

Abstract
The faithful and rapid translation of genetic information into peptide sequences is an indispensable property of the ribosome. Mechanistic understanding of strategies utilized by the ribosome in achieving both speed and fidelity during translation results from nearly a half century of biochemical and structural studies. Emerging from these studies is the common theme that the ribosome utilizes local as well as remote conformational switches to govern induced-fit mechanisms that ensure accuracy in codon recognition during both tRNA selection and translation termination.
Introduction
The ability of all living organisms to efficiently and accurately translate genomic information into functional proteins is a remarkable process that is the result of billions of years of evolution. This flow of information from DNA to protein requires that the three polymerization reactions fundamental to life -- DNA replication, transcription, and translation -- proceed with optimized levels of fidelity and speed. This optimization requirement stems from the fact that one of these two parameters is typically compromised at the expense of the other (Thompson and Karim, 1982). Thus, the accuracy of the three polymerization reactions can be ranked according to their importance in maintaining the integrity of the organism. DNA replication proceeds with an impressive level of accuracy, where an incorrect nucleotide is incorporated only once in 108–1010 events (Kunkel and Bebenek, 2000), whereas transcription and translation proceed with considerably lower levels of fidelity, with misincorporation rates of 1 in 104 and 1 in 103–104, respectively (Bouadloun et al., 1983; Edelmann and Gallant, 1977; Kramer and Farabaugh, 2007; Laughrea et al., 1987; Rosenberger and Foskett, 1981).
Each of the three polymerization processes utilizes the complementarity of nucleotides to choose the correct substrate. DNA and RNA polymerases select the precursor nucleotide triphosphate (dNTP and NTP) that is complementary to the DNA template for direct incorporation into the growing nucleic acid chain. Similarly, the ribosome selects the cognate aminoacyl-tRNA (aa-tRNA) based on the complementarity of its anticodon with the mRNA codon and extends the polypeptide chain by one amino acid. Shortly after their discovery of the DNA double-helical structure, Watson and Crick hypothesized that the selectivity of polymerases could be simply explained by the Watson-Crick hydrogen bonding between complementary nucleotides, an idea that Crick later extended to the ribosome. Later studies estimated that the energetics of hydrogen bonding could contribute at most ~40 fold to the selectivity of DNA polymerases, and that most of the selectivity must instead result from correct recognition of the geometry of a Watson-Crick base-pair (Kunkel and Bebenek, 2000). DNA polymerases, for example, have evolved molecular calipers that precisely measure the invariant properties of Watson-Crick pairs, such as the distance between the purine N3 and pyrimidine O2.
In contrast to polymerases, the recognition site (decoding center) of the ribosome is ~70 Å away from the site where actual polymerization takes place (peptidyl transferase center). In the ribosome, two distinct subunits perform the disparate tasks of tRNA recognition and polypeptide chain elongation. The 30S (40S in eukaryotes) subunit contains the decoding site where the codon-anticodon interaction is deciphered, whereas the 50S (60S in eukaryotes) subunit contains the active site where the peptidyl transfer and hydrolysis reactions occur. During translation initiation, the two subunits come together to form the 70S (80S in eukaryotes) ribosome and launch the elongation cycle. The ribosome carries three tRNA binding sites: the aminoacyl (A) site, the peptidyl (P) site and the exit (E) site. During the elongation cycle (Figure 1), a ternary complex comprised of aa-tRNA, the elongation factor EF-Tu (eEF1A in eukaryotes), and GTP is delivered to the A site where it reacts with the peptidyl-tRNA in the P site, elongating the nascent peptide by one amino acid. In order to complete the cycle, the peptidyl-transfer reaction is followed by translocation, a reaction catalyzed by elongation factor EF-G (EF-2 in eukaryotes), which uses the energy of GTP hydrolysis to promote movement of the peptidyl-tRNA and the deacylated tRNA into the P and E sites, respectively. These movements free the A site for the next round of elongation. Polypeptide chain elongation ends when a stop codon in the A site is recognized by a class I release factor (RF1 or RF2 in bacteria and eRF1 in eukaryotes). This results in the hydrolysis of peptidyl-tRNA and release of the growing polypeptide chain. An additional GTPase factor is involved in the termination process in both bacteria (RF3) and in eukaryotes (eRF3), though their roles appear to be quite distinct. The substantial differences in the function of these factors during the final steps of protein synthesis are consistent with the fact that the class I release factors in bacteria and eukaryotes are not evolutionarily related (reviewed in Youngman et al., 2008).
Figure 1. Elongation and termination steps of bacterial translation.
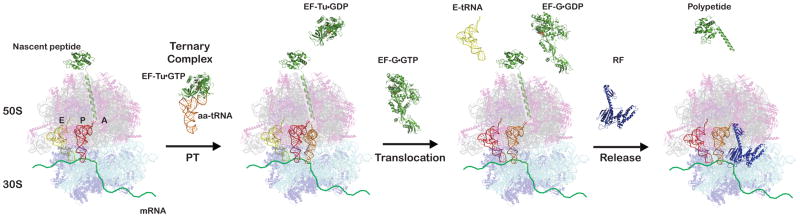
During the elongation cycle, peptidyl transfer (PT) takes place in which a ternary complex composed of the elongation factor EF-Tu, aminoacyl-tRNA (aa-tRNA), and GTP is deposited into the aminoacyl (A) site and reacts with the peptidyl-tRNA, elongating the nascent peptide by one amino acid. Subsequent translocation of the mRNA in the ribosome is mediated by elongation factor EF-G, which couples the energy of GTP hydrolysis to directional movement of the mRNA-tRNA complex. As a result, the peptidyl-tRNA and the deacylated tRNA move from the A and peptidyl (P) sites into the P and exit (E) sites, respectively. Termination of protein synthesis occurs when a stop codon enters the A site. Stop codons are recognized by class I release factors (RFs), which trigger a hydrolytic reaction that results in the release of the growing polypeptide chain from the tRNA. Molecular figures shown here were constructed and modified from PDB files (3D5A, 1GIX, 1COS, 1DAR, 1TTT, 1TUI, 2BV3 and 2VB1) using Pymol (DeLano Scientific) to depict the desired portions of the translation cycle. The E-site tRNA was omitted from the crystal structure in panel 2 and faded in panel 4 and to indicate the ambiguity of its status.
The specificity of enzyme-catalyzed reactions typically results from an active site that is tailored for the correct substrate, where favorable molecular interactions take advantage of differences in the free energy (ΔΔG) of binding between cognate and non-cognate substrates. Although inherent energetic differences are often sufficient to promote specificity in enzymatic reactions, they are not enough for template-directed polymerization synthesis. Both polymerases and the ribosome must distinguish among very similar substrates with small differences in the free energy of binding in order to achieve the level of fidelity observed (reviewed in Cochella and Green, 2005b). Though this seems like a physical improbability, these enzymes have evolved mechanisms that utilize the small energetic differences multiple times (kinetic proofreading) to exponentially increase the accuracy directly proportional to the number of times the discrimination step is used (Hopfield, 1974; Ninio, 1975). Editing mechanisms that take place after the actual chemical incorporation step also exist for each polymerization reaction, providing additional contributions to overall fidelity (Brutlag and Kornberg, 1972; Thompson and Stone, 1977). The termination of protein synthesis is catalyzed by the class I release factors, which recognize stop codons with high fidelity. In contrast to tRNA selection that depends on the exact geometry of nucleotide base pairing to determine selectivity, Release factors depend on RNA-protein interactions to discriminate between highly similar sense and nonsense codons. Interestingly, the accuracy of this reaction is more than one order of magnitude greater than that for tRNA selection, with release factors recognizing sense codons at a frequency of ~ 1 in 105 (Jorgensen et al., 1993).
In this review, we focus on the fidelity of codon recognition in tRNA selection and translation termination, beginning with a discussion of the biological implications of translational fidelity for organismal fitness. Focusing our attention exclusively on bacterial protein synthesis, we synthesize recent work in the field and describe how biochemical and structural studies have come together to form a unified understanding of translation mechanism.
Translational Fidelity in Health and Disease
Although this review focuses on the intricate molecular systems that implement high fidelity protein synthesis, it is worth pausing to think about the cellular requirements for accuracy in translation. High accuracy is important, as proteins with mistakes typically both fold and function less effectively with possibly detrimental physiological consequences, but how much accuracy is enough? Because the mechanisms that have evolved to increase the fidelity of protein synthesis inevitably depend on the expenditure of energy, accuracy comes at a cost. It then follows that cells will tune accuracy to the point where it is optimal – both too little and too much accuracy will adversely affect organismal growth and propagation under a given set of conditions (Kurland and Ehrenberg, 1984).
There has been some analysis of what can go awry in cells when translational accuracy is diminished. In these studies, the consequences of decreased fidelity were explored by assessing mutations in the editing domain of the aminoacyl-tRNA synthetases that are responsible for loading tRNAs with the appropriate amino acid (reviewed in Schimmel, 2008). Most dramatically, seemingly mild defects in the overall fidelity of protein synthesis led to severe neurodegeneration and ataxia in mice (Lee et al., 2006). More detailed molecular understanding came with the observation that low fidelity protein synthesis results in increased amounts of unfolded proteins in the cell, activating protein quality control mechanisms and eventually leading to apoptosis (Nangle et al., 2006). In bacteria, similar mutations in the editing mechanism of a synthetase result in the accumulation of defects in proteins of the DNA replication machinery and ultimately in error-prone replication of the genetic material (Bacher and Schimmel, 2007). Though each of these examples only follows the consequences of defects in the aminoacylation process, it is easy to imagine that mutations in the ribosome or other factors important for translational fidelity may trigger similar pathologies. Simply put, accuracy is important because genes have evolved to encode a specific product with optimal function.
It has also been argued, however, that translational infidelity in some instances can be beneficial, as it enables organisms under adaptive pressure to sample new landscapes of protein sequences (Shorter and Lindquist, 2005). Direct evidence for this proposal has come from studies of the non-mendelian transmission of the [PSI+] prion trait in the budding yeast, Saccharomyces cerevisiae. The [PSI+] state is induced by a self-replicating conformation of the termination factor eRF3 (encoded by the gene Sup35) and results in the reduction of translation termination and thus in stop codon read-through (Paushkin et al., 1996). Lindquist and colleagues have argued that the read-through of stop codons in [PSI+] yeast strains increases their phenotypic diversity and allows them to better adapt to a variety of challenging environments (True et al., 2004; True and Lindquist, 2000). A functioning prion domain in Sup35 is conserved among yeast species across 100 million years of evolution (Chernoff et al., 2000; Nakayashiki et al., 2001), consistent with the idea that a reduction in translational fidelity can confer a selective advantage. Functionally relevant read-through of stop codons appears to also be employed by retroviruses that sequester eRF1 to enhance this process and allow the expression of key viral factors (Orlova et al., 2003).
There are also instances where fidelity loss during translation is co-opted to facilitate a regulatory process. For example, certain proteins are only expressed as a result of mistranslation under specific conditions. The level of bacterial termination factor RF2 in the cell is modulated by such a mistranslation-regulated feedback loop. When there is little RF2 protein present to promote translation termination, a “programmed” frameshifting event occurs on a stop codon in the prfB gene transcript to allow production of full-length RF2 protein (Craigen and Caskey, 1986). In the presence of sufficient amounts of RF2 protein, translational termination occurs at the premature stop codon in the prfB transcript and a functional, full-length protein is not made. The production of key gene products in many retroviruses also depend on similar frameshifting events (for example see Jacks and Varmus, 1985). The extent to which such irregular events of mistranslation contribute to normal biological processes remains a question of considerable interest.
An Active Role for the Ribosome in Fidelity
Soon after the discovery of the ribosome, the tRNA (and its anticodon), and the codon triplets, it became clear that codon-anticodon interaction was key in dictating the sequence of nascent proteins. Early studies showed that the stability of trinucleotide codon-anticodon interactions in solution is weak (Lipsett et al., 1960), and it was suggested that the ribosome must stabilize the association between the tRNA and mRNA (McLaughlin et al., 1966). However, it remained ambiguous whether the ribosome merely added to the stability of all codon-anticodon pairings or whether it provided further specificity to cognate interactions.
The first clue hinting at the ribosome playing a more sophisticated role in modulating tRNA and mRNA interactions came from the elegant study of Gorini and Kataja that uncovered a class of E. coli auxotrophic mutants displaying a conditional streptomycin-dependent phenotype (Gorini and Kataja, 1964). In the presence of streptomycin, an antibiotic thought to affect ribosomal structure (Spotts and Stanier, 1961), variant cells produced significant amounts of an essential enzyme that was normally lacking because of a premature stop codon in the gene encoding the protein. Similarly, addition of streptomycin to in vitro translation reactions resulted in significant misreading of the mRNA template (Davies et al., 1964). Thus, ribosomal structural modulations by streptomycin appeared to allow read-through of a nonsense mutation as well as alter codon recognition, leading Gorini and colleagues to make the almost prophetic suggestion that “the ribosomal structure could influence the accuracy of the reading of the code during translation.”
