

Abstract
Objective:
To examine the effect of education (a surrogate measure of cognitive reserve) on FDG-PET brain metabolism in elderly cognitively healthy (HC) subjects with preclinical Alzheimer disease (AD).
Methods:
Fifty-two HC subjects (mean age 75 years) with FDG-PET and CSF measurement of Aβ1-42 were included from the prospective Alzheimer's Disease Neuroimaging Initiative biomarker study. HC subjects received a research classification of preclinical AD if CSF Aβ1-42 was <192 pg/mL (Aβ1-42 [+]) vs HC with normal Aβ (Aβ1-42 [−]). In regression analyses, we tested the interaction effect between education and CSF Aβ1-42 status (Aβ1-42 [+] vs Aβ1-42 [−]) on FDG-PET metabolism in regions of interest (ROIs) (posterior cingulate, angular gyrus, inferior/middle temporal gyrus) and the whole brain (voxel-based).
Results:
An interaction between education and CSF Aβ1-42 status was observed for FDG-PET in the posterior cingulate (p < 0.001) and angular gyrus ROIs (p = 0.03), but was not significant for the inferior/middle temporal gyrus ROI (p = 0.06), controlled for age, sex, and global cognitive ability (Alzheimer’s Disease Assessment Scale–cognitive subscale). The interaction effect was such that higher education was associated with lower FDG-PET in the Aβ1-42 (+) group, but with higher FDG-PET in the Aβ1-42 (−) group. Voxel-based analysis showed that this interaction effect was primarily restricted to temporo-parietal and ventral prefrontal brain areas.
Conclusions:
Higher education was associated with lower FDG-PET in preclinical AD (Aβ1-42 [+]), suggesting that cognitive reserve had a compensatory function to sustain cognitive ability in presence of early AD pathology that alters FDG-PET metabolism.
According to the cognitive reserve hypothesis, persons with higher cognitive reserve can sustain cognitive function in presence of more brain pathology than subjects with lower cognitive reserve.1,2 In Alzheimer disease (AD) dementia, patients with higher education exhibited a stronger brain pathology including reduced temporo-parietal FDG-PET or SPECT measures when compared to patients with AD dementia with low education at similar levels of dementia severity.3 Such an association between higher cognitive reserve and lower FDG-PET metabolism in patients with AD dementia has been replicated when cognitive reserve was assessed with alternative measures including IQ,4 schooling,5 occupation, and lifetime activities,4,6 supporting the robustness of the findings in AD dementia. These results are consistent with the cognitive reserve hypothesis in AD, i.e., at a given level of cognitive performance, subjects with higher education can tolerate stronger reduction of FDG-PET metabolism compared to subjects with lower cognitive reserve (education).
The major aim of the current study was to test the association between cognitive reserve (education) and temporo-parietal FDG-PET metabolism in individuals who meet research criteria for preclinical AD7 (i.e., cognitively healthy [HC] with abnormal CSF biomarker levels of Aβ). We hypothesized that, controlling for cognitive performance, higher cognitive reserve would be associated with lower temporo-parietal FDG-PET. The rationale for focusing on Aβ as the measure of primary AD pathology was based on previous results from a brain autopsy study showing that education reduced the impact of Aβ pathology but not tau pathology on cognitive performance.8
METHODS
Subjects.
The study included 52 HC subjects recruited within the North American multicenter Alzheimer's Disease Neuroimaging Initiative (ADNI; for database, see www.loni.ucla.edu/ADNI). In ADNI (phase I, 2005–2010), a total of 229 HC subjects were recruited. The study design, however, was such that only a subset of subjects received both a FDG-PET scan and CSF Aβ1-42 measurement at baseline, and thus the current sample included a total of 52 subjects (one subject was excluded due to misregistration of FDG-PET scans; figure e-1 on the Neurology® Web site at www.neurology.org). For the classification as HC, subjects had to show normal performance on the Logical Memory II Subscale adjusted for education as follows: 0–7 years: ≥3, 8–15 years: ≥5, 16 or more years: ≥9, and absence of significant impairment on cognitive function or activities of daily living.9
HC subjects were dichotomized based on pre-established cutoff points derived from postmortem-verified AD dementia patients vs living HC subjects.10 Normal CSF Aβ1-42 levels were considered ≥192 pg/mL (Aβ1-42 [−]) and abnormal levels were <192 pg/mL (Aβ1-42 [+]).10 In the ADNI study, it was previously reported that HC subjects showed a bimodal distribution of CSF Aβ1-42 levels, where a cutoff value of 192 pg/mL separated well subjects into high and low Aβ1-42 groups,11,12 thus providing a good basis for distinguishing groups with normal and abnormal CSF Aβ1-42 levels. HC subjects with CSF Aβ1-42 (+) were classified as preclinical AD according to recently proposed research criteria for preclinical AD.7
General inclusion criteria were an age between 55 and 90 years, a modified Hachinski score ≤4, education of at least 6 grade level, and stable treatment of at least 4 weeks in case of treatment with permitted medication (for full list see http://www.adni-info.org, Procedures Manual). Subjects who did not have MRI, FDG-PET, and CSF assessment were excluded by list-wise exclusion.
