

Abstract
FXR (farnesoid X receptor, NRIH4), a nuclear receptor, plays a major role in the control of cholesterol metabolism. FXR ligands have been investigated in preclinical studies for targeted therapy against metabolic diseases, but have shown limitations. Therefore, there is a need for new agonist or antagonist ligands of FXR, both for potential clinical applications, as well as to further elucidate its biological functions. Here we describe the use of the X-ray crystal structure of FXR complexed with the potent small molecule agonist GW4064 to design and synthesize a novel fluorescent, high-affinity probe (DY246) for time resolved fluorescence resonance energy transfer (TR-FRET) assays. We then used the TR-FRET assay for high throughput screening of a library of over 5,000 bioactive compounds. From this library, we identified 13 compounds that act as putative FXR transcriptional antagonists.
Keywords: FXR, farnesoid X receptor, NRs, nuclear receptors, HTS, High-Throughput Screen, LBD, ligand-binding domain, TR-FRET, time resolved fluorescence resonance energy transfer
1. Introduction
Farnesoid X receptor (FXR) is a member of bile acid nuclear hormone receptor superfamily, and is highly expressed in the liver and intestine.1 FXR is a ligand-dependent transcription factor that regulates gene networks involved in bile acid homeostasis. Since bile acids represent the end-product of cholesterol metabolism, FXR has also a role in regulating lipid and cholesterol homeostasis. Consistent with this role of FXR, bile acids are the primary activating endogenous ligands of FXR.2 Recent findings suggest that FXR is a key metabolic regulator in the liver that acts to maintain the homeostasis of liver metabolites.3 Recent reports also indicate that FXR has significant effects on vasculature as well.4 Thus, FXR represents an attractive pharmacological target for the development of novel therapeutic agents to treat lipid metabolism disorders, hyperlipidemia and cholestatic disease, as well as atherogenic disease.5
Pharmaceutical control of activity of FXR with synthetic and natural ligands having agonistic or antagonistic activity is a powerful chemical tool for managing various clinical conditions.6 Several potent and selective FXR agonists have been reported recently and are well documented in the literature.7 FXR agonists decrease plasma triglyceride levels and increase the synthesis of high-density lipoprotein cholesterol.8 This suggests that highly selective and potent agonists for the FXR might be useful for the treatment of dyslipidemia and cholestasis. However, the preclinical development of FXR agonists has been limited because activation of FXR leads to complex responses and potentially undesirable side effects, such as inhibition of bile acid synthesis by indirectly repressing the expression of cytochrome 7α1 (Cyp7α1), the rate-limiting enzyme of the bile acid synthesis pathway.6
On the other hand, an antagonist of FXR, if selective for Cyp7α could be useful therapeutically to increase the conversion of cholesterol to bile acids, resulting in lower low density lipoprotein levels in hyperlipidemic patients. FXR regulates the expression of small heterodimer partner (SHP).1 SHP attenuates the expression of CYP7A1 by inhibiting the activity of liver receptor homologue 1 (LRH-1), which is known to augment CYP7A1 expression. FXR antagonism would be expected to lead to decreased SHP levels which, in turn, should increase CYP7α1 activity, enhance cholesterol metabolism in vivo and reduce serum levels of total cholesterol. From this point, a FXR antagonist might serve as a useful drug and should be developed.9
Although the search for potent, selective FXR antagonists has intensified over the past few years, few antagonists have been identified. Recent efforts involving a coactivator assay have identified guggulsterone (Figure 1) as the first example of a direct FXR antagonist.10 However, use of guggulsterone is limited because it is extremely non-specific and targets many other receptors, not just FXR, and probably targets other pathways as well. Additionally, guggulsterone has very low potency (IC50 of 12 –25 µM),10 and its mechanism of antagonism is unclear.11 Thus, guggulsterone is not a true antagonist of FXR; it is a unique FXR ligand with antagonistic activity in coactivator association assays but with the ability to enhance the action of FXR agonists in vivo.12
Figure 1.

Chemical structures of reported FXR antagonists.
Sulfated polyhydroxy sterols isolated from marine invertebrates were also recently identified as FXR antagonists.13 One of the sulfated polyhydroxy sterols, sulfated sterol 8 (Figure 1), had an antagonistic activity toward the expression of a subset of FXR-regulated genes in liver cells and abrogated the release of nuclear corepressor from the promoter of these genes. Most recently, theonellasterol (Figure 1), a marine FXR antagonist demonstrated as a promising lead in cholestasis has been reported.14
Nonsteroidal antagonist, exemplified by AGN34 (Figure 1), antagonizes FXR in transient reporter assays but acts in a gene-selective manner in vivo: it displays agonistic effect on the expression of CYP7α, antagonistic effect on the expression of the ileal bile acid-binding protein (IBABP ), and is neutral on SHP expression.15 Recent ligand-based virtual screening of a ibrary of 12480 compounds identified one nonsteroidal compound, 1,3,4-trisubstituted-pyrazolone 12 u as an FXR antagonist (Figure 1).16 These findings further confirm that selective FXR modulators can be identified that regulate a specific subset of FXR responsive genes in a gene-specific fashion.