This idea was later supported by the isolation of streptomycin-resistant (restrictive) mutations, as well as mutations that can suppress the streptomycin-dependent mutation phenotype and cause extensive miscoding (ribosomal ambiguity, ram). The restrictive mutations were ultimately mapped to genes encoding the small ribosomal protein S12 (rpsL), whereas the ram mutations were found to alter the small subunit ribosomal proteins S4 and S5 (rpsD and rpsE). These initial studies thus provided clear evidence that the ribosome controls the accuracy of decoding through multiple distinct loci, an idea now well supported by a great range of ram and restrictive mutations since identified in various ribosome components (reviewed in Triman, 2007). The mechanistic implications of these initial genetic clues are to a great extent revealed by current high-resolution structures (also discussed in Ogle and Ramakrishnan, 2005).
Kinetic Models for Fidelity in tRNA Selection
The overall in vivo rate of misincorporation during protein synthesis has been estimated to be in the range of 6 × 10−4 to 5 × 10−3 per amino acid incorporated (Bouadloun et al., 1983; Edelmann and Gallant, 1977). Given that RNA transcription proceeds with a higher level of accuracy, this value likely reflects the two processes that are fundamentally responsible for protein synthesis: Aminoacylation of tRNAs by the cognate amino acids and correct tRNA selection by the ribosome. The aminoacylation step, carried out by the aa-tRNA synthetases (aaRSs), has been demonstrated to proceed with a remarkable level of accuracy; the incorrect amino acid is attached to the tRNA once in 104–105 events, owing to kinetic discrimination and “double-sieve” editing mechanisms utilized by the enzymes (reviewed in Francklyn, 2008). As a result, it is generally agreed upon that the in vivo value of fidelity is largely dictated by occasional mistakes in the decoding of the mRNA by the ribosome. Safeguarding against such mistakes are remarkably sophisticated ribosome-based systems that depend on kinetic proofreading, induced fit, and post-peptidyl transfer quality control, as we will discuss in detail in this review.
Early kinetic models
The term “kinetic proofreading”, introduced in the 1970s in independent papers by Hopfield and Ninio (Hopfield, 1974; Ninio, 1975), proposed that a given selection process could be separated into distinct steps (by irreversible reactions) to increase the specificity of enzyme-substrate interaction through repeated exploitation of the difference in free energy (ΔΔG). Kinetic proofreading during translation is, in principle, possible because the aa-tRNA is delivered to the A site of the ribosome in a ternary complex with the elongation factor EF-Tu and GTP. GTP hydrolysis presents the required functionally irreversible reaction that separates two independent encounters between the ribosome and the aa-tRNA. In such a scenario, the ΔΔG of binding between cognate and non-cognate tRNAs are first utilized in the context of the encounter between the ribosome and the GTP form of the ternary complex. ΔΔG of binding are again exploited following GTP hydrolysis, through the association of the ribosome with either the GDP state of the ternary complex or free form of the tRNA (i.e. after the dissociation of EF-Tu). These two independent steps of evaluation for the ribosome and aa-tRNA interaction can theoretically lead to greater discrimination, especially if equilibrium is rapidly attained in the steps preceding the relatively slow steps of GTPase activation and accommodation (Cochella and Green, 2005b).
Initial support for the proofreading mechanism came from in vitro data demonstrating that certain tRNAs (that typically carry a single mismatch to the codon in the A site) resulted in a significant increase in GTP consumption relative to the amount of amino acids incorporated (Thompson and Stone, 1977) – these tRNAs we refer to as near-cognate. In these same studies, it was observed that other tRNAs (typically with more than a single mismatch) did not appear to stimulate the hydrolysis reaction – these tRNAs we refer to as non-cognate. These observations are consistent with two steps of selection separated by GTP hydrolysis. Non-cognate tRNAs are rejected during the initial phase of the selection, but near-cognate ones escape this screening process some of the time and are instead rejected during a second selection phase following GTP hydrolysis. This kinetic proofreading model predicts that slowing down GTP hydrolysis would allow greater time for equilibrium to be reached during the initial selection phase and could thus result in an overall higher fidelity. Indeed, slow hydrolysable analogues of GTP, such as guanosine γ-thiotriphosphate (GTP-γS), increase by orders of magnitude the fidelity of in vitro translation reactions (Thompson and Karim, 1982). Similarly, certain ribosome mutants exhibiting hyperaccurate phenotypes in fidelity assays displayed reduced rates of GTP hydrolysis by EF-Tu (Bilgin et al., 1992).
A more detailed model for tRNA selection
These early observations laid the foundation for our current understanding of the overall process of tRNA selection. The recent application of higher resolution approaches, including pre-steady state kinetics and single-molecule fluorescence techniques, has expanded and somewhat altered these views. Unlike earlier analysis of translation using steady-state approaches, pre-steady state kinetics strives to utilize assays that monitor each independent molecular event in isolation using a variety of fluorescent and radioactive probes. To facilitate the dissection of the process, the usual toolbox of inhibitors is used to selectively block the tRNA selection pathway at specific stages. The many parameters determined by these approaches, coupled with computational global-fitting techniques, allowed Rodnina and colleagues to present a detailed initial picture of the kinetic and thermodynamic framework governing tRNA selection (Rodnina et al., 2005) An elaborated version of this scheme (discussed below) is shown in Figure 2.
Figure 2. The tRNA selection pathway.

The view follows a tRNA through the selection process with defined steps indicated. The scheme is largely based on accumulated pre-steady state kinetic data (reviewed in Rodnina et al., 2005) from experiments performed in the absence of E-site tRNA. Predicted correlated FRET values are indicated at the top of each intermediate (Blanchard et al., 2004a). Green arrows indicate rates that are accelerated for cognate tRNA, whereas red arrows depict rates that are higher for near-cognate tRNA. We note that the occupancy and the role of the E-site tRNA following the codon-recognition intermediate is controversial and as such, the E-site tRNA is shown in a lighter color subsequent to this stage.
We briefly outline here the features of the kinetic framework as determined for model cognate and near-cognate tRNA species. The initial step in the tRNA selection process is a codon-independent, labile interaction between the ternary complex and the ribosome that is governed by rate constants k1 and k−1 (monitored by fluorescence changes in a proflavin-labeled tRNAPhe derivative) (Rodnina et al., 1994). The codon-independent nature of this step is supported by the observation that all ternary complexes (cognate, near-cognate, or non-cognate) exhibit the same low amplitude fluorescence change in their initial ribosome encounter, as well as similar k1 and k−1 values of ~ 100 μM−1 s−1 and 85 s−1, respectively (Pape et al., 1998). The rate of binding between the ternary complex and the ribosome (k1) is unusually high and fully dependent on EF-Tu, suggesting an active mechanism for initial tRNA loading. A recent study proposes that the very large size and net positive charge of the L7/L12 stalk region of the ribosome are key to the observed fast rate of binding (Diaconu et al., 2005). We note that this codon-independent step has been difficult to observe experimentally, and as such remains the subject of some controversy (Johansson et al., 2008).
The next step in the selection process that can be readily followed – again by fluorescence changes in labeled tRNAs (Eisinger et al., 1970; Rodnina et al., 1994) – is codon-dependent. It is observed for both cognate and near-cognate tRNAs, but not for non-cognate species (Pape et al., 1998). The overall rate of codon-recognition (k2) is nearly invariant for cognate and near-cognate ternary complexes (190 s−1), whereas the dissociation rate constants (k−2) are considerably different. The k−2 for complexes carrying a single mismatch is almost 1000 fold faster than for cognate ones under high fidelity conditions (80 vs. 0.23 s−1) (Gromadski and Rodnina, 2004a). This striking difference is larger than expected based on the difference in free energy (ΔΔG) of binding between cognate and near-cognate tRNA codon-anticodon interactions in solution. This suggests that the ribosome plays an active role in stabilizing cognate interactions relative to near-cognate ones.
In contrast to the differences in binding observed between cognate and near-cognate tRNAs, all cognate tRNAs exhibit surprisingly similar affinities for the A site (Fahlman et al., 2004). This uniform binding is unexpected as certain codon-anticodon interactions are expected to be more stable than others due to factors such as the codon guanosine-cytidine (GC) content. The emerging view from biochemical and structural studies suggests that the specific sequence and post-transcriptional modification status of the tRNA in the region near the anticodon is “tuned” to ensure nearly indistinguishable binding of tRNAs during tRNA selection (Murphy et al., 2004; Olejniczak and Uhlenbeck, 2005).
The next steps in the selection pathway involve EF-Tu and GTP hydrolysis, and as such are key in establishing the irreversible step essential to the mechanism of proofreading. First, the EF-Tu active-site undergoes a conformational change (k3), as monitored using the environmentally-sensitive fluorescence analogue mant-dGTP. This structural change (referred to as GTPase activation) appears to limit the rate of the subsequent chemical step of GTP hydrolysis step (kGTP). The fact that k3 limits GTP hydrolysis allowed researchers to follow GTP hydrolysis as a reporter of this key conformational rearrangement (Rodnina et al., 1995). Several other steps can also be inferred following initial tRNA selection, including inorganic-phosphate release (kPi), rearrangement of EF-Tu into a GDP-bound state (k4), and the irreversible dissociation of EF-Tu from the aa-tRNA (k6). These latter steps, however, do not appear to be critical features for understanding discrimination during tRNA selection (Pape et al., 1999).
Initial views of the proofreading model for tRNA selection would have predicted that the GTPase activation step (k3) of the selection pathway proceeds at a constant rate, serving as an “internal clock” (Thompson, 1988). Discrimination in this situation arises simply from differences in the dissociation rates between cognate and near-cognate tRNAs. In direct conflict with this prediction, several critical studies by Rodnina and colleagues demonstrated that the rate of GTPase activation strongly depends on the properties of the decoding helix (pairing interaction between the codon-anticodon). For example, k3 was 120–500 s−1 for the cognate species and 0.06–1.3 s−1 for the near-cognate (Gromadski et al., 2006; Gromadski and Rodnina, 2004a; Pape et al., 1999). As it turns out, these differences in forward rate constants are most essential in ensuring fidelity during the initial selection phase.
Following the dissociation of EF-Tu, the selection pathway reaches the critical branch point known as proofreading where the tRNA either moves into the A site (accommodation, k5) of the large ribosome subunit and participates in peptidyl transfer (kpep), or dissociates from the ribosome (rejection, k7). Strikingly, the same kinetic study by Rodnina’s group that identified GTPase activation as codon-anticodon pairing sensitive indicated that the accommodation step (k5) is similarly regulated. Measured values for cognate and near-cognate tRNAs were 7 s−1 and 0.1 s−1, respectively, under conditions where GTPase activation does not limit accommodation (Pape et al., 1999). Cognate tRNAs are apparently accelerated uniformly through the tRNA selection pathway at two distinct steps (k3 and k5) (Kothe and Rodnina, 2007; Ledoux and Uhlenbeck, 2008), thereby allowing for both rapid and high fidelity protein synthesis.
The data gathered from multiple fluorescent reporters (proflavin- and wybutine- labeled tRNA, mant-dGTP) and chemical assays (GTP hydrolysis and peptidyl transfer), together with global fitting approaches, have allowed for reasonable estimates of the rate constants for nearly all of the identified steps in the tRNA selection pathway. These rates for both cognate and near-cognate ternary complexes can be used to estimate the contribution of each phase of the process (initial selection and proofreading) to the overall accuracy of selection, and to evaluate whether the calculated predictions match in vivo measurements. Selectivity during initial selection is dictated by the relative kcat/Km values. From these values, the contribution of selectivity in the initial selection phase is calculated to be ~30–60 fold. The selectivity of the proofreading stage is easier to determine. For a typical near-cognate species, Rodnina and colleagues determined that the proofreading contributes a factor of about 15 to selectivity. Overall selectivity was measured to be about 450-fold under competitive conditions, nicely matching the product of the initial selection and proofreading parameters (30 × 15) measured in the absence of competition (Gromadski and Rodnina, 2004a).
In the end, although there are striking differences in the dissociation rates (k−2 and k7) of cognate and near-cognate tRNAs during the selection process, they are not fully utilized by the ribosome to increase selectivity as initially proposed. While such a two-step selection process can in principle yield the observed fidelity, the problem arises with the amount of time required to reach equilibrium at each stage. Even though selectivity does appear to be achieved by the ribosome in two steps, the specific acceleration of forward rate constants (k3 and k5) for cognate tRNAs relative to near-cognate ones is predominantly responsible for the selectivity. Such mechanisms, generally referred to as “induced fit,” are important contributors to selectivity throughout biology, but are especially reminiscent of earlier observations from template-driven polymerases (Johnson, 1993).