Standard protocol approvals, registrations, and patient consents.
The study was approved after ethical review and written patient consent was obtained by the ADNI investigators.
Neuropsychological assessment.
Episodic memory performance was quantified by a composite episodic memory score based on different neuropsychological tests including the Rey Auditory Verbal Learning Test (word list learning trials, recall, and recognition), Alzheimer’s Disease Assessment Scale (ADAS, word list learning, recall, and recognition), Mini-Mental State Examination (word recall), and Logical Memory I and II, as previously described,13 and were downloaded from the ADNI Web page (http://adni.loni.ucla.edu/). The executive function test score included a range of different test scores as described in reference 13a. Briefly, scores were subjected in an iterative process to a confirmatory factor analysis and a flexible recoding of test scores to preserve variability at the extremes of the distribution.13
CSF measurement.
All CSF samples collected at the different centers were shipped on dry ice to the Penn ADNI Biomarker Core Laboratory at the University of Pennsylvania, Philadelphia, for storage at −80°C until further analysis at the laboratory. More details on data collection of the CSF samples can be found at http://www.adni-info.org, under “ADNI study procedures.” The CSF concentrations of Aβ1-42 were measured in the baseline CSF samples using the multiplex xMAP Luminex platform (Lumnix Corp., Austin, TX) at the Penn ADNI Biomarker Core Laboratory. For detailed description, see reference 10.
FDG-PET acquisition and region of interest measurement.
FDG scans were collected as 6 × 5–minute frames beginning 30 minutes after injection of approximately 5 mCi of tracer. Attenuation correction was performed either via transmission scan or CT. Images were downloaded from the ADNI data bank (http://adni.loni.ucla.edu/) after they had been preprocessed to provide standard orientation, voxel size, and resolution.
FDG-PET regions of interest (ROIs) were constructed based on a meta-analysis of the location of FDG-PET changes in the brain that are typically affected in AD, as described previously.14,15 For the voxel-based analysis of FDG uptake, spatial normalization parameters were estimated via high-dimensional diffeomorphic mapping using the tool DARTEL of the software program SPM8 (Wellcome Trust Centre for Neuroimaging, University College London, London, UK). The FDG-PET scans were coregistered to the T1 MRI scans and the spatial normalization parameters were applied to the coregistered FDG-PET scans. Subsequently, the signal intensity of FDG-PET scans was normalized to a reference region including the pons (dividing each voxel value by the average FDG-PET within the pons) and spatially smoothed with an 8-mm full-width-at-half-maximum Gaussian kernel.
Statistics.
For the FDG-PET ROI-based analysis, linear regression was used to test the interaction effect between education (years) × CSF Aβ1-42 status on FDG-PET. Global cognitive ability was controlled for through ADAS–cognitive subscale (ADAS-cog) scores in all regression models. We repeated the regression analysis controlling either for the composite score of episodic memory or executive function. Prior to building regression models, the influence of potential confounding variables, i.e., age, sex, and APOE genotype (APOE ε4 carriers vs APOE ε4 noncarriers), was tested in simple regression models to predict FDG-PET in each ROI. Only age was significantly associated with FDG-PET in at least one of the ROIs and age was therefore included in all regression models as a covariate.
The interaction term education × CSF Aβ1-42 status was considered significant if the model fit was significantly improved by including the interaction term compared to the alternative model where CSF Aβ1-42 status and education were only additive terms. The difference between the fit of both regression models was tested in likelihood ratio tests. In the main regression analysis, we controlled for cognition as measured by ADAS-cog, but we computed additional regression analyses to control for more specific cognitive differences including composite scores of episodic memory or executive function. Residuals of the regression models were tested for deviance from the normal distribution, using the Shapiro-Wilk test. For inferior/middle temporal lobe ROI of FDG-PET (when controlling for ADAS-cog), and, in addition, for the angular gyrus (when controlling for episodic memory score), the residuals of the regression analysis were not normally distributed (p < 0.05). Therefore, the variables were log-transformed and the regression analysis repeated. The distribution of the residuals was no longer different from a normal distribution after log transformation. If an interaction term was significant, the robustness of the statistical significance with regard to the potential influence of outliers was tested by robust regression, using iterated reweighted least squares, which reduces the weights for data points with large residuals.16 All statistical ROI analyses were done with the statistical software R 2.12 (freely available at http://www.r-project.org/).
In order to explore the spatial distribution of the interaction effect between education and Aβ status in brain areas other than the temporo-parietal ROIs, voxel-wise multiple regression models including age, ADAS-cog, education, CSF Aβ1-42 status, and education × CSF Aβ1-42 status as predictors were computed. The resulting t statistic maps of the interaction effect were thresholded at the voxel level at p = 0.01 and false discovery rate corrected at the cluster level at p = 0.05. In addition, we computed the effect size d derived from the t statistic of the interaction term17 in order to render the distribution of descriptive effect sizes of the interaction term independent of a significance threshold and sample size–related statistical power. All voxel-wise regression analyses were computed in SPM 8.