Members of the nuclear receptor family, to which FXR belongs, is among the most successful “drugged” targets for the pharmaceutical industry, in part because it easily binds and responds to small, orally bioavailable molecules.17 To date, enormous effort has been put forth to develop screens for agents that modulate activity of nuclear receptors. Several robust high-throughput assays with high Z’-factor are readily available,18 including fluorescence polarization assays for direct ligand binding and FRET/Alpha-Screen assays for ligand-induced recruitment of coregulator peptides.19 Z’-factor was routinely used to assess assay performance. An assay with Z’ > 0.5 is considered robust and reproducible.20
TR-FRET assay has been previously used to screen for modulators of other nuclear receptors with a Z’-factor of > 0.75.21 To better understand the antagonism of FXR and its implication in disease treatment, with a goal of identifying novel small molecule antagonists of FXR activity that might have pharmacological relevance, we developed of a TR-FRET assay suitable for high-throughput screening (HTS). In this TR-FRET assay, we employed a novel fluorescent FXR ligand (DY246), which we developed based on the chemical structure of a potent FXR ligand GW4064. The assay has a high dynamic signal range and Z’ value. We used this assay to screen for all possible FXR ligands (agonists or antagonists) within a bioactive library of 5,600 compounds, which have been previously described.22–23 We then characterized for cellular activity of selected inhibitors identified from the TR-FRET assay. Our results demonstrated that the TR-FRET assay is suitable for identifying FXR modulators with cellular activities.
2. Results and discussion
2.1. Probe design
To successfully use the TR-FRET assay for HTS to identify novel FXR ligands, we required a novel fluorescent FXR ligand that had high affinity and potency. This required that the FXR fluorescent ligand contain a potent small molecule ligand that recognizes FXR, fluorescent tag useful for detection and quantitation (e.g., a fluorophore), and a suitable linker to connect the two (Figure 2A).
Figure 2.


(A). Design strategy of a fluoresent probe for FXR and hypothetical binding mode of the probe to FXR. (B). Ligand binding domain of the X-ray co-crystal structure of GW4064 in complex with FXR. Carbon atoms for FXR are shown in cyan. Carbon atoms for GW4064 are shown as green sticks. The semi-transparent gray surface represents the molecular surface, while hydrogen bonds are depicted as yellow dashed lines.26
We chose the small molecule GW4064 as the small molecule ligand because of its high binding potency for FXR (EC50 = 45 nM) as assayed by fluorescence resonance energy transfer for recruitment of the SRC1 peptide to human FXR.24 Similar potency of GW4064 (EC50 value of 90 nM) has also been reported in another study.25
An X-ray co-crystal structure of GW4064 with the ligand binding domain of FXR and the SRC-1 co-activator peptide has been solved.26 This structure shed light on the agonist recognition elements of the FXR ligand binding domain and on the binding modes, and also further revealed some open space adjacent to the terminal side-arm of GW4064 that could be exploited in the design of FXR probe. We used this X-ray crystal structure of the co-complex of FXR/GW4064 to design a FXR fluorescent probe for FXR.
The high-resolution X-ray structure of the protein-bound complex (FXR-GW4064) indicated potential side arms of GW4064 could be modified between the carbonyl acid group and isoxazole hetero atoms, respectively (Fig. 2B). However, because a dichlorophenyl oxazole moiety of GW4064 was recognized to provide considerable conformational rigidity to the agonist,27 we kept this moiety intact in the probe design. It is well established that side-arm of GW4064 bearing the carboxylate head group is important for activating FXR by forming a network of hydrogen bonds with 331Arg.28 Further examination revealed that the carboxylic acid group of GW4064 is co-planar with its phenyl ring, and that the two oxygen atoms of the carboxylate coordinate with one NH2 and the s-NH of the guanidine group of 331Arg. Analysis of the FXR–GW4064 binding revealed that the electrostatic interaction mimics the binding mode of the carboxylic acid of the natural bile acid ligands, such as GCDCA (Glyco-CDCA).29 Furthermore, crystal analysis of FXR co-complex indicates that the glycine-moiety of GCDCA already extends outside the receptor such that linkage at this position via an extended linker cannot interfere with ligand binding.29 Because the carboxyl-moiety of GW4064 echoes the binding mode of the carboxylic acid of the natural bile acid ligands, it could be hypothesized that the flexible guanidine side chain of 331Arg could move to facilitate the interaction with the negatively charged carboxylic oxygen moiety on GW4064. The potential binding mode of designed linker for the probe could be twisted out-of the plane of the aryl ring and extended outside the receptor, similar to the binding mode of GCDCA, and therefore would be unlikely to interfere with ligand binding (Figure 2A). In addition, molecular modeling studies30 suggest that the prolonged distance to the acidic function should be incompatible with the formation of the known ionic interaction to 331Arg, suggesting that there is a ‘hole’ in the structure of the FXR ligand binding pocket that marks an exit vector for substituents ranging out from the p-position of the terminal aryl passing along the 331Arg that forms an important salt bridge with the benzoic acid ‒COO” of the ligand towards the aqueous surface of the receptor.31 Note that when benzoic acid of GW4064 was changed into –COOH bioisosteres that has similar acidity such as a linker acylsulfonamide,31 its FXR agonism and potency correlation is still maintained, suggesting that use of an amide linker could be suitable for the probe. On the basis of this information, we placed a linear, rigid linker using hydrazine hydrate as an amide bridge that was coupled to the carboxyl moiety of GW4064.
