by e-mail, and on a national neuromuscular online forum. Survey respondents reviewed medical records to obtain information about Coats diagnosis and FSHD genetic testing. The Coats stage was modified from the literature and includes the following categories: 1 = retinal telangiectasia only; 2 = retinal telangiectasia and exudation; 3 = exudative retinal detachment; 4 = total retinal detachment and glaucoma; and 5 = advanced end-stage disease.2
Literature review.
We searched PubMed using the following search terms: facioscapulohumeral muscular dystrophy, FSHD, Coats disease, retinal vascular disease, and retinopathy. Thirty-one articles contained the search terms; 4 articles reported D4Z4 contraction size.
Statistical considerations.
We performed analysis using SAS version 9.3 (SAS Institute Inc., Cary, NC). Unless otherwise stated, all descriptive analysis is presented as median with quartile data in parenthesis (first quartile = Q1 = 25% of data, third quartile = Q3 = 75% of data). We presented sex, hearing loss, Coats stage, and sidedness of eye involvement as population frequencies. Prevalence is taken as the number affected divided by the number at risk, with Exact binomial confidence interval (CI).
RESULTS
Database review.
The Registry population (n = 357, table 1) was equally divided between the sexes, with a median age at diagnosis of 29 years, and median D4Z4 fragment size of 25 kb, including 18 participants with an allele size ≤15 kb. Two participants reported a history of Coats disease (both female, allele sizes 26 kb and 30 kb). The UR-FSHD database (n = 51) included more men than women (70.6% male). The median age at diagnosis was 32 years, with a median D4Z4 fragment size of 21 kb, and 9 participants with allele size ≤15 kb. One participant reported a history of Coats disease (female, survey case 5).
Table 1.
Frequency of Coats syndrome in the FSHD National Registry and UR-FSHD database
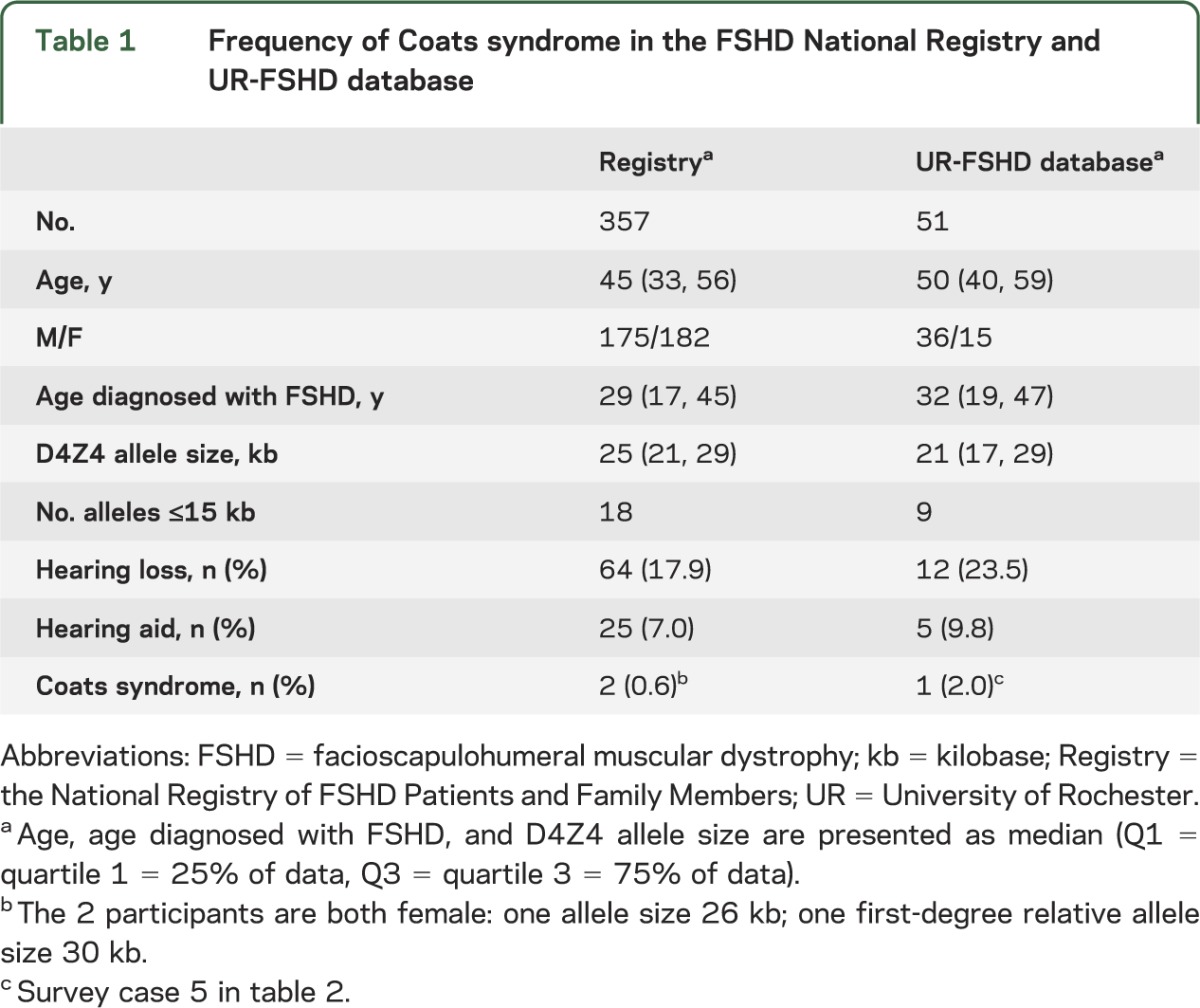