It should be noted that overall selectivity is highly dependent on experimental conditions, with Mg2+ and polyamine concentrations playing an especially critical role (Gromadski and Rodnina, 2004a; Jelenc and Kurland, 1979; Thompson et al., 1981). Under reduced fidelity conditions, where Mg2+ is high and polyamines are absent, the initial selection stage is less effective, resulting in a relatively high error frequency (Pape et al., 1999). Other in vitro studies using several different higher fidelity buffer systems have yielded misincorporation rates that more closely approach those reported in vivo (Gromadski and Rodnina, 2004a; Johansson et al., 2008). These measurements are generally carried out by comparing the rate constants for one particular near-cognate tRNA with those of the cognate species and assuming that their concentrations are equal in vivo. As such, these calculations do not take into account that for each tRNA, multiple near-cognate tRNAs that compete with similar efficiencies also exist (Gromadski et al., 2006). These near-cognant tRNAs will in turn increase the level of misincorporation by a factor dependent on their overall concentration. Recent in vivo experiments by Farabaugh and colleagues document the importance of tRNA competition in specifying the fidelity of tRNA selection (Kramer and Farabaugh, 2007). Our in vitro experiments conducted with a complete competitor tRNA population mimicking the in vivo milieu yield somewhat higher error frequencies ranging from 2–10 misincorporations in 103 events (Zaher and Green, 2009).
New Intermediates Revealed by Single-Molecule Approaches
In a relatively recent set of advances, the tRNA selection process has been studied using single-molecule Förster resonance energy transfer techniques (smFRET). The power of this approach comes from its inherent ability to follow individual behaviors in the dynamic multi-step pathway, especially those that are easily lost by averaging when studying the bulk properties of these same molecules. The feasibility of using these techniques for the study of protein synthesis is the result of recent methodological advances in the labeling, immobilization and detection of single ribosomal complexes (reviewed in Marshall et al., 2008).
Overall, the smFRET studies with Cy3 and Cy5 fluorescent dye-labeled P- and A-site tRNAs have yielded results that reinforce and extend our core understanding of tRNA selection obtained from more traditional bulk approaches. Encounters between a ternary complex (EF-Tu, GTP, and Cy5-labeled Phe-tRNAPhe) and ribosome complexes loaded with Cy3-labeled fMet-tRNAfMet revealed three distinct FRET states (low at 0.35, mid at 0.5 and high at 0.75, Figure 2), suggesting three different modes of interactions (Blanchard et al., 2004a). To date, smFRET studies have failed to reveal a codon-independent interaction between the ternary complex and the ribosome, as was previously observed in bulk studies (Pape et al., 1999)., perhaps due to the longer distance between the two labeled tRNAs during this early stage of the interaction that prevents it from being detected by the technique. Both the low- and mid-FRET states are associated with ternary complex-ribosome interactions preceding GTP hydrolysis. The low FRET state has been proposed to represent a previously uncharacterized interaction between ternary complex and ribosome called “codon-dependent sampling.” This state is thought to occur after initial binding and prior to true codon recognition. Examination of this FRET state may provide explanation for how non-cognate tRNAs that do not proceed to GTPase activation can be discriminated against by the codon-anticodon interaction that must underlie this discrimination. This is notable given that non-cognate tRNAs fail to yield signal for the codon-recognition step (k2) in bulk studies. The mid-FRET state can be stabilized by the non-hydrolysable analogue GDPNP, indicating that this step is a component of the initial selection phase, that is, prior to GTP hydrolysis but likely representing a state of GTPase activation. As previously observed with pre-steady state kinetic analysis, near-cognate tRNAs are clearly discriminated against during this stage as a greater proportion of cognate tRNAs proceed from the low- to the mid-FRET state than do the near-cognate species. Careful examination of the mid-FRET phase for cognate- and near-cognate-tRNA species also suggests that these species are in a somewhat distinct conformation on the ribosome – cognate tRNAs have an average FRET of 0.43 while the near-cognate average is 0.39 (Blanchard et al., 2004b). Additionally, these two states have different properties – the mid-FRET state is longer lived for the cognate species, and is more likely to proceed to the higher FRET state that follows. Transition to the high-FRET state (0.75) requires GTP hydrolysis and the distances calculated from the FRET value are consistent with the tRNA being fully accommodated in the A site. As for the initial selection step, quantification of the transition efficiency from the mid- to the high-FRET state relative to the total number of FRET events, allows for an estimate of the contribution of “proofreading” to overall fidelity. These quantitative evaluations indicate that the overall misincorporation rate in this single molecule system is ~ 7.1 × 10−3, with the contribution from initial selection being ~ 20% (Blanchard et al., 2004a). These values (measured under relatively high magnesium, low fidelity conditions) are strikingly consistent with earlier bulk studies (Pape et al., 1999) where initial selection only modestly contributed to overall fidelity under similar conditions. Subsequent bulk tudies indicated that greater contributions to fidelity by the initial selection phase can be observed in experiments using more physiological buffer conditions (Gromadski et al., 2004a).
It is evident is that bulk and single-molecule approaches can each make important contributions to the understanding of mechanistic details in the complex process of tRNA selection. Different probes and assays can reveal different intermediates and reactions. What will be important for the field in moving forward is for the practitioners of each approach to make a concerted effort to reconcile their studies (and rate constants) with those that came before them. This is a daunting task. Indeed, it is not yet clear how the initial selection and proofreading parameters from bulk and single molecule studies correspond to one another.
Antibiotics as Probes of Ribosome Function
Since the earliest studies of streptomycin resistance in E. coli by Gorini and colleagues, the aminoglycoside class of antibiotics has been known to affect the overall fidelity of translation (Davies et al., 1965). Recent biochemical and structural studies of this class of antibiotics has revealed much about the core mechanisms of translation as well as their mode of action. This antibiotic class encompasses a broad range of molecules including streptomycin, neomycin, kanamycin, paromomycin, and gentamycin. The extent and the spectrum of the misreading events that these antibiotics induce are correspondingly broad (Davies and Davis, 1968). These observations immediately suggested that the compounds utilize at least somewhat distinct mechanisms to alter the decoding process, perhaps by binding to different sites on the ribosome.
With the development of pre-steady state approaches, the effects of several of the aminoglycosides on specific steps of the tRNA selection pathway were recently elucidated. In an initial pre-steady state analysis, Rodnina and colleagues not only confirmed previous findings (Karimi and Ehrenberg, 1994) that paromomycin reduces the rate of near-cognate tRNA dissociation from the A site (k−2 and k7), but also that the antibiotic accelerates both of the critical forward reaction rates in the tRNA selection pathway (GTPase activation, k3, and accommodation, k5) (Pape et al., 2000). As these steps are also accelerated during cognate-tRNA binding, it is likely that paromomycin might induce similar structural rearrangements. In contrast to paromomycin, streptomycin substantially reduces the forward reaction rates of GTPase activation (k3) for cognate tRNA (by two orders of magnitude), while modestly stimulating these values for near-cognate tRNA. As a result, the rates of GTPase activation for cognate and near cognate tRNAs are closely matched and rate limiting for the overall process (Gromadski and Rodnina, 2004b).. Selectivity is strongly diminished as a result. These data suggest that the two antibiotics induce somewhat distinct conformational changes in the small ribosome subunit. Interestingly, the effects of streptomycin are dominant to those of paromomycin; the rates of GTPase activation with cognate tRNAs are diminished in the presence of both antibiotics. Paromomycin appears to switch the ribosome into a highly activated state regardless of the codon-anticodon interaction, whereas streptomycin induces a state of intermediate activation that precludes normal communication from the decoding center to the GTPase-activating domains upon cognate-tRNA binding (Gromadski and Rodnina, 2004b).
Structural Insights into the Decoding Process
Early biochemical studies identified the decoding center of the ribosome to be located on the small 30S ribosomal subunit at the interface with the large 50S subunit, encompassing 16S rRNA nucleotides 1400–1500 of helix 44, the 530 loop (helix 18), and residues 1050–1500. Chemical modification protection analysis showed that the bases of the conserved nucleotides guanosine 529 (G529), guanosine 530 (G530), adenosine 1492 (A1492) and adenosine 1493 (A1493) are protected by the binding of an A-site tRNA (Moazed and Noller, 1990). Moreover, the aminoglycoside paromomycin induces protection of nucleotides 1408 and 1494, just across from 1492 and 1493 in helix 44 (Moazed and Noller, 1987). Mutational experiments later demonstrated that these nucleotides are critical for A-site tRNA binding (Powers and Noller, 1990, 1994; Yoshizawa et al., 1999). Moreover, an NMR structure of an oligonucleotide corresponding to this region of helix 44 bound to paromomycin revealed that the aminoglycoside stabilized a structure of A1408, A1492 and A1493 that is distinct from that observed in the absence of ligand (Fourmy et al., 1996; Fourmy et al., 1998). The authors suggested that the observed conformational changes might mimic those induced by the binding of cognate A-site tRNA to the ribosome.
The beginning of the millennium saw great breakthroughs in the mechanistic understanding of translation as high-resolution crystal structures of 30S and 50S ribosome subunits, as well as that of the 70S ribosome, were solved (Ban et al., 2000; Schluenzen et al., 2000; Wimberly et al., 2000; Yusupov et al., 2001). The crystal structures of the 30S subunit in the absence and presence of an anticodon stem-loop (ASL) in the A site revealed the identities of the elements -- 16S rRNA and ribosomal proteins (r-proteins) -- that interact with the codon-anticodon helix (Ogle et al., 2001). The previously identified nucleotides A1492 and A1493 appear to directly monitor the geometry of the first two base pairs in the codon-anticodon helix through well-characterized A-minor interactions with the minor groove. Additional contributions to the monitoring of the second and third codon positions are made by cytidine 518 (C518), G530, and portions of the r-protein S12. The molecular details of the interactions between the ribosome and the decoding helix, in particular at the first and second codon positions, readily demonstrate how the geometric commonalities of all four Watson-Crick base pairs are the criterion for selection of the incoming tRNA. The structures also reveal how these positions of the codon are monitored more precisely (only Watson-Crick pairings are allowed) than the third position, where certain wobble pairing interactions are accepted. The caliper-like measurement of the minor groove made by the ribosome is reminiscent of the way in which RNA polymerases (and DNA polymerases) monitor fidelity during nucleotide polymerization, remembering of course that their mechanism depends on recognition of the minor groove by amino acids, rather than nucleotides (Figure 3A, B).
Figure 3. Molecular recognition of cognate pairing interactions by T7 RNA polymerase and the ribosome.
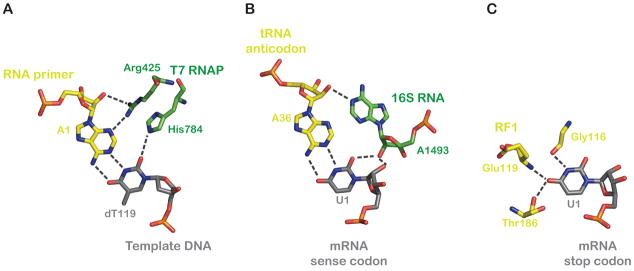
(Left) T7 RNAP (green) recognizes correct base-pairs between RNA (yellow) and DNA (grey) by a “molecular-caliper” mechanism, wherein side groups of specific residues measure the invariant properties of the Watson-Crick base-pair geometry (PDB 1MSW) (Yin and Steitz, 2002). (Center) The type-I A minor interaction at the first base-pair between the codon and anticodon (PDB 2J00) (Selmer et al., 2006). (Right) Recognition of U1 of the UAA stop codon by release factor 1 (RF1) (PDB 3D5A) (Laurberg et al., 2008). All molecular representations were generated using PyMol (DeLano Scientific).
Structural studies of the 30S subunit bound to different aminoglycosides have provided important insights into the molecular basis of their action. Paromomycin binds in the internal loop of helix 44 of 16S rRNA, where A1492 and A1493 are positioned in the apo-structure (no ligand in the A site) of the 30S subunit (Figure 4A). Thus, binding of paromomycin induces a conformational rearrangement of A1492 and A1493 that results in their displacement from helix 44 to a position where they can engage the minor-groove of the codon-anticodon helix in the A site (Ogle et al., 2001). Interestingly, these same residues (in addition to others) are similarly (but not identically) rearranged upon cognate-tRNA binding (Figure 4B, C). These structural observations reconcile earlier biochemical data showing that addition of paromomycin stimulates forward reaction rate constants in the tRNA selection pathway (Pape et al., 2000).
Figure 4. Ligand-dependent local conformational changes in the ribosome decoding center.

A comparison of the relative positions of key decoding center nucleotides (green) in an A site-vacant 30S ribosome subunit (A) (PDB 1J5E) (Wimberly et al., 2000), (B) in an ASL (yellow) -bound 30S subunit (PDB 1IBM) (Ogle et al., 2001), (C) in the presence of paromomycin (orange) (PDB 1IBK) (Ogle et al., 2001), and (D) in an RF1 (yellow) -bound 70S ribosome (PDB 3D5A) (Laurberg et al., 2008). 23S rRNA A1913: brick red. The movements of the nucleotides is described in details in the text. All molecular representations were generated using PyMol (DeLano Scientific).