After designing the linker, we introduced a fluorophore onto the probe scaffold. We chose fluorescein isothiocyanate (FITC) as the fluorophore because it is the most widely used fluorophore for labeling and sensing biomolecules,32 and specifically, it is reactive towards amino group of the linker connected to the GW4064. The chemical structure of final probe, which we named DY246, is shown in Figure 3.
Figure 3.

Synthesis of the novel fluorescent probe DY246 (compound 3) for the TR-FRET assay.
2.2. Synthesis of DY246 for TR-FRET assay
Our synthetic strategy started with commercially available GW4064. To prepare the carboxyl hydrazide, the carboxylic acid group of GW4064 was esterified with methyl bromoacetate to afford compound 1 in a quantitative yield. Compound 1 was subsequently treated with excess hydrazine hydrate in methanol to produce compound 2, which was crystallized from EtoAc at a 48% of yield. The fluorescent moiety (FITC) was added onto 2 in THF/EtOH, followed by purification with flash chromatography, producing the fluorescein adduct compound 3 as a yellowish-orange solid at a 31% yield. Compound 3 (DY246) was fully characterized by 1H NMR, 13C NMR, and HRMS. Thus, a synthetic procedure (Figure 3) to produce the FXR fluorescent probe DY246 has been achieved in a three-step process starting from GW4064 and led to 14% overall yield.
2.3. Components of the DY246-based TR-FRET assay
We used DY246 as a fluorescent probe (acceptor moleculae) in the TR-FRET assay, in which energy transfer can be detected by an increase in the fluorescence emission of the acceptor molecule and a decrease in the fluorescence emission of the donor molecule. In our TR-FRET assay, the components are Terbium (Tb) labeled-anti-GST antibody, DY246 and GST-hFXR-LBD. The established TR-FRET assay from Invitrogen uses a fluorescein labeled coactivator (fluorescein-SRC 2-2) as a fluorescent acceptor. The disadvantage of screening by established Invitrogen assay is that it narrows the range of ligands that can be identified. However, use of DY246 can overcome this disadvantage. For testing DY246, Tb in the Tb labeled anti-GST antibody was the donor species, and FITC in DY246 was the acceptor species. Excitation of the Tb-labeled anti-GST antibody by an energy source would trigger an energy transfer to DY246, which would then emit light at 520 nm. This energy transfer is distance dependent, and could only occur in the presence of GST-FXR, which brings the Tb-labeled anti-GST and DY246 into close proximity. The ratio between the donor and acceptor wavelengths was measured by instruments with TR-FRET detecting capacity, such as a PHERAstar plate reader.
2.4. Pre-screening characterization of DY246 as an FXR fluorescent ligand for TR-FRET binding assays
The new fluorescein labeled ligand DY246 had an EC50 value of 550 nM in a cell-based transfection assay. To assess the utility of DY246 as an hFXR fluorescent probe for TR-FRET binding assays, we determined the binding interactions between 10 nM DY246, 10 nM GST-hFXR-LBD, and 1.5 nM Tb-anti-GST in the presence or absence of various concentrations of GW4064 at 15, 20, and 30 min. Both total binding (with DMSO) and non-specific binding (with 5 µM GW4064) were relatively stable (Figure 4A) at 15 and 20 min and slightly decreased at 30 min. Correspondingly, the signal/background ratios (DMSO/5 µM GW4064 ratios, for which the 5 µM GW4064 signals were normalized to 1) had similar trends to the total binding and nonspecific binding signals (Figure 4B), with values of 9.52, 9.28, and 8.33 for 15, 20 and 30 min, respectively. The Z’-factor remained constant at 15, 20, and 30 min, with corresponding values of 0.75, 0.70, and 0.78 (Figure 4C). An acceptable assay typically has a Z’-factor value of more than 0.5.20 The EC50 values for GW4064 were constant for 15 and 20 min (317 and 373 nM, respectively) and then increased to 753 nM at 30 min (Figure 4D). The DY246-based hFXR TR-FRET assay data for Figure 4 was from assays using 20 µl/well, but assays using 30 µl/well had similar results (data not shown).
Figure 4.