RESULTS
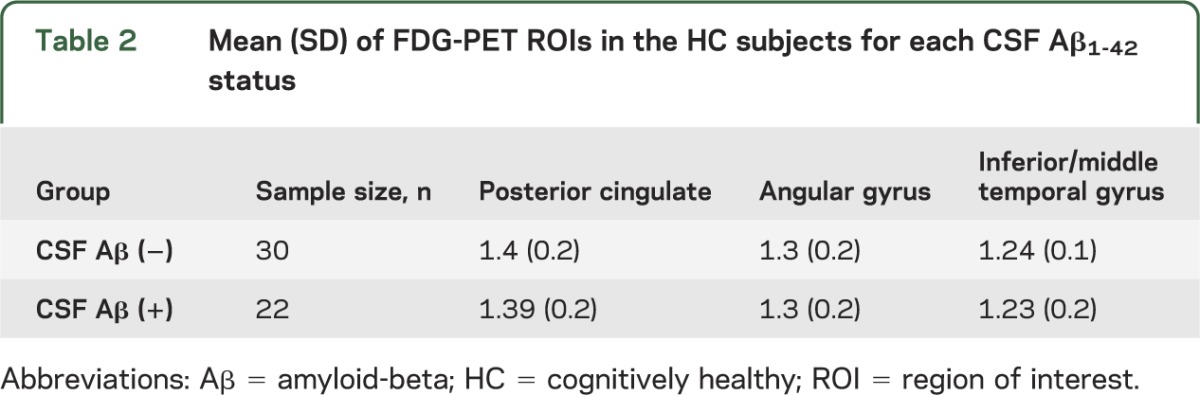
Descriptive statistics of the mean and SD on demographic, clinical, and APOE genotype data for both HC Aβ1-42 (−) and HC Aβ1-42 (+) groups and the total sample are displayed table 1. Mean FDG-PET ROI values are displayed for both HC groups in table 2. The association between education and cognitive variables in the whole sample is described in appendix e-1.
Table 1.
Demographic, neuropsychological, and genetic characteristics of HC subjects split by CSF Aβ1-42 statusa
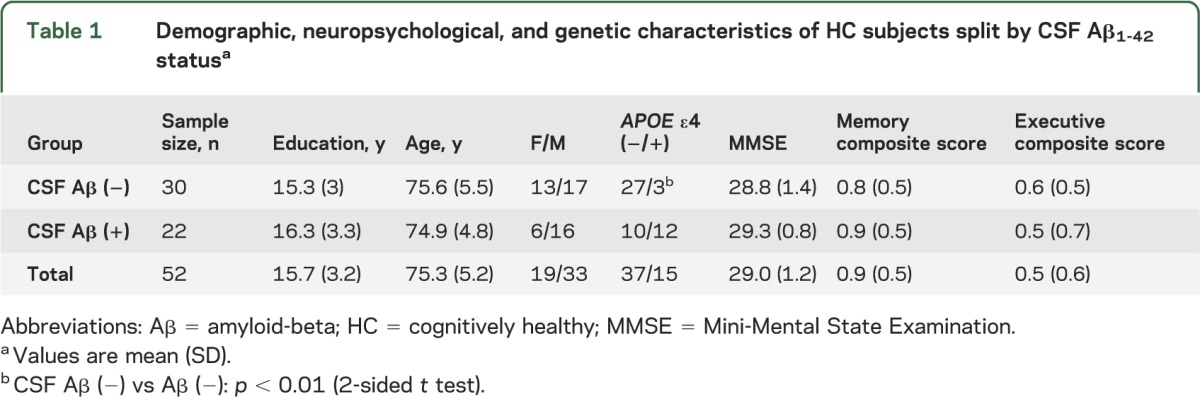
Table 2.
Mean (SD) of FDG-PET ROIs in the HC subjects for each CSF Aβ1-42 status
ROI-based analysis of interaction effect of education × CSF Aβ1-42 status on FDG-PET.
The results of the regression analysis are presented in table 3. For the posterior cingulum FDG-PET ROI, regression analysis showed significant main effects for age (p < 0.01), education (p < 0.01), and CSF Aβ1-42 status (p < 0.01). The hypothesized interaction effect of education × CSF Aβ1-42 status was significant (p = 0.001, figure 1A). When including the interaction term, the overall model fit was improved (χ2 = 11.7, p < 0.001). For the angular gyrus FDG-PET ROI, the main effect of age was significant (p = 0.01), but not the main effect of education (p = 0.16). The hypothesized interaction effect between education and CSF Aβ status was significant (p = 0.03, figure 1B) and improved the overall model fit (χ2 = 4.9, p = 0.03). Repeating the analysis with robust regression to control for potential outliers showed similar regression coefficients of the interaction terms for both the posterior cingulum FDG-PET ROI (B = −0.03, SE = 0.01, p = <0.01) and the angular gyrus FDG-PET ROI (−0.03, SE = 0.01, p = 0.05). For the inferior/middle temporal gyri FDG-PET ROI, the interaction effect (B = −0.01, SE = 0.01, p = 0.06) and the change in the overall model fit (χ2 = 3.6, p = 0.06) failed to reach the signficance threshold. As shown in figure 1, the interactions between education and CSF Aβ status (controlled for ADAS-cog) for the posterior cingulate (figure 1A) and angular gyrus FDG-PET ROIs (figure 1B) were such that a higher number of years of education was associated with lower FDG-PET metabolism in Aβ1-42 (+) subjects, but a higher number of years of education was associated with higher FDG-PET metabolism in Aβ1-42 (−) subjects.