While an equivalent 30S structure with streptomycin alone is lacking, a structure of the 30S subunit complexed with three antibiotics (spectinomycin, streptomycin and paromomycin) revealed that streptomycin makes contact with the phosphate backbone of five different helices of the 16S RNA (helices 1, 18, 27, 28 and 44) (Carter et al., 2000). Streptomycin also interacts directly with the S12 protein. As in the paromomycin-alone structure, A1492 and A1493 move into their extra-helical conformation (swinging out of h44), but unlike in the paromomycin-alone structure, G530 rotates around its glycosidic bond (from a syn to an anti conformation). This structure thus most closely resembles that of a cognate tRNA bound decoding center. It is unclear whether streptomycin alone can induce the rearrangements of A1492 and A1493, since their conformational change is also observed in the presence of only paromomycin. These structural data provide molecular rational for the restrictive streptomycin-dependent ribosome mutants discussed earlier; these ribosomes carry mutations in an interface region of the S12 protein that is stabilized by the binding of streptomycin. How the stability of this interface specifies the overall conformation of the subunit, and in turn dictates selectivity, will be discussed below.
Reconciling Structures with Rate Constants
What does not emerge from simply looking at static structures of the decoding center in the process of recognizing a cognate helix is why near-cognate species are so effectively discriminated against. Biochemical studies showed that the substantial difference in free energy (ΔΔG) of binding for cognate and near-cognate tRNA species (~1000 fold) cannot simply be explained by stability differences in the decoding helix. In comparing the apo-ribosome structure with an anticodon stemloop (ASL)-loaded structure, clues begin to emerge regarding how the ribosome brings about such impressive levels of discrimination. Three key decoding center nucleotides (G530, A1492, and A1493) undergo substantial conformational rearrangements upon binding of cognate ASL. A1492 and A1493 move from an intrahelical position (in helix 44) to an extrahelical position, whereas G530 flips from a syn to an anti conformation (Figure 4B) (Ogle et al., 2001). These nucleotides thus congregate together for inspection of the minor groove of the decoding helix. The geometry-dependent interactions between these three nucleotides and the minor groove of the codon-anticodon helix enhance the specificity for Watson-Crick pairs to an extent much greater than would be attained from the stability of base pairing alone. Structures of the 30S subunit carrying a first position mismatch between the near-cognate ASLLeu (anticodon GAG) and Phe codon (UUU) revealed that the wobble pairing results in the displacement of the 1st codon position U into the minor groove, thus preventing it from forming the appropriate hydrogen bond with A1493 (Ogle et al., 2002). Moreover, there is no space for water to solvate the polar groups on the distorted U-G base-pair – this uncompensated desolvation is associated with large energetic losses that are reflected in differential off-rates of the cognate and near-cognate tRNAs (k−2 or k7), as discussed earlier.
Biochemical analysis also shows that the ribosome does not fully utilize the available thermodynamic differences between cognate- and near-cognate-tRNA species, but relies instead on accelerated forward reaction rate constants for GTPase activation (k3) and tRNA accommodation (k5) to achieve high levels of discrimination. The dramatic conformational rearrangements that are observed in the decoding center provide a compelling explanation for how these kinetic effects may be initiated at the molecular level. Why initiated and not facilitated? This distinction comes from the realization that the key kinetic steps that are accelerated by cognate tRNA binding (k3 and k5) occur principally in association with the large ribosome subunit. The GTPase domain of the elongation factor EF-Tu is positioned near the sarcin-ricin loop (SRL) region of the large ribosome subunit (Moazed et al., 1988), and accommodation essentially involves the release of the acceptor end of the aa-tRNA from EF-Tu and full entry of the aa-tRNA into the large ribosome subunit A site. Both of these events, thus, occur at a distance far from the decoding center where cognate and near-cognate species are distinguished. How do the decoding events then facilitate the tRNA selection process? A likely clue to this conundrum comes from the observation of global structural changes in the small subunit specifically in response to the binding of cognate ASLs (Ogle et al., 2002). These conformational changes result in a more closed state of the small subunit through concerted rotations of the head and shoulder domains. Whereas near-cognate ASLs do not induce the closed conformation even when the A site is fully occupied, the addition of paromomycin (known to induce the extrahelical conformation of A1492/1493) to this complex induces full domain closure. These observations suggest that A1492/93 movement is a key contributor to the global structural state. As paromomycin causes misreading during elongation through the acceleration of these same forward reaction rate constants, closure of the 30S subunit likely takes place during the initial codon-recognition state prior to GTPase activation (Ogle et al., 2003).
Visualizing the transition from the open to closed conformation of the 30S subunit during the decoding process has aided our mechanistic understanding of ribosomal mutants that affect the fidelity of protein synthesis. The error-prone ram mutants typically carry altered versions of the small subunit ribosomal proteins S4 and S5. These proteins form an interface in the 30S subunit that is broken during domain closure – mutations in these proteins disrupt salt bridges at their mutual interface (Figure 5a). By reducing the number of bonds that must be broken for domain closure to take place, these mutants decrease the energy barrier needed for this transition to occur and so facilitate the acceptance of tRNAs during the selection process. Restrictive mutants that are resistant to the fidelity-loss induced by streptomycin typically carry mutations in the S12 protein, located on the opposite side of the shoulder relative to the S4/S5 interface. Many of these mutations alter contact points between the S12 protein and 16S RNA helix 44 and helix 27 that are important for domain-closure (Figure 5b) (Ogle et al., 2002). These changes destabilize the closed conformation and promote accuracy during tRNA selection. Together, these interactions function as tethering points to trap ribosomal motions critical to tRNA selection.
Figure 5. Global changes in ribosome structure on binding cognate anticodon stemloop or RF1.

A. The S4/S5 small subunit proteins interface of cognate anticodon stemloop (ASL)-bound 30S ribosome subunit (PDB 1IBM) superimposed on the S4/S5 interface of near-cognate ASL-bound 30S ribosome subunit (PDB 1N34) (Ogle et al., 2002). S4 and S5 move apart as a result of cognate-ASL binding, breaking salt-bridges that favor their interaction.
B. Superimposed structures as in A, now showing the relative positions of small subunit protein S12 and helix 44 of 16S RNA, where closer interactions are seen for the cognate structure.
C. The S4/S5 interface of apo (PDB 2OW8) (Korostelev et al., 2006) superimposed on that of RF1-bound 70S ribosomes (PDB 3D5A) (Laurberg et al., 2008). In this case, distinct, more lateral movements are observed as a result of RF1 binding.
D. Superimposed structures as in C, now showing the relative positions of S12 and helix 44, where in the context of RF1 binding, S12 moves away from helix 44.
Although domain-closure is important to tRNA selection, we do not yet have a clear understanding of how EF-Tu is activated for GTP hydrolysis by these movements. Moreover, the path of communication that leads from localized changes in the decoding center to the more global ones of domain closure is unresolved. Whereas several studies have argued that signaling occurs in part through the tRNA structure (Cochella and Green, 2005a; Piepenburg et al., 2000; Valle et al., 2003), other studies have argued for multiple independent paths being important in inducing the remote structural changes (Cochella et al., 2007; Liiv and O’Connor, 2006). Other clues to this problem come from cryo-EM reconstructions of kirromycin-stalled ternary complexes (thought to represent a GTPase-activated state) bound to the ribosome (Stark et al., 2002; Valle et al., 2002). In these reconstructions, the ternary complex makes multiple contacts with both the 50S and 30S subunits to stabilize distinct configurations for the EF-Tu domains and the aa-tRNA as compared to the structure of free ternary complex. Visible distortion of the tRNA in the region between the anticodon and D stem of the tRNA (Valle et al., 2002) provides some insight into why certain mutations in this region might result in miscoding through the specific acceleration of GTPase activation and accommodation (Cochella and Green, 2005a).
The Specificity of Peptide Release
The termination of protein synthesis occurs when one of the three nearly universal stop codons (UAA, UAG and UGA) enters the A site, signaling the end of the coding region. In contrast to the elongation cycle, where sense codons are decoded by aa-tRNAs, stops codons are recognized by specialized protein factors called class I release factors (RFs) that trigger the release of the growing polypeptide chain (reviewed in Youngman et al., 2008). In bacteria, there are two such factors with overlapping specificities for the stop codons they recognize; RF1 decodes UAG and UAA, whereas RF2 decodes UAA and UGA. In eukaryotes, a single factor (eRF1) recognizes all three codons. Strikingly, the bacterial and eukaryotic class I release factors have no structural similarity apart from a universally-conserved GGQ motif, indicating that they evolved independently to perform their related tasks (reviewed in Youngman et al., 2008). In addition to the class I release factor, both bacteria and eukaryotes depend on a GTPase class II release factor (RF3 and eRF3, respectively) to complete termination. In bacteria, RF3 appears to make no contribution to the catalysis of peptide release on authentic stop codons, though it does stimulate release on sense codons (Freistroffer et al., 2000). Instead, RF3 appears to be principally involved in downstream events in termination, coupling the energy of GTP hydrolysis to the removal of the class I RF following peptide release (Freistroffer et al., 1997). The role of the eukaryotic eRF3 is clearly distinct from that of RF3: eRF1 and eRF3 form a heterodimer in the cell to catalyze peptide release on all codons (Pisareva et al., 2006).
Class I release factors are in essence functionally similar to the aa-tRNAs involved in the elongation step of protein synthesis. Both species are bifunctional in nature, with a domain responsible for recognizing the appropriate codons with high specificity in the small subunit of the ribosome (domain 2 and the anticodon region, respectively) and another domain involved in promoting catalysis in the peptidyl transfer center (PTC) of the large subunit (domain 3 and the acceptor stem, respectively). Like tRNA selection, the decoding process is highly accurate with premature termination (whereby release factors recognize sense codons) having an error frequency of 1 in 105 in vivo (Jorgensen et al., 1993). The recognition of the stop codons by release factors is distinct from that of sense codons by tRNAs, as Watson-Crick RNA-RNA base pairing interactions cannot be employed. Instead, RNA-protein interactions are central to the “decoding” process. Genetic studies identifying regions of the class I release factors that are responsible for distinguishing between stop and sense codons uncovered mutations in specific regions of RF1 and RF2 that alter the specificity of these factors for stop codons (Ito et al., 2000). These studies identified “tripeptide anticodons” that are critical for stop codon recognition: proline-any amino acid-threonine (PxT) for RF1 and serine-proline-phenylalanine (SPF) for RF2. Initial cryo-EM and X-ray crystal structures provided clear evidence that these regions occupy the ribosome decoding center near the mRNA in the A site (Klaholz et al., 2003; Petry et al., 2005; Rawat et al., 2003), recently reinforced by higher resolution structures (Korostelev et al., 2008; Laurberg et al., 2008; Weixlbaumer et al., 2008).
Biochemical insights
Detailed in vitro biochemical analysis has provided further insights into the mechanism of peptide release and its specificity. In one particularly informative study, Ehrenberg and colleagues used a well-defined in vitro system to evaluate the kcat and apparent binding (K1/2) contributions to the fidelity of RF1- and RF2-mediated hydrolysis reactions in the absence and presence of RF3 (Freistroffer et al., 2000). The discrimination in these experiments nicely recapitulated what had been observed in vivo, and thus provided experimental support for several important conclusions. First, class I release factors appear to achieve high specificity in the absence of kinetic proofreading. This is supported by the observation that the inclusion of RF3 and its associated GTP hydrolysis activity does not increase the fidelity of codon recognition. Thus, RF3 cannot provide an irreversible step essential for the iteration of selection (as occurs with EF-Tu’s participation in tRNA selection). A second key point to emerge is that class I release factor specificity derives from a relatively large apparent binding (Km) contribution (2 to 3 orders of magnitude over sense-codon recognition), indicating that all sense codons trigger decreased class I release factor binding. However, additional contributions to specificity also derive from kcat effects that vary considerably depending on the sense codon and the release factor (ranging from 2 to 1000-fold). These latter results suggested that the class I release factors bind in a qualitatively different fashion to stop versus sense codons, thus evoking models of induced fit akin to those proposed and documented for tRNA selection. A recent study provided clear structural evidence for stop codon-specific conformational rearrangements induced by class I release factor binding (Youngman et al., 2007).