Longitudinal signal stability of the interaction of DY246 with GST-FXR-LBD and Tb-anti-GST. (A) Interaction of 10 nM DY246 with 10 nM GST-FXR-LBD and 1.5 nM Tb-anti-GST at the indicated time points in the presence of DMSO or 5 µM GW4064. (B) Signal-to-background ratio of the interaction of the data shown in A. (C) Z’-factor values of the interaction of the data shown in A and B. The Z’-factor was calculated from the total binding signal (DMSO) and background signal (5 µM GW4064) by using equation 2 (see Experimental section). (D) GW4064 dose-response curves in the presence of 10 nM DY246 with 10 nM GST-FXR-LBD and 1.5 nM Tb-anti-GST at the indicated time points.
DY246 exhibited spectroscopic properties that were well-suited for TR-FRET. The constant EC50 values for GW4064 in the DY246-based FXR TR-FRET assays suggest that a short incubation time, such as 15 or 20 min, is preferred, which is also compatible with HTS because a shorter incubation time contributes to higher throughput. In an automated large-scale screen, which would use scheduling software, the signal from each plate would be measured exactly at a pre-set incubation time, and therefore there should not be variation on incubation time between plates. We chose to use a 20 min incubation time for further HTS because this incubation time best fits the automation system. For the same reason, we used a 20 µl/well assay volume for the primary HTS and a 30 µl/well assay volume for the following high throughput dose response screening. Another FXR agonist fexaramine was used instead of GW4064 to evaluate the DY246-based TR-FRET assay. Similar high Z’ value (0.82) was obtained (Figure 5). Taken together, these results indicate that the DY246-based TR-FRET assay is suitable for HTS to evaluate a variety of FXR ligands that have differing affinities.
Figure 5. TR-FRET assay for identification of FXR ligands.

The assay was performed with the FXR probe ligand DY246 (GW-FL) alone giving high signal output. However, signal was lost in the presence of unlabeled competitor ligand Fexaramine. The assay was repeated 22 times yielding a calculated Z’-factor of 0.82 indicating an excellent assay.
2.5. HTS to identify FXR ligands
Current screening strategies for FXR (and many other nuclear receptors) use TR-FRET, AlphaScreen or other homogeneous assays to detect ligand-induced interactions between receptors and small coregulator peptides. A “hit” is identified when the test compound promotes or disrupts the receptor-peptide interaction. Antagonists can be identified when the assay, typically a cell-based assay, is set under antagonistic mode in which the assay system includes a putative agonist such that the assay readout will be high in the absence of an antagonist and low in the presence of an antagonist. Co-activator recruitment assays might have the advantage of detecting more ligands, but cannot distinguish whether the ligands directly bind to the LBD. Although these assays are useful, they are biased in that they rely on interactions with a single coregulator peptide. Therefore, by design, this strategy is inherently limited as it excludes the detection of selective FXR modulators that fail to recruit the particular peptide used in the assay. To overcome this obstacle we developed a TR-FRET assay that directly detects ligand binding without a bias for any downstream conformation change or biochemical activity. We subsequently used the DY246-based TR-FRET assay in a 384-well format to screen a collection of 5,600 bioactive compounds (3,200 unique compounds) representing drugs, drug candidates, and other molecules with characterized biological activities from a variety of sources, including MicroSource, Prestwick, and Sigma as previously described.22–23 For this screen, we used the putative hFXR agonist GW4064 (5 µM) and DMSO as positive and negative controls, respectively. The compounds were screened at 15µM. To exclude false-positive compounds that non-specifically interfered with the assay, including compounds that self-absorb fluorescence, we counter-screened by replacing GST-FXR+DY246 with a GST-FXR-Fluorescein covalent conjugate.
We arbitrarily selected 50% inhibition as the hit criterion because we are interested in further characterizing relatively potent compounds (the screening concentration was 15 µM). Using this criterion, the screen identified 249 chemicals (a hit rate of 249/5600 = 4.44%), of which 193 were unique chemical entities (an actual hit rate of 193/3200 = 6.03%). The 249 compounds were then tested in a dose-responsive analysis with the TR-FRET assay, using compound concentrations ranging from 1.2 nM-23.3 µM, in triplicate. Only compounds that displayed dose-responsive inhibitory activity were selected for further characterization. Among the 249 chemicals tested in the dose-responsive analysis, we selected 17 compounds for further follow- up (Table 1).
Table 1.