Table 3.
Regression models for the interaction between CSF Aβ1-42 status and education on FDG-PET controlled for ADAS-cog
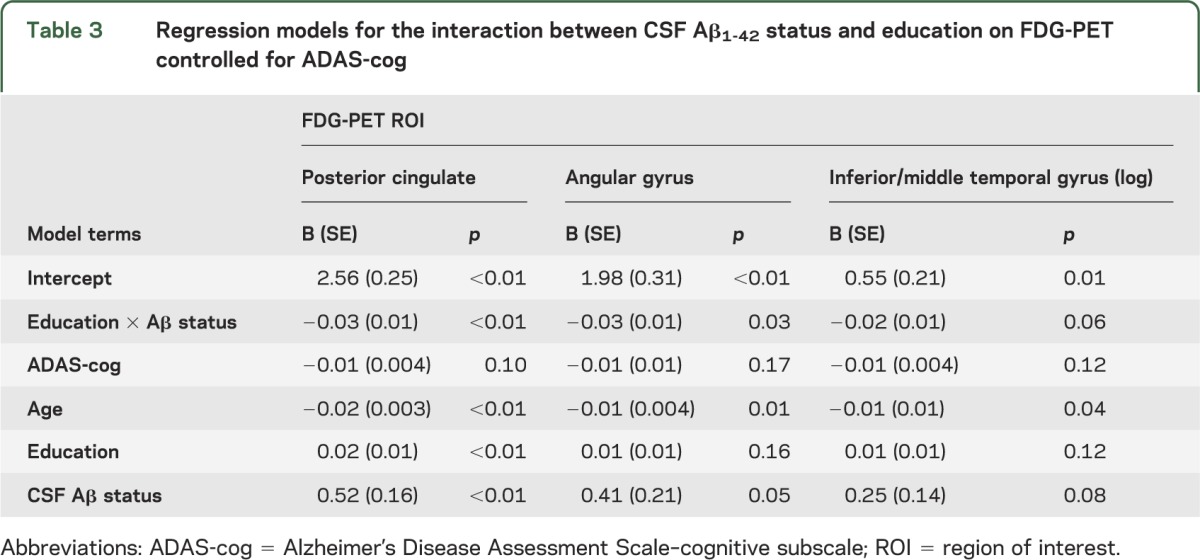
Figure 1. Scatterplot of the interaction between education and CSF Aβ1-42 group for predicting FDG-PET.
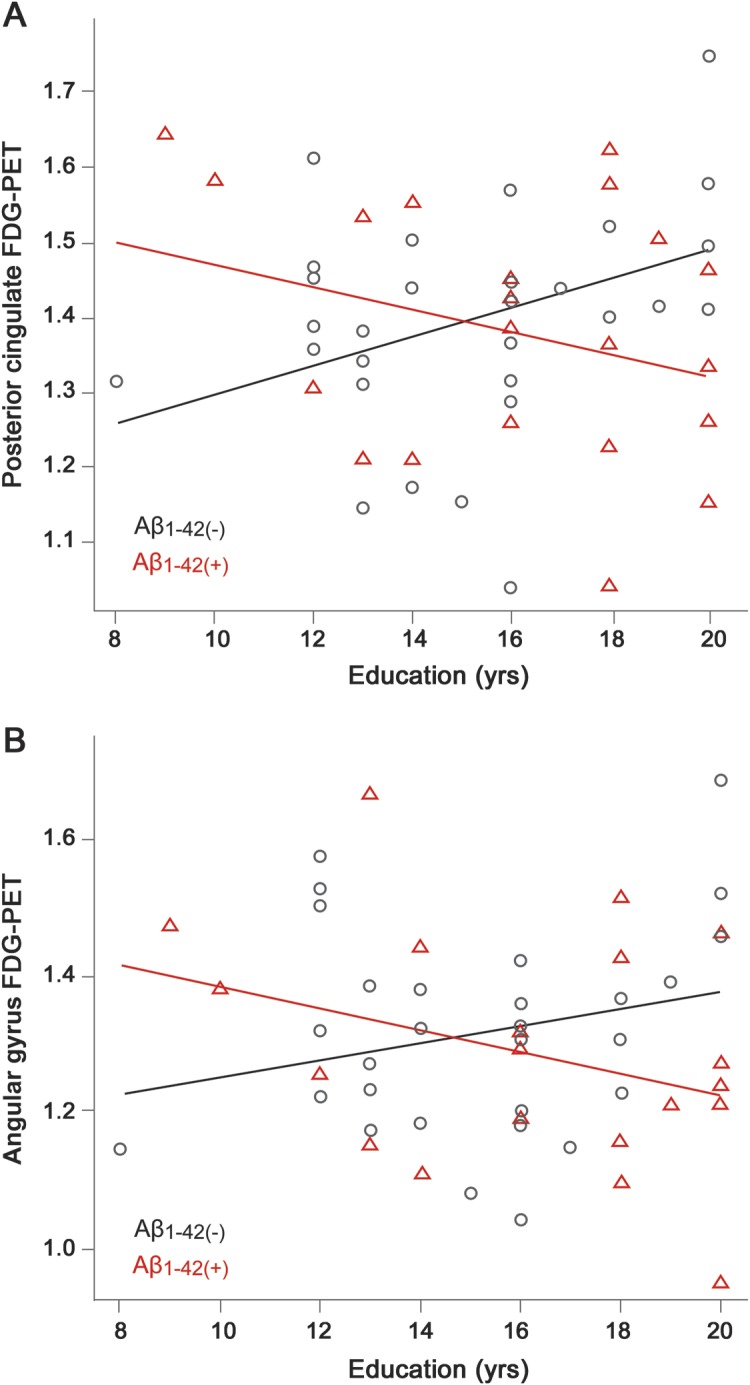
The number of years of education is plotted against FDG-PET for the regions of interest (ROIs) of posterior cingulate gyrus (A) and angular gyrus (B). Regression lines are plotted for groups including CSF Aβ1-42 (−) indicated by black circles and CSF Aβ1-42 (+) indicated by red triangles.
To ensure that subtle cognitive deficits in specific cognitive domains, such as memory and executive function, did not influence the findings, we repeated the analyses of the interaction between CSF Aβ1-42 status and education on FDG-PET, this time controlling for the composite measure of episodic memory or executive function. The results on the interaction effect remained virtually the same (tables e-1 and e-2).
In a secondary analysis, we conducted a selective subgroup analysis on education, FDG-PET (posterior cingulate ROI), and composite episodic memory in the Aβ1-42 (+) group in order to confirm the directionality of the associations between the different variables in the preclinical AD group. The results are presented in appendix e-1.
Voxel-based mapping of interaction effect of education and CSF Aβ1-42 status on FDG-PET.
Voxel-based mapping of interaction effect of education and CSF Aβ1-42 status on FDG-PET is detailed in table e-3. Significant interaction effects (education × CSF Aβ1-42 status) were observed within the bilateral temporal lobe, including the hippocampus region and lateral temporal lobe structures, and the ventral prefrontal cortex (figure 2A). The voxel-wise interaction effects were in the same direction as observed in the ROI analysis reported above, i.e., for the Aβ1-42 (+) subject group, an increase in years of education was associated with