The large apparent binding (Km) contribution to specificity for class I release factor recognition is of interest, as this property is different from that of the tRNA selection pathway. Release factors may resort to such binding strategies for specificity because of inherently larger differences in the free energy (ΔΔG) of binding available for protein-RNA interactions (relative to the RNA-RNA interactions), or because they do not have proofreading mechanisms. Freistroffer et al. (2000) raised the possibility that the relatively slow rates of release, when compared with those for peptidyl transfer, may be useful in allowing equilibrium to be reached and thus greater utilization of the large ΔΔG of binding. As for tRNA binding, it seems possible that induced-fit rearrangements of ribosome structure upon release factor binding to cognate stop codons may increase productive binding interactions, thus resulting in the large ΔΔG of binding. Site-directed mutagenesis studies have provided some insights into the importance of specific molecular features for this induced-fit mechanism. Strikingly, mutations in elements critical to tRNA selection (nucleotides A1492/93 and G530 and the 2′OH groups of the mRNA A-site codon) had essentially no effect on release factor function (Youngman et al., 2007). Moreover, aminoglycoside antibiotics, known to stimulate various steps in the tRNA selection process, strongly inhibit release factor recognition of stop codons (Brown et al., 1993). These biochemical results clearly indicate that the ribosome decoding center must work in fundamentally different modes for these two seemingly related processes of tRNA and stop codon selection. This notion is well supported by recent high-resolution structural studies (Korostelev et al., 2008; Laurberg et al., 2008; Weixlbaumer et al., 2008).
Despite our increasing knowledge of the molecular details of stop-codon recognition, we still lack basic understanding of the steps of interaction between class I release factors and the ribosome – no kinetic and thermodynamic framework has been established. The development of fluorescent reporters to follow the peptide-release reaction using stopped-flow techniques, along with single-molecule approaches, are likely to shed light on biologically-relevant intermediates encountered along the pathway, and thus ultimately on the process of termination.
Structural insights
Bacterial RF1 and RF2 are homologous proteins composed of four distinct domains. The N-terminal domain 1 is required for association with the GTPase RF3 but is dispensable for the core events of peptide release (Mora et al., 2003). Domain 2 contains the anticodon tripeptide motif (PxT and SPF for RF1 and RF2, respectively) required for codon recognition in the small ribosome subunit. The structure of this domain is further stabilized by packing against domain 4. Domain 3 carries the GGQ motif that is critical for catalysis of peptide release in the large ribosome subunit. Early crystal structures of non-ribosome-bound RF1 and RF2 revealed that the two proteins adopt a closed conformation incompatible with positioning the functional motifs on the appropriate ribosomal sites (Shin et al., 2004; Vestergaard et al., 2001). However, low-resolution cryo-EM and crystal structures of ribosome-bound release factors later revealed ribosome-binding of both class I release factors in the extended form (Figure 6a) (Klaholz et al., 2003; Petry et al., 2005; Rawat et al., 2003). The three relevant release factor domains (domains 2–4) superimpose surprisingly well with an A-site bound tRNA, with the release factor functional motifs PxT/SPF and GGQ occupying equivalent sites to those occupied by the anticodon and CCA ends of the tRNA, respectively (Figure 6b).
Figure 6. Functional mimicry of tRNAs and RF1.
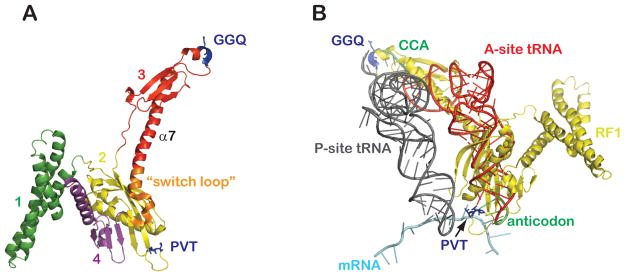
(A) Structure of release factor 1 (RF1) as bound to the 70S ribosome (PDB 3D5A) (Laurberg et al., 2008). Domains of RF1 are colored and numbered as indicated with the functional motifs highlighted in blue.
(B) Ribosome-bound A-site tRNA (PDB 1GIX) (Yusupov et al., 2001) superimposed onto the structure of the same 70S termination complex (where P-site tRNA, mRNA, and RF1 are shown).
In exciting recent advances, high-resolution structures of “post-termination” ribosome complexes (with either RF1 or RF2 bound in the A site and a deacylated tRNA in the P site) have provided substantial insight into how the three stop codons are recognized with high fidelity (Korostelev et al., 2008; Laurberg et al., 2008; Weixlbaumer et al., 2008). Additionally, these structures have begun to provide clues about how “reading” of the codon leads to catalysis of the termination reaction in the large subunit. As anticipated, the structures reveal that the stop codon is recognized in a pocket formed by conserved elements of the class I release factors and the 16S RNA. Consistent with earlier biochemical data, the structures reveal that the PxT and SPF motifs are important, though not wholly responsible, for the specificity of stop codon recognition. Moreover, despite the obvious overlaps in the binding sites of the tRNAs and class I release factors on the ribosome, the molecular binding determinants for recognition/function are strikingly distinct, at least in the small ribosome subunit. These observations are consistent with biochemical predictions based on mutational analysis and antibiotic sensitivity profiles for the two processes (Youngman et al., 2006).
Both RF1 and RF2 must specifically recognize a uridine at the first position of the codon (U1, which is shared by all three stop codons). RF1 and RF2 use nearly identical mechanisms for recognition that involve the packing of a specific residue against the Watson-Crick face of U1, an interaction that would not be possible with a purine due to steric clash. Another set of interactions with U1 involves hydrogen bonds between specific residues on the protein factors and the base of the nucleotide. These interactions are also only possible with a U at this position (shown for RF1 in Figure 3c). At the second position of the stop codon, RF1 exhibits specificity towards adenosine, whereas RF2 recognizes both purines (adenosine or guanosine). RF1 utilizes an amino acid residue that is only capable of accepting a hydrogen bond from the N6 group of adenosine and as such cannot interact with the hydrogen-accepting O6 group of guanosine. In contrast, RF2 carries a residue that can both donate and accept hydrogen bonds, and so can form interactions with either N1/N6 of adenosine or N1/O6 of guanosine. The third position of the stop codon in the RF1 and RF2 structures is in a distinct conformation from that observed in tRNA bound structures; the third nucleotide of the codon stacks on G530 of the 16S RNA, rather than on the second codon nucleotide. RF1 recognizes both adenosines and guanosines at this position through a bifunctional amino acid that forms hydrogen bonds with both nucleotides. An equivalent residue is lacking in RF2, instead an amino acid only capable of accepting a hydrogen bond from N6 of adenosine is found, explaining the discrimination by RF2 against guanosine at this position.
Although the molecular details above explain in large part the specificity of stop codon recognition by release factors, these structures must also be reconciled with the biochemical data. Biochemical studies indicate that near-cognate stop codons are discriminated against both at the level of apparent binding affinity (Km) and catalysis (kcat). These observations suggested that stop codon recognition results in particular conformational rearrangements ultimately productive for catalysis. The published structures have thus far provided information on recognition of cognate stop codons by the class I release factors, but do not provide insight into how recognition might be different on a near-cognate (sense) complex. Some clues do emerge, however, as to how communication might be transmitted from the small to the large ribosome subunit. As previously mentioned, in ribosome structures with A site-bound tRNAs, both A1492 and A1493 unstack from helix 44 of the 16S RNA, and G530 rotates from a syn to an anti conformation to engage the minor groove of the codon-anticodon helix. In structures with release factors bound to stop codons, the observed conformational changes are different – only A1492 unstacks from helix 44, whereas A1493 remains stacked within the helix. Interestingly, this positioning of A1493 is stabilized by the movement of A1913 of the 23S RNA into the region to provide a stacking interaction (Figure 4d). G530 also seems to play an important role in forming stacking interactions with the third nucleotide of the codon to stabilize its markedly splayed configuration.
In addition to these localized structural changes, there are some hints about how such rearrangements might be communicated to the remainder of the ribosome. Specifically, an element connecting domains 3 and 4 of RF1, recently termed the “switch loop”, forms an extended conformation relative to the structure observed in free RF1. This conformation results in a helical extension that allows domain3, including its catalytically important GGQ motif, to comfortably reach the active site in the PTC (Figure 6) (Laurberg et al., 2008). The rearranged “switch loop” appears to be stabilized by interactions with the ribosome and the relase factor itself, thus connecting stop codon-induced conformational changes in the decoding center to more long range effects, possibly including subtle rearrangements in the peptidyl transfer center that affect the rates of peptide release. Although there has been no discussion in the literature of closure-like movements in the head-shoulder regions of the small ribosome subunit upon release factor binding, it is possible that such movements may play similar roles in facilitating selectivity during stop codon recognition (Figure 5c, d).
Contributions to Fidelity from Possible Remote, and not so Remote, Locales
At this point we have considered the core mechanisms that contribute to codon recognition as deciphered in the A site by tRNAs and class I release factors. In addition to these central A-site elements, there are other ribosomal regions that have been implicated in the accuracy of codon recognition. Most significantly, but not without controversy, it has long been argued that the E site (and its occupation status) is critical to fidelity during translation. Nierhaus and colleagues have proposed an allosteric model for ribosome function wherein the affinity of the A site for incoming tRNA depends on the occupancy of the E site (Nierhaus, 2006). According to their studies, occupancy of the E site with a cognate tRNA species results in a low affinity A site that discriminates effectively against non-cognate tRNA species, whereas an empty E site results in a high affinity and low fidelity A site. These conclusions were made based on the observation that the rate of A-site occupation is the same as that of E-site dissociation (Rheinberger and Nierhaus, 1986) and that the activation energy for A-site binding is larger when the E site is occupied with a cognate tRNA (Schilling-Bartetzko et al., 1992). We note that these binding studies were carried out in the absence of EF-Tu (so called non-enzymatic loading) and thus may not be physiologically relevant. In other experiments, acceptance of non-cognate tRNA species was substantially reduced in the presence of a cognate E-site tRNA (Geigenmuller and Nierhaus, 1990). Recent work by Nierhaus and colleagues further suggest that during the initiation phase of translation (when the E site is unoccupied), the Shine-Dalgarno helix may functionally substitute for the E-site codon:anticodon pairing in the maintenance of fidelity (Di Giacco et al., 2008). Nierhaus and colleagues propose that such negative allostery between the E and A sites provides a mechanism to allow easy discrimination against the many non-cognate tRNAs in the cell, thus reducing the tRNA selection challenge to just a few near-cognate tRNAs (Nierhaus, 2006). The near-cognate tRNAs must still be discriminated against, of course, and this is arguably the principle role of kinetic proofreading and induced fit, discussed previously.
Although the idea of coupling fidelity to negative allostery on the ribosome is an appealing one, some of the key kinetic and thermodynamic observations supporting this model have been called into question. Rodnina and colleagues failed to observe allosteric interactions between the A- and E-site tRNAs in a highly purified in vitro system and have instead long argued that the role of the E site is to facilitate the exit of the deacylated tRNA from the P site during translocation (Semenkov et al., 1996). (We would like to point out that in Figure 2, although an E-site tRNA has been incorporated into the tRNA selection scheme, and retained in the ribosome up until the accommodation step, it has been specifically highlighted to emphasize that many controversies surround its functional role in the process.) While the data surrounding E-site occupancy and tRNA selection fidelity remain unresolved, a role for the E-site tRNA in frame maintenance is less controversial. For example, Fredrick and colleagues deleted a portion of the E-site tRNA binding site, by truncating a β-strand from S7, and observed clear effects on frameshifting in their in vivo reporter system; interestingly, they observed no effects on tRNA selection (Devaraj et al., in press). The observed stimulation of frameshifting is also consistent with data suggesting that perturbations of E-site codon-anticodon pairing interactions promotes frameshifting (Marquez et al., 2004). Yet in a different study, mutations in the 23S rRNA forming the E-site tRNA binding site affect both frameshifting and certain tRNA selection events (Sergiev et al., 2005). These studies highlight a common problem in thinking about fidelity in the ribosome: what is the relationship between frameshifting, missense and nonsense suppression? Should losses in fidelity necessarily affect all three phenomena, or is each process dictated by distinct features of the ribosome?
In thinking about whether or not tRNA interactions in the E site might affect fidelity, it is worth asking what is known at the structural level. Structural studies have amply documented E-site tRNA binding on the ribosome (Jenner et al., 2007; Korostelev et al., 2006; Selmer et al., 2006; Yusupov et al., 2001). Cryo-EM studies have even documented intermediate states of tRNA binding where tRNAs occupy different states on the two subunits, forming so called “hybrid states” (Agirrezabala et al., 2008; Julian et al., 2008). tRNAs occupancies in these distinct states, coupled with the dynamics of the ribosome itself, provides an extremely complicated set of variables that must be deciphered in order to have a complete understanding of translation. Single-molecule studies may be the best approach for characterizing these many parameters. For the moment, however, we can ask whether the static ribosome structures that are available provide insights into how the E-site codon-anticodon helix might contribute to ribosome function. It is interesting to note that most ribosome structures do contain E-site-bound tRNAs, not because they were supplied by the researcher, but because they naturally co-purified with the ribosomes. As such, the tRNA species are neither homogeneous nor cognate (e.g. Yusupov et al., 2001). It is not surprising then that in these structures, no codon-anticodon pairing interactions were observed. What is less anticipated is that the conformation of the E-site codon was contorted to preclude any such codon-anticodon interactions. In a recent study by Yusopova and colleagues, a comparison of two distinct functional ribosome complexes yielded some new insights (Jenner et al., 2007). An “initiation” complex (with tRNAfMet in the P site and a nearby Shine-Dalgarno sequence) resembled previous structures where no E-site tRNA interaction could be seen. However, an “elongation” complex (with tRNAPhe in the P and A sites, and a more distant Shine-Dalgarno sequence) showed a conformation of the E site codon compatible with E-site tRNA anticodon interaction. Indeed, the authors observed “continuous density” in this structure between the 1st nucleotide of the E-site codon and the 3rd nucleotide of the E-site tRNA anticodon.. It should be noted that as the E-site tRNA in this structure was co-purified during the ribosome preparation, it was presumed to be non-cognate. Comparison of multiple E-site-occupied ribosome structures makes it clear that these issues are far from resolved, as each exhibits different conformations of the mRNA and the E-site tRNA (Figure 7). Although not conclusive, these data suggest that such interactions may be possible and may provide support for some of the biochemical data (Nierhaus, 2006). Some of the confusion and discrepancies in the E-site literature may be resolved by consideration of two slightly different E sites, E′ and E, where only the E′ site relies on codon:anticodon interactions, as first proposed by Paulsen and Wintermeyer (1986).