The prevalence of Coats syndrome was 0.8% of the total population (95% CI 0.2%–2.2%).
Survey and literature review.
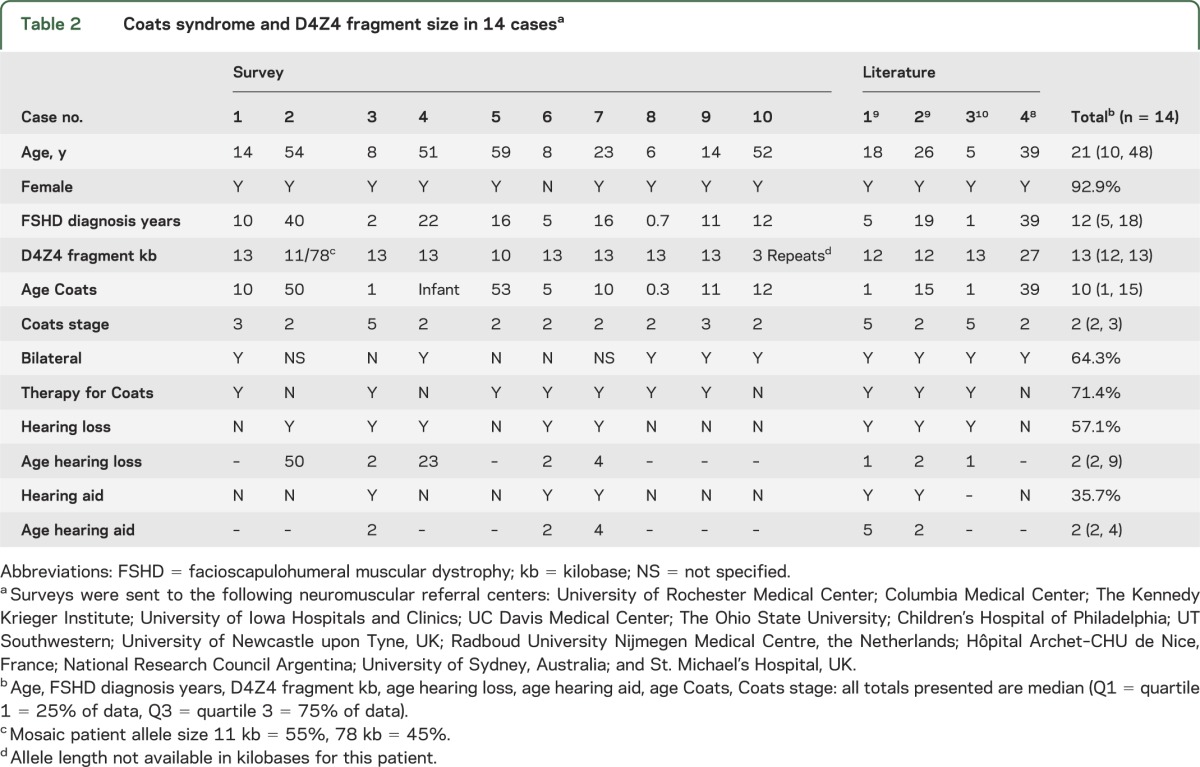
Ten survey cases and 4 cases in the literature reported D4Z4 contraction size in patients with FSHD and Coats syndrome confirmed on funduscopy (table 2). Thirteen out of 14 of these cases were female (92.9%). The median age at diagnosis of FSHD was 12 years, and the median D4Z4 fragment length was 13 kb. One subject was mosaic with 11 kb (55%), 78 kb (45%, survey case 2). The median age at diagnosis for Coats syndrome was 10 years, but 2 respondents developed symptoms at ≥50 years. Both eyes were affected in 64.3% of cases and 71.4% had received therapy. Hearing loss was reported in 57.1% of cases, with hearing aid use in 35.7% of cases, median age at first use 2 years.
Table 2.
Coats syndrome and D4Z4 fragment size in 14 casesa
DISCUSSION
Prevalent rates of retinal abnormalities for FSHD1 in the literature varied greatly between studies, from 7% to 75%; however, only a single patient had visual loss (0.7%).3–6 In addition, a Dutch population survey found 3/256 (1.7%) patients reporting Coats disease.4 This yields a conservative range for symptomatic disease between 0.7% and 1.7%.
The median allele size in patients with FSHD1 with Coats syndrome was 13 kb, considerably smaller than the median reported in the Registry or UR-FSHD database. Approximately 5%–10% of patients with FSHD type 2 (FSHD2) develop disease by a D4Z4 contraction–independent pathway. Common to both FSHD1 and FSHD2 is relaxation of chromatin structure in the D4Z4 region of chromosome 4q35 and a specific polymorphism, which leads to release of repression of a putative retrogene, DUX4, believed to be directly toxic to cells.7 To our knowledge, no cases of Coats syndrome have been described in patients with FSHD2, although the literature on FSHD2 is limited. Patients with FSHD1 with the largest contractions typically have more severe