Summary of compounds identified by TR-FRET
| Compound | Name | Structure | clog Pa | FXR TR-FRET Activity | PXR Transactivation Activity | ||
|---|---|---|---|---|---|---|---|
| %Inhibitionb | IC50 (µM)c | %Activationd | EC50 (µM)e | ||||
| 1 | Felodipine | 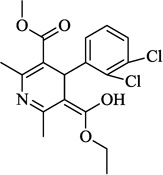 |
5.29 | 58.2 | 4.96 ± 1.4 | 30.6 ± 1.3 (40 µM) | NA |
| 2 | Thyoxine | 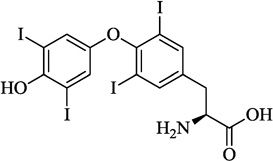 |
3.51 | 58.2 | 7.86 ± 1.7 | 13.9 ± 0.4 (40 µM) | NA |
| 3 | Fulvestrant | 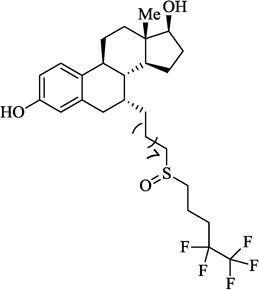 |
8.5 | 62.1 | 0.79 ± 0.28 | 102.8 ± 3.9 (20 µM) | 1.95 ± 0.01 |
| 4 | Flutrimazole | 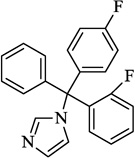 |
4.47 | 66.7 | 13.8 ± 4.9 | 120.4 ± 2.1 (40 µM) | 6.10 ± 0.16 |
| 5 | Nimodipine |  |
3.01 | 67.5 | 8.96 ± 1.95 | 125.9 ± 4.1 (2.5 µM) | 0.39 ± 0.01 |
| 6 | Sulconazole | 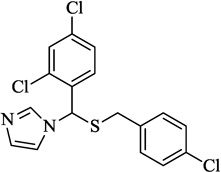 |
5.66 | 71.1 | 6.88 ± 1.59 | 50.0 ± 1.6 (10 µM) | NA |
| 7 | Raloxifene | 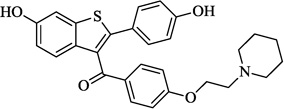 |
6.86 | 71.4 | 11.56 ± 2.99 | 81.5 ± 0.2 (20 µM) | 11.71 ± 0.33 |
| 8 | Loratadine | 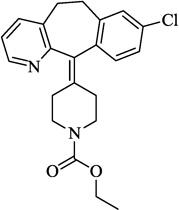 |
5.05 | 72.5 | 3.07 ± 0.76 | 57.4 ± 1.7 (10 µM) | 3.00 ± 0.05 |
| 9 | 6-HO-DL-DOPA | 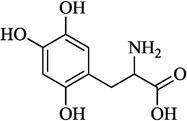 |
−3.53 | 73.0 | 7.92 ± 2.34 | Inactive | NA |
| 10 | JWH-015 | 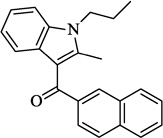 |
6.55 | 75.5 | 3.22 ± 0.85 | 125.0 ± 1.1 (40 µM) | 2.64 ± 0.12 |
| 11 | Ivermectin | 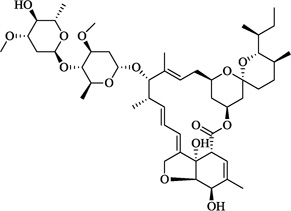 |
5.96 | 77.4 | 0.26 ± 0.02 | Inactive | NA |
| 12 | NBI 27914 | 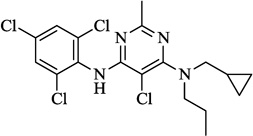 |
7.65 | 77.7 | 2.77 ± 1.00 | 114.8 ± 1.2 (20 µM) | 4.95 ± 0.46 |
| 13 | Clotrimazole | 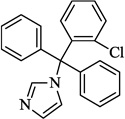 |
5.25 | 81.4 | 3.24 ± 0.87 | 35.6 ± 1.9 (10 µM) | NA |
| 14 | (R,R)-cis-Diethyl tetrahydro-2,8-chrysenediol |  |
6.11 | 81.9 | 1.31 ± 0.31 | 53.1 ± 2.8 (20 µM) | 8.60 ± 0.98 |
| 15 | GW7647 |  |
7.08 | 84.8 | 4.91 ± 0.92 | 25.9 ± 1.4 (40 µM) | NA |
| 16 | Palmitoyl-DL-Carnitine |  |
0.53 | 93.6 | 9.75 ± 1.86 | 40.7 ± 0.1 (40 µM) | NA |
| 17 | Bio |  |
4.27 | 96.3 | 3.4 ± 0.58 | 42.6 ± 1.5 (1.25 µM) | NA |
| Z-guggulsterone | 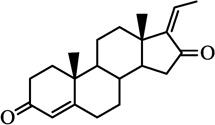 |
3.91 | NA | NA | 26.9 ± 0.6 (40 µM) | NA | |
| Lithocholic acid | 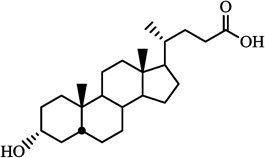 |
6.60 | NA | NA | 19.4 ± 1.4 (40 µM) | NA | |
log P values were calculated using Chemdraw.
The %Inhibition was calculated using the equation as described in section 4.2.4, for compound screened at 15 µM.
The IC50 value was derived as described in section 4.2.4. Values are presented as the means ± SE of three independent experiments. NA indicates no IC50 could be determined.
the %Activation was calculated using the equation described in section 4.2.5. The value shown in the table represents the maximum percent activation observed, and its corresponding compound concentration is noted in the parentheses. Values are presented as the means ± SE of three independent experiments.