Figure 7. E-site-bound ribosome structures reveals heterogeneity in mRNA and tRNA conformations.
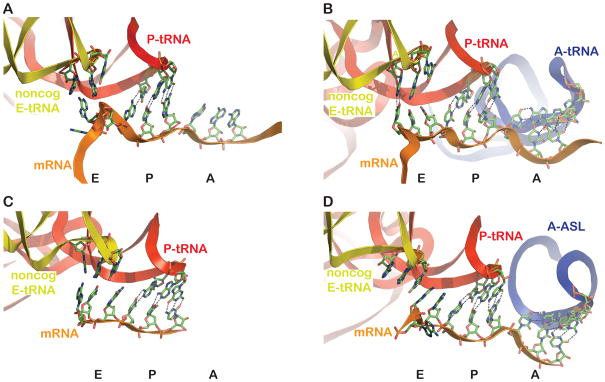
A. Structure of initiation-like complex, with a Shine-Dalgarno sequence upstream, from Jenner et al. (2007) (PDB 2HGR) containing non-cognate E-site tRNA, cognate P-site tRNA, and an empty A site. The mRNA adopts a conformation not appearing compatible with codon-anticodon interaction in the E site.
B. Structure of elongation-like complex from same study as A (PDB 2HGP), also with non-cognate tRNA in the E site, and cognate P- and A- sites tRNAs. Continuous electron density, from the X-ray diffraction data, is observed between position 1 of the E site codon and the corresponding position of the E-site tRNA anticodon, as indicated with the dashed lines.
C. Structure from Korostelev et al. (2006) (PDB 2OW8) with non-cognate E-site tRNA, cognate P-site tRNA and an empty A site (and no Shine-Dalgarno sequence). The mRNA adopts a conformation that appears to be compatible with codon-anticodon interactions, though none are documented with the non-cognate tRNA species bound.
D. Structure from Selmer et al. (2006) (PDB 2J00) with ligands as in C, except that the A site is occupied with a cognate anticodon stemloop (ASL). The mRNA (most notably the first position of the E-site codon) and the E-site tRNA positions are distinct from those in C.
In recent results from our own lab, we have documented other new contributions to fidelity that likely intersect with the E-site models (Zaher and Green, 2009). In these studies, we found that mismatches located in both the P and E site decoding helices result in dramatic losses in fidelity during codon recognition in the A site. For example, single mismatches in the P-site codon-anticodon helix increase the rate of peptide release on sense codons by ~ 2 orders of magnitude and result in substantial losses in the fidelity of tRNA selection. Even more strikingly, when mismatches are formed in both the P and E sites, as might result from iterated errors caused by the initial P-site mismatch, the rates of release on sense codons are stimulated by as much as 4 orders of magnitude. We have argued that these dramatic enhancements of the rate of peptide release on sense codons lead to premature termination, thus functioning to increase the overall fidelity of protein synthesis in a post peptidyl transfer quality control mechanism. These studies are not the first to suggest that perturbations in the P site lead to losses in fidelity. Farabaugh and colleagues have described the stimulation of frameshifting as a result of mismatches in the P site decoding helix (Sundararajan et al., 1999). The very large synthetic effects on fidelity that we attribute to E-site mismatches in our system may be mechanistically related to the allosteric model for E- and A-site function on the ribosome (reviewed in Nierhaus, 2006).
Among the questions that need to be addressed next is how perturbations in the adjacent P site and the more distant E site of the small ribosome subunit can alter A-site behavior so dramatically. More generally, how is ribosome function impacted by long-range signaling? A number of studies in recent years have highlighted the existence of extended signaling networks within the ribosome that allow for communication between the interior and the exterior of the ribonucleoprotein complex. For example, the growing polypeptide chain, positioned within the exit tunnel, appears to communicate with the peptidyl transfer center to control catalysis and with the exterior of the ribosome to control external factor interactions (reviewed in Tenson and Ehrenberg, 2002). The molecular triggers of such events, the paths of signal transduction, and the long-range effects on structure and function all remain to be determined. These questions are directly related to those previously posed for tRNA and release factor selection in the A site for which we now have considerable molecular understanding of the initiating events in the functional centers for decoding and peptidyl transfer. There now remains much to learn about signaling throughout the ribosome and the resulting downstream consequences for translation fidelity.
Acknowledgments
The authors thank K. Fredrick for valuable comments on the manuscript. R.G. is funded by the NIH and HHMI. HSZ is supported by a Natural Sciences and Engineering Research Council of Canada (NSERC) fellowship.
References
- Agirrezabala X, Lei J, Brunelle JL, Ortiz-Meoz RF, Green R, Frank J. Visualization of the hybrid state of tRNA binding promoted by spontaneous ratcheting of the ribosome. Mol Cell. 2008;32:190–197. doi: 10.1016/j.molcel.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacher JM, Schimmel P. An editing-defective aminoacyl-tRNA synthetase is mutagenic in aging bacteria via the SOS response. Proc Natl Acad Sci USA. 2007;104:1907–1912. doi: 10.1073/pnas.0610835104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ban N, Nissen P, Hansen J, Moore PB, Steitz TA. The complete atomic structure of the large ribosomal subunit at 2.4 Å resolution. Science. 2000;289:905–920. doi: 10.1126/science.289.5481.905. [DOI] [PubMed] [Google Scholar]
- Bilgin N, Claesens F, Pahverk H, Ehrenberg M. Kinetic properties of Escherichia coli ribosomes with altered forms of S12. J Mol Biol. 1992;224:1011–1027. doi: 10.1016/0022-2836(92)90466-w. [DOI] [PubMed] [Google Scholar]
- Blanchard SC, Gonzalez RL, Kim HD, Chu S, Puglisi JD. tRNA selection and kinetic proofreading in translation. Nat Struct Mol Biol. 2004a;11:1008–1014. doi: 10.1038/nsmb831. [DOI] [PubMed] [Google Scholar]
- Blanchard SC, Kim HD, Gonzalez RL, Jr, Puglisi JD, Chu S. tRNA dynamics on the ribosome during translation. Proc Natl Acad Sci USA. 2004b;101:12893–12898. doi: 10.1073/pnas.0403884101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouadloun F, Donner D, Kurland CG. Codon-specific missense errors in vivo. EMBO J. 1983;2:1351–1356. doi: 10.1002/j.1460-2075.1983.tb01591.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CM, McCaughan KK, Tate WP. Two regions of the Escherichia coli 16S ribosomal RNA are important for decoding stop signals in polypeptide chain termination. Nucleic Acids Res. 1993;21:2109–2115. doi: 10.1093/nar/21.9.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brutlag D, Kornberg A. Enzymatic synthesis of deoxyribonucleic acid. 36 A proofreading function for the 3′ leads to 5′ exonuclease activity in deoxyribonucleic acid polymerases. J Biol Chem. 1972;247:241–248. [PubMed] [Google Scholar]
- Carter AP, Clemons WM, Brodersen DE, Morgan-Warren RJ, Wimberly BT, Ramakrishnan V. Functional insights from the structure of the 30S ribosomal subunit and its interactions with antibiotics. Nature. 2000;407:340–348. doi: 10.1038/35030019. [DOI] [PubMed] [Google Scholar]
- Chernoff YO, Galkin AP, Lewitin E, Chernova TA, Newnam GP, Belenkiy SM. Evolutionary conservation of prion-forming abilities of the yeast Sup35 protein. Mol Microbiol. 2000;35:865–876. doi: 10.1046/j.1365-2958.2000.01761.x. [DOI] [PubMed] [Google Scholar]
- Cochella L, Brunelle JL, Green R. Mutational analysis reveals two independent molecular requirements during transfer RNA selection on the ribosome. Nat Struct Mol Biol. 2007;14:30–36. doi: 10.1038/nsmb1183. [DOI] [PubMed] [Google Scholar]
- Cochella L, Green R. An active role for tRNA in decoding beyond codon:anticodon pairing. Science. 2005a;308:1178–1180. doi: 10.1126/science.1111408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochella L, Green R. Fidelity in protein synthesis. Curr Biol. 2005b;15:R536–540. doi: 10.1016/j.cub.2005.07.018. [DOI] [PubMed] [Google Scholar]
- Craigen WJ, Caskey CT. Expression of peptide chain release factor 2 requires high-efficiency frameshift. Nature. 1986;322:273–275. doi: 10.1038/322273a0. [DOI] [PubMed] [Google Scholar]
- Davies J, Davis BD. Misreading of ribonucleic acid code words induced by aminoglycoside antibiotics. The effect of drug concentration. J Biol Chem. 1968;243:3312–3316. [PubMed] [Google Scholar]
- Davies J, Gilbert W, Gorini L. Streptomycin, Suppression, and the Code. Proc Natl Acad Sci USA. 1964;51:883–890. doi: 10.1073/pnas.51.5.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies J, Gorini L, Davis BD. Misreading of RNA codewords induced by aminoglycoside antibiotics. Mol Pharmacol. 1965;1:93–106. [PubMed] [Google Scholar]
- Di Giacco V, Marquez V, Qin Y, Pech M, Triana-Alonso FJ, Wilson DN, Nierhaus KH. Shine-Dalgarno interaction prevents incorporation of noncognate amino acids at the codon following the AUG. Proc Natl Acad Sci USA. 2008;105:10715–10720. doi: 10.1073/pnas.0801974105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaconu M, Kothe U, Schlunzen F, Fischer N, Harms JM, Tonevitsky AG, Stark H, Rodnina MV, Wahl MC. Structural basis for the function of the ribosomal L7/12 stalk in factor binding and GTPase activation. Cell. 2005;121:991–1004. doi: 10.1016/j.cell.2005.04.015. [DOI] [PubMed] [Google Scholar]
- Edelmann P, Gallant J. Mistranslation in E. coli. Cell. 1977;10:131–137. doi: 10.1016/0092-8674(77)90147-7. [DOI] [PubMed] [Google Scholar]
- Eisinger J, Feuer B, Yamane T. Luminescence and binding studies on tRNA-Phe. Proc Natl Acad Sci USA. 1970;65:638–644. doi: 10.1073/pnas.65.3.638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahlman RP, Dale T, Uhlenbeck OC. Uniform binding of aminoacylated transfer RNAs to the ribosomal A and P sites. Mol Cell. 2004;16:799–805. doi: 10.1016/j.molcel.2004.10.030. [DOI] [PubMed] [Google Scholar]
- Fourmy D, Recht MI, Blanchard SC, Puglisi JD. Structure of the A site of Escherichia coli 16S ribosomal RNA complexed with an aminoglycoside antibiotic. Science. 1996;274:1367–1371. doi: 10.1126/science.274.5291.1367. [DOI] [PubMed] [Google Scholar]
- Fourmy D, Yoshizawa S, Puglisi JD. Paromomycin binding induces a local conformational change in the A-site of 16 S rRNA. J Mol Biol. 1998;277:333–345. doi: 10.1006/jmbi.1997.1551. [DOI] [PubMed] [Google Scholar]