neuromuscular disease, raising the possibility of a contraction-dependent increase in DUX4 expression. Somatic mosaics tend to have mild neuromuscular manifestations, and so Coats syndrome in a mosaic patient carrying a large D4Z4 contraction (case 2) suggests a differential susceptibility of the retinal vasculature to large contractions. All cases were ≤13 kb, except for literature case 4 (27 kb).8 This case is atypical with no symptoms or signs of FSHD and retinal changes that were noted incidentally on routine funduscopy. Further testing to exclude a rare false-positive genetic test is warranted given this unusual presentation.7
The predominance of females in the cases presented is striking given the male predominance reported in idiopathic Coats disease, and suggests a unique (genetic, hormonal) susceptibility of the retinal vasculature in females with FSHD1. Although other conditions lead to secondary Coats-like syndromes (retinitis pigmentosa, CTC1 mutation), the pathologic mechanisms and clinical presentations are distinct.2
Limitations to this study include small sample size and self-reported disease status (Registry). The rarity of Coats syndrome in an already rare disease makes definitive association studies impracticable.
Coats syndrome occurs most frequently in female patients with FSHD with large contractions (allele size <15 kb) and with variable age at onset (range <1–53 years). Recent expert opinion–based FSHD guidelines recommend a single dilated screening eye examination in adults and yearly examinations in infantile-onset disease.1 The data presented here suggest that rather than patient age or disease severity, contraction size should determine the need for repeated retinal examinations. Consequently, although we still recommend all patients be screened for retinal vascular involvement at the time of diagnosis, patients with large contractions should be examined yearly afterwards, with a low threshold for fluorescein angiography.