lower FDG-PET metabolism, whereas for the Aβ1-42 (−) group, an increase in years of education was associated with an increase in FDG-PET metabolism. No significant interaction effect in the opposite direction was observed. Mapping of the spatial distribution of the effect sizes of the interaction effect showed that the largest effect sizes were predominantly distributed within the medial temporal lobe, inferior frontal lobe, and parietal lobe (figure 2B).
Figure 2. Voxel-wise assessment of the interaction effect education × CSF Aβ1-42 status for predicting FDG-PET.

The t statistics projected onto the surface (A) and the effect sizes projected onto axial slices (B) are displayed. In A, red shows those brain regions with significant interaction effects (false discovery rate corrected at the cluster level at p = 0.05). The interaction effects were all in the same direction as reported for the region of interest (ROI) analysis, i.e., higher number of years of education was associated with lower FDG-PET in Aβ1-42 (+) subjects, but the reverse was true for Aβ1-42 (−) subjects. There were no significant interaction effects in the opposite direction. In B, colors ranging between green/yellow and red indicate the size of the interaction effects that were in the same direction as reported for the ROI analysis.
DISCUSSION
FDG-PET hypometabolism within temporo-parietal brain areas has been consistently observed to develop early in the course of AD.18,19 The main finding of the current study including the interaction effect of education × Aβ1-42 status on temporo-parietal FDG-PET showed that in preclinical AD subjects (Aβ1-42 [+]) but not normal elderly subjects (Aβ1-42 [−]) higher education was associated with lower temporo-parietal FDG-PET metabolism at a given level of cognitive performance. This finding suggests that in the early stage of AD (abnormal Aβ) cognitive reserve (higher education) compensates FDG-PET hypometabolism to maintain cognitive performance.