- Francklyn CS. DNA polymerases and aminoacyl-tRNA synthetases: shared mechanisms for ensuring the fidelity of gene expression. Biochemistry. 2008;47:11695–11703. doi: 10.1021/bi801500z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freistroffer DV, Kwiatkowski M, Buckingham RH, Ehrenberg M. The accuracy of codon recognition by polypeptide release factors. Proc Natl Acad Sci USA. 2000;97:2046–2051. doi: 10.1073/pnas.030541097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freistroffer DV, Pavlov MY, MacDougall J, Buckingham RH, Ehrenberg M. Release factor RF3 in E.coli accelerates the dissociation of release factors RF1 and RF2 from the ribosome in a GTP-dependent manner. EMBO J. 1997;16:4126–4133. doi: 10.1093/emboj/16.13.4126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geigenmuller U, Nierhaus KH. Significance of the third tRNA binding site, the E site, on E. coli ribosomes for the accuracy of translation: an occupied E site prevents the binding of non-cognate aminoacyl-tRNA to the A site. EMBO J. 1990;9:4527–4533. doi: 10.1002/j.1460-2075.1990.tb07904.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorini L, Kataja E. Phenotypic repair by streptomycin of defective genotypes in E. Coli. Proc Natl Acad Sci USA. 1964;51:487–493. doi: 10.1073/pnas.51.3.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gromadski KB, Daviter T, Rodnina MV. A uniform response to mismatches in codon-anticodon complexes ensures ribosomal fidelity. Mol Cell. 2006;21:369–377. doi: 10.1016/j.molcel.2005.12.018. [DOI] [PubMed] [Google Scholar]
- Gromadski KB, Rodnina MV. Kinetic determinants of high-fidelity tRNA discrimination on the ribosome. Mol Cell. 2004a;13:191–200. doi: 10.1016/s1097-2765(04)00005-x. [DOI] [PubMed] [Google Scholar]
- Gromadski KB, Rodnina MV. Streptomycin interferes with conformational coupling between codon recognition and GTPase activation on the ribosome. Nat Struct Mol Biol. 2004b;11:316–322. doi: 10.1038/nsmb742. [DOI] [PubMed] [Google Scholar]
- Hopfield JJ. Kinetic proofreading: a new mechanism for reducing errors in biosynthetic processes requiring high specificity. Proc Natl Acad Sci USA. 1974;71:4135–4139. doi: 10.1073/pnas.71.10.4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Uno M, Nakamura Y. A tripeptide ‘anticodon’ deciphers stop codons in messenger RNA. Nature. 2000;403:680–684. doi: 10.1038/35001115. [DOI] [PubMed] [Google Scholar]
- Jacks T, Varmus HE. Expression of the Rous sarcoma virus pol gene by ribosomal frameshifting. Science. 1985;230:1237–1242. doi: 10.1126/science.2416054. [DOI] [PubMed] [Google Scholar]
- Jelenc PC, Kurland CG. Nucleoside triphosphate regeneration decreases the frequency of translation errors. Proc Natl Acad Sci USA. 1979;76:3174–3178. doi: 10.1073/pnas.76.7.3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenner L, Rees B, Yusupov M, Yusupova G. Messenger RNA conformations in the ribosomal E site revealed by X-ray crystallography. EMBO Rep. 2007;8:846–850. doi: 10.1038/sj.embor.7401044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson M, Bouakaz E, Lovmar M, Ehrenberg M. The kinetics of ribosomal peptidyl transfer revisited. Mol Cell. 2008;30:589–598. doi: 10.1016/j.molcel.2008.04.010. [DOI] [PubMed] [Google Scholar]
- Johnson KA. Conformational coupling in DNA polymerase fidelity. Annu Rev Biochem. 1993;62:685–713. doi: 10.1146/annurev.bi.62.070193.003345. [DOI] [PubMed] [Google Scholar]
- Jorgensen F, Adamski FM, Tate WP, Kurland CG. Release factor-dependent false stops are infrequent in Escherichia coli. J Mol Biol. 1993;230:41–50. doi: 10.1006/jmbi.1993.1124. [DOI] [PubMed] [Google Scholar]
- Julian P, Konevega AL, Scheres SH, Lazaro M, Gil D, Wintermeyer W, Rodnina MV, Valle M. Structure of ratcheted ribosomes with tRNAs in hybrid states. Proc Natl Acad Sci USA. 2008;105:16924–16927. doi: 10.1073/pnas.0809587105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karimi R, Ehrenberg M. Dissociation rate of cognate peptidyl-tRNA from the A-site of hyper-accurate and error-prone ribosomes. Eur J Biochem / FEBS. 1994;226:355–360. doi: 10.1111/j.1432-1033.1994.tb20059.x. [DOI] [PubMed] [Google Scholar]
- Klaholz BP, Pape T, Zavialov AV, Myasnikov AG, Orlova EV, Vestergaard B, Ehrenberg M, van Heel M. Structure of the Escherichia coli ribosomal termination complex with release factor 2. Nature. 2003;421:90–94. doi: 10.1038/nature01225. [DOI] [PubMed] [Google Scholar]
- Korostelev A, Asahara H, Lancaster L, Laurberg M, Hirschi A, Zhu J, Trakhanov S, Scott WG, Noller HF. Crystal structure of a translation termination complex formed with release factor RF2. Proc Natl Acad Sci USA. 2008;105:19684–19689. doi: 10.1073/pnas.0810953105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korostelev A, Trakhanov S, Laurberg M, Noller HF. Crystal structure of a 70S ribosome-tRNA complex reveals functional interactions and rearrangements. Cell. 2006;126:1065–1077. doi: 10.1016/j.cell.2006.08.032. [DOI] [PubMed] [Google Scholar]
- Kothe U, Rodnina MV. Codon reading by tRNAAla with modified uridine in the wobble position. Mol Cell. 2007;25:167–174. doi: 10.1016/j.molcel.2006.11.014. [DOI] [PubMed] [Google Scholar]
- Kramer EB, Farabaugh PJ. The frequency of translational misreading errors in E. coli is largely determined by tRNA competition. RNA. 2007;13:87–96. doi: 10.1261/rna.294907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkel TA, Bebenek K. DNA replication fidelity. Annu Rev Biochem. 2000;69:497–529. doi: 10.1146/annurev.biochem.69.1.497. [DOI] [PubMed] [Google Scholar]
- Kurland CG, Ehrenberg M. Optimization of translation accuracy. Prog Nucleic Acid Res Mol Biol. 1984;31:191–219. doi: 10.1016/s0079-6603(08)60378-5. [DOI] [PubMed] [Google Scholar]
- Laurberg M, Asahara H, Korostelev A, Zhu J, Trakhanov S, Noller HF. Structural basis for translation termination on the 70S ribosome. Nature. 2008;454:852–857. doi: 10.1038/nature07115. [DOI] [PubMed] [Google Scholar]
- Laughrea M, Latulippe J, Filion AM, Boulet L. Mistranslation in twelve Escherichia coli ribosomal proteins. Cysteine misincorporation at neutral amino acid residues other than tryptophan. Eur J Bioch / FEBS. 1987;169:59–64. doi: 10.1111/j.1432-1033.1987.tb13580.x. [DOI] [PubMed] [Google Scholar]
- Ledoux S, Uhlenbeck OC. Different aa-tRNAs are selected uniformly on the ribosome. Mol Cell. 2008;31:114–123. doi: 10.1016/j.molcel.2008.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JW, Beebe K, Nangle LA, Jang J, Longo-Guess CM, Cook SA, Davisson MT, Sundberg JP, Schimmel P, Ackerman SL. Editing-defective tRNA synthetase causes protein misfolding and neurodegeneration. Nature. 2006;443:50–55. doi: 10.1038/nature05096. [DOI] [PubMed] [Google Scholar]
- Liiv A, O’Connor M. Mutations in the intersubunit bridge regions of 23 S rRNA. J Biol Chem. 2006;281:29850–29862. doi: 10.1074/jbc.M603013200. [DOI] [PubMed] [Google Scholar]
- Lipsett MN, Heppel LA, Bradley DF. Complex formation between adenine oligonucleotides and polyuridylic acid. Biochim Biophys Acta. 1960;41:175–177. doi: 10.1016/0006-3002(60)90394-2. [DOI] [PubMed] [Google Scholar]
- Marquez V, Wilson DN, Tate WP, Triana-Alonso F, Nierhaus KH. Maintaining the ribosomal reading frame: The influence of the E site during translational regulation of release factor 2. Cell. 2004;118:45–55. doi: 10.1016/j.cell.2004.06.012. [DOI] [PubMed] [Google Scholar]
- Marshall RA, Aitken CE, Dorywalska M, Puglisi JD. Translation at the single-molecule level. Annu Rev Biochem. 2008;77:177–203. doi: 10.1146/annurev.biochem.77.070606.101431. [DOI] [PubMed] [Google Scholar]
- McLaughlin CS, Dondon J, Grunberg-Manago M, Michelson AM, Saunders G. Stability of the messenger RNA-sRNA-ribosome complex. Cold Spring Harb Symp Quant Biol. 1966;31:601. doi: 10.1101/sqb.1966.031.01.078. [DOI] [PubMed] [Google Scholar]
- Moazed D, Noller HF. Interaction of antibiotics with functional sites in 16S ribosomal RNA. Nature. 1987;327:389–394. doi: 10.1038/327389a0. [DOI] [PubMed] [Google Scholar]
- Moazed D, Noller HF. Binding of tRNA to the ribosomal A and P sites protects two distinct sets of nucleotides in 16 S rRNA. J Mol Biol. 1990;211:135–145. doi: 10.1016/0022-2836(90)90016-F. [DOI] [PubMed] [Google Scholar]
- Moazed D, Robertson JM, Noller HF. Interaction of elongation factors EF-G and EF-Tu with a conserved loop in 23S RNA. Nature. 1988;334:362–364. doi: 10.1038/334362a0. [DOI] [PubMed] [Google Scholar]
- Mora L, Zavialov A, Ehrenberg M, Buckingham RH. Stop codon recognition and interactions with peptide release factor RF3 of truncated and chimeric RF1 and RF2 from Escherichia coli. Mol Microbiol. 2003;50:1467–1476. doi: 10.1046/j.1365-2958.2003.03799.x. [DOI] [PubMed] [Google Scholar]
- Murphy FV, Ramakrishnan V, Malkiewicz A, Agris PF. The role of modifications in codon discrimination by tRNA(Lys) UUU. Nat Struct Mol Biol. 2004;11:1186–1191. doi: 10.1038/nsmb861. [DOI] [PubMed] [Google Scholar]
- Nakayashiki T, Ebihara K, Bannai H, Nakamura Y. Yeast [PSI+] “prions” that are crosstransmissible and susceptible beyond a species barrier through a quasi-prion state. Mol Cell. 2001;7:1121–1130. doi: 10.1016/s1097-2765(01)00259-3. [DOI] [PubMed] [Google Scholar]
- Nangle LA, De Crecy Lagard V, Doring V, Schimmel P. Genetic code ambiguity. Cell viability related to the severity of editing defects in mutant tRNA synthetases. J Biol Chem. 2002;277:45729–45733. doi: 10.1074/jbc.M208093200. [DOI] [PubMed] [Google Scholar]
- Nierhaus KH. Decoding errors and the involvement of the E-site. Biochimie. 2006;88:1013–1019. doi: 10.1016/j.biochi.2006.02.009. [DOI] [PubMed] [Google Scholar]
- Ninio J. Kinetic amplification of enzyme discrimination. Biochimie. 1975;57:587–595. doi: 10.1016/s0300-9084(75)80139-8. [DOI] [PubMed] [Google Scholar]
- Ogle JM, Brodersen DE, Clemons WM, Tarry MJ, Carter AP, Ramakrishnan V. Recognition of cognate transfer RNA by the 30S ribosomal subunit. Science. 2001;292:897–902. doi: 10.1126/science.1060612. [DOI] [PubMed] [Google Scholar]
- Ogle JM, Carter AP, Ramakrishnan V. Insights into the decoding mechanism from recent ribosome structures. Trends Bioch Sci. 2003;28:259–266. doi: 10.1016/S0968-0004(03)00066-5. [DOI] [PubMed] [Google Scholar]
- Ogle JM, Murphy FV, Tarry MJ, Ramakrishnan V. Selection of tRNA by the ribosome requires a transition from an open to a closed form. Cell. 2002;111:721–732. doi: 10.1016/s0092-8674(02)01086-3. [DOI] [PubMed] [Google Scholar]
- Ogle JM, Ramakrishnan V. Structural insights into translational fidelity. Ann Rev Biochem. 2005;74:129–177. doi: 10.1146/annurev.biochem.74.061903.155440. [DOI] [PubMed] [Google Scholar]
- Olejniczak M, Dale T, Fahlman RP, Uhlenbeck OC. Idiosyncratic tuning of tRNAs to achieve uniform ribosome binding. Nat Struct Mol Biol. 2005;12:788–793. doi: 10.1038/nsmb978. [DOI] [PubMed] [Google Scholar]
- Orlova M, Yueh A, Leung J, Goff SP. Reverse transcriptase of Moloney murine leukemia virus binds to eukaryotic release factor 1 to modulate suppression of translational termination. Cell. 2003;115:319–331. doi: 10.1016/s0092-8674(03)00805-5. [DOI] [PubMed] [Google Scholar]
- Pape T, Wintermeyer W, Rodnina M. Induced fit in initial selection and proofreading of aminoacyl-tRNA on the ribosome. EMBO J. 1999;18:3800–3807. doi: 10.1093/emboj/18.13.3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pape T, Wintermeyer W, Rodnina MV. Complete kinetic mechanism of elongation factor Tu-dependent binding of aminoacyl-tRNA to the A site of the E-coli ribosome. EMBO J. 1998;17:7490–7497. doi: 10.1093/emboj/17.24.7490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pape T, Wintermeyer W, Rodnina MV. Conformational switch in the decoding region of 16S rRNA during aminoacyl-tRNA selection on the ribosome. Nat Struct Biol. 2000;7:104–107. doi: 10.1038/72364. [DOI] [PubMed] [Google Scholar]