ACKNOWLEDGMENT
The authors thank all the people who took the time to complete the survey, in particular Pauline Lahaut, Kathy Mathews, John Kissel, Robin Fitzsimons, and Carsten Bonnemann, the National FSHD Registry, and all the people who gave their time and opinions.
GLOSSARY
- CI
confidence interval
- FSHD1
facioscapulohumeral muscular dystrophy type 1
- kb
kilobase
- UR
University of Rochester
AUTHOR CONTRIBUTIONS
Jeffrey Statland and Rabi Tawil: drafting/revising the manuscript for content, study concept or design, analysis or interpretation of data, acquisition of data, and statistical analysis. Constantine Farmakidis: drafting/revising the manuscript for content, study concept or design, analysis or interpretation of data. Sabrina Sacconi: drafting/revising the manuscript for content, analysis or interpretation of data, acquisition of data. Mina Chung: drafting/revising the manuscript for content, analysis or interpretation of data, acquisition of data. Colleen Donlin-Smith: study concept or design, study coordination.
STUDY FUNDING
No targeted funding reported.
DISCLOSURE
J. Statland is supported by the MDA Clinical Research Training Grant, and is a consultant for Cytokinetics. S. Sacconi, C. Farmakidis, C. Donlin-Smith, and M. Chung report no disclosures. R. Tawil received research support from the NIH National Institute of Neurological Disorders and Stroke 1P01NS069539-01 (co-PI), NIH R01 AR054366 (coinvestigator), NIH 2U54NS048843-08 (coinvestigator), R01 NS061795-01A2 (co-PI), the Fields Center for FSHD, and Neuromuscular Research, and is a consultant for Cytokinetics. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Tawil R, van der Maarel S, Padberg GW, van Engelen BG. 171st ENMC international workshop: standards of care and management of facioscapulohumeral muscular dystrophy. Neuromuscul Disord 2010;20:471–475 [DOI] [PubMed] [Google Scholar]
- 2.Shields JA, Shields CL. Review: coats disease: the 2001 LuEsther T. Mertz lecture. Retina 2002;22:80–91 [DOI] [PubMed] [Google Scholar]
- 3.Fitzsimons RB, Gurwin EB, Bird AC. Retinal vascular abnormalities in facioscapulohumeral muscular dystrophy: a general association with genetic and therapeutic implications. Brain 1987;110:631–648 [DOI] [PubMed] [Google Scholar]
- 4.Padberg GW, Brouwer OF, de Keizer RJ, et al. On the significance of retinal vascular disease and hearing loss in facioscapulohumeral muscular dystrophy. Muscle Nerve 1995;2:S73–S80 [PubMed] [Google Scholar]
- 5.Funakoshi M, Goto K, Arahata K. Epilepsy and mental retardation in a subset of early onset 4q35-facioscapulohumeral muscular dystrophy. Neurology 1998;50:1791–1794 [DOI] [PubMed] [Google Scholar]
- 6.Nakagawa M, Matsuzaki T, Higuchi I, et al. Facioscapulohumeral muscular dystrophy: clinical diversity and genetic abnormalities in Japanese patients. Intern Med 1997;36:333–339 [DOI] [PubMed] [Google Scholar]
- 7.van der Maarel SM, Tawil R, Tapscott SJ. Facioscapulohumeral muscular dystrophy and DUX4: breaking the silence. Trends Mol Med 2011;17:252–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bass SJ, Sherman J, Giovinazzo V. Bilateral Coats' response in a female patient leads to diagnosis of facioscapulohumeral muscular dystrophy. Optometry 2011;82:72–76 [DOI] [PubMed] [Google Scholar]
- 9.Bindoff LA, Mjellem N, Sommerfelt K, et al. Severe fascioscapulohumeral muscular dystrophy presenting with Coats' disease and mental retardation. Neuromuscul Disord 2006;16:559–563 [DOI] [PubMed] [Google Scholar]
- 10.Ganesh A, Kaliki S, Shields CL. Coats-like retinopathy in an infant with preclinical facioscapulohumeral dystrophy. J AAPOS 2012;16:204–206 [DOI] [PubMed] [Google Scholar]