Specifically, we interpret the findings as follows: at higher levels of cognitive reserve, relatively low levels of temporo-parietal FDG-PET metabolism (indicative of more brain damage) can be tolerated to maintain normal cognition in preclinical AD. In contrast, at lower levels of cognitive reserve, temporo-parietal FDG-PET metabolism must be relatively high in preclinical AD subjects (compared to the HC Aβ1-42 [−]) in order to maintain normal cognition. The increased FDG-PET metabolism in preclinical AD with low cognitive reserve can be explained by a selection effect: subjects with low FDG-PET and low education are more likely to show mild cognitive impairment (MCI) or AD dementia than to stay cognitively normal.15 Results of our post hoc analysis in the preclinical AD subjects showed that both higher posterior cingulate FDG-PET and higher education were associated with higher memory performance, but higher education was associated with lower posterior cingulate FDG-PET, suggesting a compensatory role of education with regard to FDG-PET hypometabolism to maintain cognitive performance in preclinical AD.
Our findings on the association between higher education and lower FDG-PET metabolism in subjects with preclinical AD (CSF Aβ1-42 [+]) are in line with previous studies in patients with clinically manifest AD dementia,3,5,20 which reported an association between higher education and lower FDG-PET in the temporo-parietal brain areas while controlling for global cognitive ability. Together the current and previous results suggest that across different clinical stages of AD, subjects with high cognitive reserve can tolerate more FDG-PET hypometabolism to maintain a certain level of cognitive performance when compared to subjects with low cognitive reserve.
A further implication of the current results is that subjects with higher levels of cognitive reserve show better cognitive performance at similar levels of brain pathology. Such a notion is supported also by results from previous studies showing a lower likelihood of AD dementia or cognitive impairment in subjects with higher cognitive reserve when controlling for brain Aβ measured by Pittsburgh compound B (PiB) PET.21,22 Importantly, results from several studies suggest that cognitive reserve modifies the effect of Aβ on cognition (i.e., interaction between education and Aβ levels).21,23 In cognitively normal subjects, the interaction between education and global brain PiB PET was such that more education was associated with better global cognitive ability at abnormally high but not at normal levels of PiB PET21 (but see reference 24). These results suggest a compensatory role of cognitive reserve (education) to maintain cognitive performance in subjects with abnormal brain Aβ levels, and are in agreement with the current result of increased tolerance of FDG-PET hypometabolism associated with cognitive reserve (education) in preclinical AD (HC Aβ1-42 [+]).
Support for the hypothesis that cognitive reserve compensates localized brain damage in AD comes also from studies focusing on gray matter volume changes. Greater brain atrophy and higher levels of brain Aβ have been reported in subjects with higher cognitive reserve compared to subjects with lower cognitive reserve in a pooled sample of cognitively normal and cognitively impaired (MCI and AD dementia) subjects.25 Together the previous and current findings suggest that cognitive reserve is associated with relatively preserved cognitive performance in the face of AD pathology (Aβ) and AD-associated functional (as assessed by FDG-PET) and structural (as assessed by MRI) brain changes.
The current study includes caveats that should be kept in mind when interpreting the results. First, we have used education as a proxy of cognitive reserve, but alternative measures including IQ, schooling, life activities, and occupation have been previously used to assess cognitive reserve.3–5,20,26,27 However, across those studies using different measures of cognitive reserve in AD dementia, an association between higher scores on cognitive reserve measures and lower temporo-parietal FDG-PET was consistently reported.3–5,20,26,27 Thus, it is unlikely that the current results on the association between cognitive reserve and temporo-parietal FDG-PET are specific to education as a measure of cognitive reserve.
We note that in the current study a relatively high proportion of HC subjects with abnormal Aβ (42%) was found. However, such a proportion is still within the range of the proportion of elderly HC subjects with abnormal CSF-Aβ or PiB PET levels reported previously (18–50%).28,29 Potential factors that influence the size of the proportion of subjects with abnormal brain Aβ are age and APOE genotype.28
It should be pointed out that the current study did not address what neuronal mechanisms actively compensate AD pathology and brain dysfunction (e.g., parietal FDG-PET hypometabolism). It has been suggested that such compensatory mechanisms could consist of the increased efficiency or capacity of task-related networks (i.e., “neural reserve”), the ability to recruit compensatory task-related networks (i.e., “neural compensation”), or generalized networks that underlie cognitive support in the presence of pathology.1,30 Functional imaging studies have begun to uncover cognitive reserve–related changes of brain activity, including the prefrontal and temporal cortices.30–35 In addition to functional network changes, structural brain differences, such as bigger brain size or white matter integrity, may allow for larger extent of neuronal loss before cognitive deficits start to emerge.36–38 However, it remains unclear how such brain differences, which may be considered “brain reserve,” relate to cognitive reserve and cognitive function and remain to be described in future studies.
Supplementary Material
ACKNOWLEDGMENT
The authors thank Norbert Schuff, PhD, University of California at San Francisco, for comments on the data analysis.