The EC50 value was derived as described in section 4.2.5. Values are presented as the means ± SE of three independent experiments. NA indicates no EC50 could be determined.
2.6. Characterization of hits from HTS
The 17 selected compounds were then examined for their ability to act as antagonists of ligand activated FXR in cell-based transcription reporter assays (Figure 6). However, two of the 17 selected compounds, C16-carnitine and 6-OH-L-DOPA, are poorly permeable in cells that lack specific trans-membrane transporters.33 C16-carnitine (palmitoyl-DL-carnitine) was excluded from follow-up because of its poor cell permeability, while 6-OH-L-DOPA was included as an additional negative control. We also excluded (R,R)-cis-diethyl tetrahydro-2,8- chrysenediol) because it is toxic at high concentrations. The remaining compounds were tested for their ability to inhibit transactivation mediated by 8 µM CDCA. Compounds were tested at concentrations ranging from 3–30 µM, a range that reflects toxicity-limiting concentrations for certain compounds. Of the remaining 14 compounds, 13 inhibited transactivation by > 50%, and 9 of these inhibited transactivation by 75–100% (Figure 6). These compounds also displayed potent activity in the TR-FRET assay. For example, ivermectin revealed an IC50 of ~300 nM in the TR- FRET assay (Figure 7). The compounds that failed to inhibit transactivation by 50% or more in the cell-based reporter assay (T4, or thyroxine) only showed 54% inhibition in the TR-FRET assay. Taken together, these findings strongly suggest that use of the TR-FRET assay with a selection criterion of > 50% inhibition can accurately predict true biological activity.
Figure 6.

Abilities of compounds identified throught TR-FRET assay act as antagonists of ligand-activated FXR in cell-based reporter transcription assays. (A). Chemical structure of CDCA (chenodeoxycholic acid). (B). Inhibition of FXR/CDCA-mediated transactivation by the indicated compounds identified in the TR-FRET assay.
Figure 7.

Dose response studies for ivermectin in the TR-FRET assay.
The selected compounds were also tested for agonist activity relative to 100 µM CDCA in similar cell-based transcription reporter assays (Figure. 8). Compounds were tested at concentrations ranging from 3–30 µM, a range that reflects toxicity-limiting concentrations for certain compounds. Only one compound, nimodipine, had significant agonist activity. Because nimodipine also inhibited activation by 8 µM CDCA (Figure 6), this compound is best characterized as a weak or partial agonist. We noticed that although the vast majority of selected compounds acted as antagonists, only one acted as weak or partial agonist. The high hit rate of 6.03% for antagonists might be due to the selection of compound library. In contrast the hit rate for agonists was 0.4%, which is in line with the hit rate expected for screening nuclear receptors.18 These findings further suggest that our TR-FRET assay is appropriate for large-scale screening as it can successfully identify all ligands, regardless of whether they are agonists or antagonists.
Figure 8.

Agonist activity of compounds in transactivation assay.
2.7. A PXR transactivation assay
Pregnane X receptor (PXR) is a xenobiotic receptor that binds promiscuously to structurally diverse chemicals, including ligands for other nuclear receptors 23 Several FXR antagonists, including Z-guggulsterone and lithocholic acid, could be endowed with PXR agonistic activity.34–36 We used a PXR transactivation assay described previously23 to evaluate the 17 compounds identified by the TR-FRET assay, in addition to Z-guggulsterone and lithocholic acid, for their ability to activate PXR. In the assay, DMSO (0% activation) and 10 µM rifampicin (100% activation; rifampicin is a known PXR agonist37 was used as negative and positive control, respectively. As shown in Table 1, Z-guggulsterone and lithocholic acid showed moderate activity (27% and 19% maximum activation, respectively), which is consistent with previous observations.34–35 The 17 compounds identified by the TR-FRET displayed varying PXR agonistic activity, ranging from inactive (e.g., ivermectin) to very potent (e.g. 120% maximum activation and an EC50 of 6.1 µM for flutrimazole). Flutrimazole is a close analog of clotrimzole, and had not been previously reported as a PXR agonist. Clotrimazole, a known PXR agonist37 showed moderate PXR activity (36% maximum activation). Nimodipine and felodipine are known PXR agonists with potency comparable to rifampicin.38–39 We showed that felodipine had a maximum activation of 31%, and nimodipine had a maximum activation of 126% (EC50 of 0.4 µM). The other 13 compounds, thyoxine, fulvestrant, sulconazole, raloxifene, loratadine, 6-HO- DL-DOPA, JWH-015, ivermectin, NBI 27914, (R,R)-cis-diethyl tetrahydro-2,8-chrysenediol, GW7647, palmitoyl-DL-carnitine, and bio, have not been previously evaluated for their ability to activate PXR. These results are in agreement with previous observations for other FXR antagonists34–36 and also suggest that a PXR transactivation assay can be used to facilitate the selection of more specific FXR antagonists for further characterization.