- Paulsen H, Wintermeyer W. tRNA topography during translocation: steady-state and kinetic fluorescence energy-transfer studies. Biochemistry. 1986;25:2749–2756. doi: 10.1021/bi00358a002. [DOI] [PubMed] [Google Scholar]
- Paushkin SV, Kushnirov VV, Smirnov VN, Ter-Avanesyan MD. Propagation of the yeast prion-like [psi+] determinant is mediated by oligomerization of the SUP35-encoded polypeptide chain release factor. EMBO J. 1996;15:3127–3134. [PMC free article] [PubMed] [Google Scholar]
- Petry S, Brodersen DE, Murphy FVt, Dunham CM, Selmer M, Tarry MJ, Kelley AC, Ramakrishnan V. Crystal structures of the ribosome in complex with release factors RF1 and RF2 bound to a cognate stop codon. Cell. 2005;123:1255–1266. doi: 10.1016/j.cell.2005.09.039. [DOI] [PubMed] [Google Scholar]
- Piepenburg O, Pape T, Pleiss JA, Wintermeyer W, Uhlenbeck OC, Rodnina MV. Intact aminoacyl-tRNA is required to trigger GTP hydrolysis by elongation factor Tu on the ribosome. Biochemistry. 2000;39:1734–1738. doi: 10.1021/bi992331y. [DOI] [PubMed] [Google Scholar]
- Pisareva VP, Pisarev AV, Hellen CU, Rodnina MV, Pestova TV. Kinetic analysis of interaction of eukaryotic release factor 3 with guanine nucleotides. J Biol Chem. 2006;281:40224–40235. doi: 10.1074/jbc.M607461200. [DOI] [PubMed] [Google Scholar]
- Powers T, Noller HF. Dominant lethal mutations in a conserved loop in 16S rRNA. Proc Natl Acad Sci USA. 1990;87:1042–1046. doi: 10.1073/pnas.87.3.1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers T, Noller HF. Selective perturbation of G530 of 16 S rRNA by translational miscoding agents and a streptomycin-dependence mutation in protein S12. J Mol Biol. 1994;235:156–172. doi: 10.1016/s0022-2836(05)80023-3. [DOI] [PubMed] [Google Scholar]
- Rawat UB, Zavialov AV, Sengupta J, Valle M, Grassucci RA, Linde J, Vestergaard B, Ehrenberg M, Frank J. A cryo-electron microscopic study of ribosome-bound termination factor RF2. Nature. 2003;421:87–90. doi: 10.1038/nature01224. [DOI] [PubMed] [Google Scholar]
- Rheinberger HJ, Nierhaus KH. Allosteric interactions between the ribosomal transfer RNA-binding sites A and E. J Bio Chem. 1986;261:9133–9139. [PubMed] [Google Scholar]
- Rodnina MV, Fricke R, Kuhn L, Wintermeyer W. Codon-dependent conformational change of elongation factor Tu preceding GTP hydrolysis on the ribosome. EMBO J. 1995;14:2613–2619. doi: 10.1002/j.1460-2075.1995.tb07259.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodnina MV, Fricke R, Wintermeyer W. Transient conformational states of aminoacyl-tRNA during ribosome binding catalyzed by elongation factor Tu. Biochemistry. 1994;33:12267–12275. doi: 10.1021/bi00206a033. [DOI] [PubMed] [Google Scholar]
- Rodnina MV, Gromadski KB, Kothe U, Wieden HJ. Recognition and selection of tRNA in translation. FEBS Lett. 2005;579:938–942. doi: 10.1016/j.febslet.2004.11.048. [DOI] [PubMed] [Google Scholar]
- Rosenberger RF, Foskett G. An estimate of the frequency of in vivo transcriptional errors at a nonsense codon in Escherichia coli. Mol Gen Genet. 1981;183:561–563. doi: 10.1007/BF00268784. [DOI] [PubMed] [Google Scholar]
- Schimmel P. Development of tRNA synthetases and connection to genetic code and disease. Protein Sci. 2008;17:1643–1652. doi: 10.1110/ps.037242.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilling-Bartetzko S, Bartetzko A, Nierhaus KH. Kinetic and thermodynamic parameters for tRNA binding to the ribosome and for the translocation reaction. J Biol Chem. 1992;267:4703–4712. [PubMed] [Google Scholar]
- Schluenzen F, Tocilj A, Zarivach R, Harms J, Gluehmann M, Janell D, Bashan A, Bartels H, Agmon I, Franceschi F, et al. Structure of functionally activated small ribosomal subunit at 3.3 angstrom resolution. Cell. 2000;102:615–623. doi: 10.1016/s0092-8674(00)00084-2. [DOI] [PubMed] [Google Scholar]
- Selmer M, Dunham CM, Murphy FVt, Weixlbaumer A, Petry S, Kelley AC, Weir JR, Ramakrishnan V. Structure of the 70S ribosome complexed with mRNA and tRNA. Science. 2006;313:1935–1942. doi: 10.1126/science.1131127. [DOI] [PubMed] [Google Scholar]
- Semenkov YP, Rodnina MV, Wintermeyer W. The “allosteric three-site model” of elongation cannot be confirmed in a well-defined ribosome system from Escherichia coli. Proc Natl Acad Sci USA. 1996;93:12183–12188. doi: 10.1073/pnas.93.22.12183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sergiev PV, Lesnyak DV, Kiparisov SV, Burakovsky DE, Leonov AA, Bogdanov AA, Brimacombe R, Dontsova OA. Function of the ribosomal E-site: a mutagenesis study. Nucleic Acids Res. 2005;33:6048–6056. doi: 10.1093/nar/gki910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin DH, Brandsen J, Jancarik J, Yokota H, Kim R, Kim SH. Structural analyses of peptide release factor 1 from Thermotoga maritima reveal domain flexibility required for its interaction with the ribosome. J Mol Biol. 2004;341:227–239. doi: 10.1016/j.jmb.2004.05.055. [DOI] [PubMed] [Google Scholar]
- Shorter J, Lindquist S. Prions as adaptive conduits of memory and inheritance. Nat Rev Genet. 2005;6:435–450. doi: 10.1038/nrg1616. [DOI] [PubMed] [Google Scholar]
- Spotts CR, Stanier RY. Mechanism of streptomycin action on bacteria: a unitary hypothesis. Nature. 1961;192:633–637. doi: 10.1038/192633a0. [DOI] [PubMed] [Google Scholar]
- Stark H, Rodnina MV, Wieden HJ, Zemlin F, Wintermeyer W, van Heel M. Ribosome interactions of aminoacyl-tRNA and elongation factor Tu in the codon-recognition complex. Nat Struct Biol. 2002;9:849–854. doi: 10.1038/nsb859. [DOI] [PubMed] [Google Scholar]
- Sundararajan A, Michaud WA, Qian Q, Stahl G, Farabaugh PJ. Near-cognate peptidyl-tRNAs promote +1 programmed translational frameshifting in yeast. Mol Cell. 1999;4:1005–1015. doi: 10.1016/s1097-2765(00)80229-4. [DOI] [PubMed] [Google Scholar]
- Tenson T, Ehrenberg M. Regulatory nascent peptides in the ribosomal tunnel. Cell. 2002;108:591–594. doi: 10.1016/s0092-8674(02)00669-4. [DOI] [PubMed] [Google Scholar]
- Thompson RC. EFTu provides an internal kinetic standard for translational accuracy. Trends Bioch Sci. 1988;13:91–93. doi: 10.1016/0968-0004(88)90047-3. [DOI] [PubMed] [Google Scholar]
- Thompson RC, Dix DB, Gerson RB, Karim AM. Effect of Mg2+ concentration, polyamines, streptomycin, and mutations in ribosomal proteins on the accuracy of the two-step selection of aminoacyl-tRNAs in protein biosynthesis. The J Biol Chem. 1981;256:6676–6681. [PubMed] [Google Scholar]
- Thompson RC, Karim AM. The accuracy of protein biosynthesis is limited by its speed: high fidelity selection by ribosomes of aminoacyl-tRNA ternary complexes containing GTP[gamma S] Proc Natl Acad Sci USA. 1982;79:4922–4926. doi: 10.1073/pnas.79.16.4922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson RC, Stone PJ. Proofreading of the codon-anticodon interaction on ribosomes. Proc Natl Acad Sci USA. 1977;74:198–202. doi: 10.1073/pnas.74.1.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triman KL. Mutational analysis of the ribosome. Adv Genet. 2007;58:89–119. doi: 10.1016/S0065-2660(06)58004-6. [DOI] [PubMed] [Google Scholar]
- True HL, Berlin I, Lindquist SL. Epigenetic regulation of translation reveals hidden genetic variation to produce complex traits. Nature. 2004;431:184–187. doi: 10.1038/nature02885. [DOI] [PubMed] [Google Scholar]
- True HL, Lindquist SL. A yeast prion provides a mechanism for genetic variation and phenotypic diversity. Nature. 2000;407:477–483. doi: 10.1038/35035005. [DOI] [PubMed] [Google Scholar]
- Valle M, Sengupta J, Swami NK, Grassucci RA, Burkhardt N, Nierhaus KH, Agrawal RK, Frank J. Cryo-EM reveals an active role for aminoacyl-tRNA in the accommodation process. EMBO J. 2002;21:3557–3567. doi: 10.1093/emboj/cdf326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valle M, Zavialov A, Li W, Stagg SM, Sengupta J, Nielsen RC, Nissen P, Harvey SC, Ehrenberg M, Frank J. Incorporation of aminoacyl-tRNA into the ribosome as seen by cryo-electron microscopy. Nat Struct Biol. 2003;10:899–906. doi: 10.1038/nsb1003. [DOI] [PubMed] [Google Scholar]
- Vestergaard B, Van LB, Andersen GR, Nyborg J, Buckingham RH, Kjeldgaard M. Bacterial polypeptide release factor RF2 is structurally distinct from eukaryotic eRF1. Mol Cell. 2001;8:1375–1382. doi: 10.1016/s1097-2765(01)00415-4. [DOI] [PubMed] [Google Scholar]
- Weixlbaumer A, Jin H, Neubauer C, Voorhees RM, Petry S, Kelley AC, Ramakrishnan V. Insights into translational termination from the structure of RF2 bound to the ribosome. Science. 2008;322:953–956. doi: 10.1126/science.1164840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wimberly BT, Brodersen DE, Clemons WM, Morgan-Warren RJ, Carter AP, Vonrhein C, Hartsch T, Ramakrishnan V. Structure of the 30S ribosomal subunit. Nature. 2000;407:327–339. doi: 10.1038/35030006. [DOI] [PubMed] [Google Scholar]
- Yin YW, Steitz TA. Structural basis for the transition from initiation to elongation transcription in T7 RNA polymerase. Science. 2002;298:1387–1395. doi: 10.1126/science.1077464. [DOI] [PubMed] [Google Scholar]
- Yoshizawa S, Fourmy D, Puglisi JD. Recognition of the codon-anticodon helix by ribosomal RNA. Science. 1999;285:1722–1725. doi: 10.1126/science.285.5434.1722. [DOI] [PubMed] [Google Scholar]
- Youngman EM, Cochella L, Brunelle JL, He S, Green R. Two Distinct Conformations of the Conserved RNA-rich Decoding Center of the Small Ribosomal Subunit Are Recognized by tRNAs and Release Factors. Cold Spring Harb Symp Quant Biol. 2006;71:545–549. doi: 10.1101/sqb.2006.71.036. [DOI] [PubMed] [Google Scholar]
- Youngman EM, He SL, Nikstad LJ, Green R. Stop codon recognition by release factors induces structural rearrangement of the ribosomal decoding center that is productive for peptide release. Mol Cell. 2007;28:533–543. doi: 10.1016/j.molcel.2007.09.015. [DOI] [PubMed] [Google Scholar]
- Youngman EM, McDonald ME, Green R. Peptide release on the ribosome: mechanism and implications for translational control. Annu Rev Microbiol. 2008;62:353–373. doi: 10.1146/annurev.micro.61.080706.093323. [DOI] [PubMed] [Google Scholar]
- Yusupov MM, Yusupova GZ, Baucom A, Lieberman K, Earnest TN, Cate JHD, Noller HF. Crystal structure of the ribosome at 5.5 angstrom resolution. Science. 2001;292:883–896. doi: 10.1126/science.1060089. [DOI] [PubMed] [Google Scholar]
- Zaher HS, Green R. Quality control by the ribosome following peptide bond formation. Nature. 2009;457:161–166. doi: 10.1038/nature07582. [DOI] [PMC free article] [PubMed] [Google Scholar]