GLOSSARY
- AD
Alzheimer disease
- ADAS
Alzheimer’s Disease Assessment Scale
- ADAS-cog
ADAS–cognitive subscale
- ADNI
Alzheimer's Disease Neuroimaging Initiative
- HC
cognitively healthy
- MCI
mild cognitive impairment
- PiB
Pittsburgh compound B
- ROI
region of interest
Footnotes
AUTHOR CONTRIBUTIONS
Dr. Ewers: study concept, analysis and interpretation of data, drafting and revising the manuscript for content. Mr. Insel: analysis and interpretation of data. Dr. Stern: drafting and revising the manuscript for content. Dr. Weiner: drafting and revising the manuscript for content, study concept.
STUDY FUNDING
Data collection and sharing for this project was funded by the Alzheimer's Disease Neuroimaging Initiative (ADNI) (NIH grant U01 AG024904). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: Abbott, AstraZeneca AB, Bayer Schering Pharma AG, Bristol-Myers Squibb, Eisai Global Clinical Development, Elan Corporation, Genentech, GE Healthcare, GlaxoSmithKline, Innogenetics, Johnson and Johnson, Eli Lilly and Co., Medpace, Inc., Merck and Co., Inc., Novartis AG, Pfizer Inc, F. Hoffman-La Roche, Schering-Plough, and Synarc, Inc., as well as nonprofit partners the Alzheimer's Association and Alzheimer's Drug Discovery Foundation, with participation from the US Food and Drug Administration. Private sector contributions to ADNI are facilitated by the Foundation for the NIH (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer's Disease Cooperative Study at the University of California, San Diego. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of California, Los Angeles. This research was also supported by NIH grants P30 AG010129 and K01 AG030514.
DISCLOSURE
M. Ewers and P. Insel report no disclosures. Y. Stern has received payments from the companies Ortho-McNeil Neurologics, Merck & Co, Glaxo Smith Kline, Eli Lily, Janssen, and Bayer for consultancy. M.W. Weiner holds stocks of the companies Synarc and Elan. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Stern Y. Cognitive reserve. Neuropsychologia 2009;47:2015–2028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stern Y. What is cognitive reserve? Theory and research application of the reserve concept. J Int Neuropsychol Soc 2002;8:448–460 [PubMed] [Google Scholar]
- 3.Stern Y, Alexander GE, Prohovnik I, Mayeux R. Inverse relationship between education and parietotemporal perfusion deficit in Alzheimer's disease. Ann Neurol 1992;32:371–375 [DOI] [PubMed] [Google Scholar]
- 4.Scarmeas N, Zarahn E, Anderson KE, et al. Association of life activities with cerebral blood flow in Alzheimer disease: implications for the cognitive reserve hypothesis. Arch Neurol 2003;60:359–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Perneczky R, Drzezga A, Diehl-Schmid J, et al. Schooling mediates brain reserve in Alzheimer's disease: findings of fluoro-deoxy-glucose-positron emission tomography. J Neurol Neurosurg Psychiatry 2006;77:1060–1063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stern Y, Alexander GE, Prohovnik I, et al. Relationship between lifetime occupation and parietal flow: implications for a reserve against Alzheimer's disease pathology. Neurology 1995;45:55–60 [DOI] [PubMed] [Google Scholar]
- 7.Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging–Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement 2011;7:280–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bennett DA, Schneider JA, Wilson RS, Bienias JL, Arnold SE. Education modifies the association of amyloid but not tangles with cognitive function. Neurology 2005;65:953–955 [DOI] [PubMed] [Google Scholar]
- 9.Ewers M, Walsh C, Trojanowski JQ, et al. Prediction of conversion from mild cognitive impairment to Alzheimer's disease dementia based upon biomarkers and neuropsychological test performance. Neurobiol Aging 2012;33:1203–1214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shaw LM, Vanderstichele H, Knapik-Czajka M, et al. Cerebrospinal fluid biomarker signature in Alzheimer's disease neuroimaging initiative subjects. Ann Neurol 2009;65:403–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ewers M, Insel P, Jagust WJ, et al. CSF biomarker and PiB-PET-derived beta-amyloid signature predicts metabolic, gray matter, and cognitive changes in nondemented subjects. Cereb Cortex 2012;22:1993–2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.De Meyer G, Shapiro F, Vanderstichele H, et al. Diagnosis-independent Alzheimer disease biomarker signature in cognitively normal elderly people. Arch Neurol 2010;67:949–956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Crane PK, Carle A, Gibbons LE, et al. Development and assessment of a composite score for memory in the Alzheimer's Disease Neuroimaging Initiative (ADNI). Available at: http://www.ncbi.nlm.nih.gov/pubmed/22782295 Accessed July 11, 2012. [DOI] [PMC free article] [PubMed]