Although the above findings demonstrate that the current assay and hit selection criteria are appropriate for large-scale screening, we have identified some areas that can be further improved to reduce the false positive rate. Specifically, we found that some compounds non-specifically inhibit TR-FRET to a certain degree when tested at 10 µM or higher. This can be seen when comparing inhibition of non-specific FRET (GST-fluorescein-TbAb, Figure 9) with inhibition of specific FRET (DY246-FXR-TbAb, Figure 10). For example, of the 17 compounds tested for non-specific inhibition, three produced ~50% inhibition [Bio, (R,R)-cis-diethyl tetrahydro-2,8-chrysenediol, and 6-OH-L-DOPA] (Figure 9). Initial dose response analysis indicated that, as expected, these non-specific effects were not seen at lower doses (e.g., 2 µM; data not shown). Nonetheless, it must be noted that when compared with specific inhibition of FXR, the nonspecific inhibition was much less significant (Figure 9 and Figure 10).
Figure 9.

Non-specific effect of compounds (10 µM) on non-specific TR-FRET (GST-Fluorescein)
Figure 10.

Specific effects of compounds (10 µM) on FXR-mediated TR-FRET by DY246.
These findings suggest the following:
Although the screen is acceptable as is, screening compound at 10–15 µM concentrations may produce weak non-specific inhibition for a minority of compounds. The extent of this inhibition can be as high as 50%, the cutoff used to define a hit. Therefore, it may be useful to screen at slightly lower concentration (5–7.5 µM) to further minimize nonspecific inhibition. A follow-up dose-response study should be helpful in establishing the optimal screening concentration.
It may also be useful to increase the selection criteria to >60 or 70% inhibition. This level of inhibition appears to be higher than any non-specific inhibition observed for any compound. The cutoff used to define a hit can be selected after the large-scale screen is completed, i.e., the larger the number of hits, the more stringent the selection criteria can be. Selection of the proper screening concentration and use of proper selection criteria will help minimize false-positives and false-negatives.
If any questions do arise as to the validity of certain compounds, the GST-Fluorescein counter screen provides a rapid and accurate approach to filter out false-positives, including compounds that self-absorb fluorescence. Although this option may not be needed, it is useful to know that a rapid and affordable counter screen exists to eliminate false-positives. Comparison of FXR inhibition vs. GST-fluorescein appears to be as accurate and predictive of true activity as the cell-based transcription reporter assay.
The non-specific activity that we observed against GST-fluorescein is likely to be a general problem for all assays that rely on Tb-fluorescein-based-TR-FRET. As time and resources permit, it may be useful to screen the compound library intended to be used for TR-FRET assay against GST-fluorescein. The resulting database may be helpful in minimizing false-negatives in a broad array of related screening projects.
3. Conclusions
FXR plays a critical role in the control of cholesterol, lipid and glucose metabolism, and has become a challenging drug target for the treatment of hyperlipidemia and cholestatic disease as well as atherosclerotic heart diseases. FXR antagonists raise the exciting possibility that they may be useful as therapeutic agents for these diseases. To facilitate the search for modulators of FXR activity, we developed an FXR binding assay based on TR-FRET. We developed and synthesized the novel fluorescently-labeled FXR probe DY246, and showed that it selectively bound to the ligand binding domain of FXR. This suggests it is a promising fluorescent FXR ligand suitable for TR-FRET assays directed at the discovery of novel FXR ligands, including antagonists. We demonstrated that the TR-FRET assay we reported here using DY246 offers an efficient primary screening tool for identifying FXR modulators. Use of DY246 in TR-FRET is expected to provide further insights into ligand binding mechanisms and to improve the specificity of therapeutics targeting FXR biological functions. Studies aimed at further in vitro profiling of FXR antagonists identified in our initial HTS and expanding our knowledge of their structure activity relationships (SAR), with an ultimate goal of developing orally active FXR antagonists in vivo, are under way.
4. Experimental section
4.1. Chemistry
General procedures
Organic reagents were purchased from commercial suppliers unless otherwise noted and were used without further purification. All solvents were analytical or reagent grade. All reactions were carried out in flame-dried glassware under argon or nitrogen. Melting points were determined and reported automatically by an optoelectronic sensor in open capillary tubes and were uncorrected. 1H NMR and 13C NMR spectra were measured at 500 MHz and 125 MHz, respectively, using CDCl3 or CD3OD as the solvents and tetramethylsilane (Me4Si) as the internal standard. Flash column chromatography was performed using Sigma-Aldrich silica gel 60 (200–400 mesh), carried out under moderate pressure with columns of an appropriate size packed and eluted with appropriate eluents. All reactions were monitored by thin layer chromatography (TLC ) on precoated plates (silica gel HLF). TLC spots were visualized either by exposure to iodine vapors or by irradiation with UV light. Organic solvents were removed under vacuum by a rotary evaporator.