- 13a.Gibbons LE, Carle AC, Mackin RS, et al. A composite score for executive functioning, validated in Alzheimer's Disease Neuroimaging Initiative (ADNI) participants with baseline mild cognitive impairment. Brain Imaging Behav 2012;6:517–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jagust WJ, Landau SM, Shaw LM, et al. Relationships between biomarkers in aging and dementia. Neurology 2009;73:1193–1199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Landau SM, Harvey D, Madison CM, et al. Associations between cognitive, functional, and FDG-PET measures of decline in AD and MCI. Neurobiol Aging 2011;32:1207–1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Venables WN, Ripley BD. Modern Applied Statistics with S, 4th ed New York: Springer; 2002 [Google Scholar]
- 17.Cohen J. A power primer. Psychol Bull 1992;112:155–159 [DOI] [PubMed] [Google Scholar]
- 18.Drzezga A, Lautenschlager N, Siebner H, et al. Cerebral metabolic changes accompanying conversion of mild cognitive impairment into Alzheimer's disease: a PET follow-up study. Eur J Nucl Med Mol Imaging 2003;30:1104–1113 [DOI] [PubMed] [Google Scholar]
- 19.Minoshima S, Giordani B, Berent S, Frey KA, Foster NL, Kuhl DE. Metabolic reduction in the posterior cingulate cortex in very early Alzheimer's disease. Ann Neurol 1997;42:85–94 [DOI] [PubMed] [Google Scholar]
- 20.Kemppainen NM, Aalto S, Karrasch M, et al. Cognitive reserve hypothesis: Pittsburgh compound B and fluorodeoxyglucose positron emission tomography in relation to education in mild Alzheimer's disease. Ann Neurol 2008;63:112–118 [DOI] [PubMed] [Google Scholar]
- 21.Roe CM, Mintun MA, D’Angelo G, Xiong C, Grant EA, Morris JC. Alzheimer disease and cognitive reserve: variation of education effect with carbon 11–labeled Pittsburgh compound B uptake. Arch Neurol 2008;65:1467–1471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roe CM, Mintun MA, Ghoshal N, et al. Alzheimer disease identification using amyloid imaging and reserve variables: proof of concept. Neurology 2010;75:42–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rentz DM, Locascio JJ, Becker JA, et al. Cognition, reserve, and amyloid deposition in normal aging. Ann Neurol 2010;67:353–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roe CM, Fagan AM, Williams MM, et al. Improving CSF biomarker accuracy in predicting prevalent and incident Alzheimer disease. Neurology 2011;76:501–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vemuri P, Weigand SD, Przybelski SA, et al. Cognitive reserve and Alzheimer's disease biomarkers are independent determinants of cognition. Brain 2011;134:1479–1492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alexander GE, Furey ML, Grady CL, et al. Association of premorbid intellectual function with cerebral metabolism in Alzheimer's disease: implications for the cognitive reserve hypothesis. Am J Psychiatry 1997;154:165–172 [DOI] [PubMed] [Google Scholar]
- 27.Liao YC, Liu RS, Teng EL, et al. Cognitive reserve: a SPECT study of 132 Alzheimer's disease patients with an education range of 0-19 years. Dement Geriatr Cogn Disord 2005;20:8–14 [DOI] [PubMed] [Google Scholar]
- 28.Morris JC, Roe CM, Xiong C, et al. APOE predicts amyloid-beta but not tau Alzheimer pathology in cognitively normal aging. Ann Neurol 2010;67:122–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Klunk WE. Amyloid imaging as a biomarker for cerebral beta-amyloidosis and risk prediction for Alzheimer dementia. Neurobiol Aging 2011;32(suppl 1):S20–S36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stern Y, Habeck C, Moeller J, et al. Brain networks associated with cognitive reserve in healthy young and old adults. Cereb Cortex 2005;15:394–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Scarmeas N, Zarahn E, Anderson KE, et al. Cognitive reserve modulates functional brain responses during memory tasks: a PET study in healthy young and elderly subjects. Neuroimage 2003;19:1215–1227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stern Y, Zarahn E, Habeck C, et al. A common neural network for cognitive reserve in verbal and object working memory in young but not old. Cereb Cortex 2008;18:959–967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bosch B, Bartres-Faz D, Rami L, et al. Cognitive reserve modulates task-induced activations and deactivations in healthy elders, amnestic mild cognitive impairment and mild Alzheimer's disease. Cortex 2010;46:451–461 [DOI] [PubMed] [Google Scholar]
- 34.Scarmeas N, Zarahn E, Anderson KE, et al. Cognitive reserve-mediated modulation of positron emission tomographic activations during memory tasks in Alzheimer disease. Arch Neurol 2004;61:73–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grady CL, Furey ML, Pietrini P, Horwitz B, Rapoport SI. Altered brain functional connectivity and impaired short-term memory in Alzheimer's disease. Brain 2001;124:739–756 [DOI] [PubMed] [Google Scholar]
- 36.Perneczky R, Wagenpfeil S, Lunetta KL, et al. Head circumference, atrophy, and cognition: implications for brain reserve in Alzheimer disease. Neurology 2010;75:137–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Teipel SJ, Meindl T, Wagner M, et al. White matter microstructure in relation to education in aging and Alzheimer's disease. J Alzheimers Dis 2009;17:571–583 [DOI] [PubMed] [Google Scholar]
- 38.Bartres-Faz D, Sole-Padulles C, Junque C, et al. Interactions of cognitive reserve with regional brain anatomy and brain function during a working memory task in healthy elders. Biol Psychol 2009;80:256–259 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.