4.1.1. (E)-2-methoxy-2-oxoethyl 3-(2-chloro-4-((3-(2,6-dichlorophenyl)-5-isopropylisoxazol-4-yl)methoxy)styryl)benzoate (1)
(E)-3-(2-chloro-4-((3-(2,6-dichlorophenyl)-5-isopropylisoxazol-4-yl)methoxy)styryl)benzoic acid (GW4064) (25 mg, 0.046 mmol) was dissolved in DMF (2 mL) and treated with methyl bromoacetate (15 mg, 0.1 mmol) and powder potassium carbonate (13 mg, 0.1 mmol). The resulting mixture was heated at 75 °C overnight. After cooling to room temperature, the reaction mixture was poured into 20 mL of saturated ammonium chloride solution and then extracted with EtOAc (3 × 20 mL). The organic extracts were washed with H2O (15 mL), followed by brine (2 × 15 mL), dried over Na2SO4, and concentrated under vacuum to give the desired ester as an oil (25 mg) that was used for the next step without further purification. 1H NMR (CDCl3) 7.58 (m, 1H), 7.52 (d, 2H), 7.39 (m, 4H), 7.33 (m, 2H), 6.92 (d, 1H), 6.78 (s, 1H), 6.77 (d, 1H), 4.88 (s, 2H), 4.72 (s, 2H), 3.77 (s, 3H), 3.35 (m, 1H), 1.44 (d, 6H).
4.1.2. (E)-3-(2-chloro-4-((3-(2,6-dichlorophenyl)-5-isopropylisoxazol-4-yl)methoxy) styryl)benzohydrazide (2)
To a solution of (E)-2-methoxy-2-oxoethyl 3-(2-chloro-4-((3-(2,6-dichlorophenyl)-5-isopropylisoxazol-4-yl)methoxy)styryl)benzoate (1) (25 mg, 0.04 mmol) in methanol (2 mL), hydrazine monohydrate (50 mg, 1 mmol) was added. The reaction mixture was stirred at room temperature for 36 h. Then organic solvent and excess hydrazine were removed in vacuo. The residue was poured into 10 mL cold water and stirred for 10 min. The formed solid was washed with cold water several times and dried under vacuum. Crystallization of the crude solid from warm EtOAc generated product 2 (10 mg, 48%) as a beige solid.
1H NMR (CDCl3) 7.64 (m, 3H), 7.33 (m, 2H), 7.30 (m, 4H), 6.94 (d, 1H), 6.77 (s, 1H), 6.57 (dd, 1H), 4.73 (s, 2H), 3.46 (m, 1H), 1.43 (d, 6H); 13C NMR (CDCl3) 176.4, 159.7, 159.0, 157.1, 142.2, 135.7, 134.2, 132.0, 131.4, 130.9, 128.7, 128.1, 127.7, 127.1, 124.4, 115.7, 113.7, 109.1, 59.6, 27.1, 25.5, 25.0.
4.1.3. (E)-5-(2-(3-(2-chloro-4-((3-(2,6-dichlorophenyl)-5-isopropylisoxazol-4-yl)methoxy) styryl)benzoyl)hydrazinecarbothioamido)-2-(6-hydroxy-3-oxo-3H-xanthen-9-yl)benzoate (3)
5 fluorescein isothiocyanate, isomer I (16 mg, 0.042 mmol) was added to a solution of product 2 (10 mg, 0.010 mmol) in EtOH/THF (3:2, 2 mL). The resulting orange reaction mixture was stirred at room temperature in the dark for 24 h. The reaction mixture was then concentrated under vacuum in the dark to give an orange semi-solid that was purified by flash chromatography (95:3:2 CH2Cl2/MeOH/AcOH, v/v) to generate the product 3 (5 mg, 30%) as a yellowish-orange solid. 1H NMR (MeOD) 8.31 (s, 1H), 7.91 (m, 1H), 7.82 (d, 1H), 7.77 (s, 1H), 7.47 (m, 2H), 7.42 (m, 2H), 7.35 (d, 2H), 7.12 (m, 2H), 6.96 (d, 1H), 6.68 (s, 1H), 6.63 (m, 6H), 6.56 (m, 5H), 4.75 (s, 2H), 3.35 (m, 1H), 1.38 (d, 6H); 13C NMR (MeOD) 188.5, 176.5, 174.6, 169.9, 160.7, 159.5, 157.3, 153.5, 145.9, 143.1, 140.6, 135.8, 134.0, 131.4, 128.6, 127.3, 125.8, 124.8, 123.9, 112.8, 110.3, 102.4, 59.3, 27.1, 19.5. HRMS (ESI); Calcd for C49H35Cl3N4O8S (M+1): 944.1242. Found: 944.1259.
4.2. Biology
4.2.1. Pre-screening characterization of DY246 as FXR fluorescent ligand in a TR-FRET binding assay
In a black 384-well plate (Corning Incorporated, Corning, NY), DMSO, GW4064 (5 µM) or serial dilutions of GW4064 (Tocris Bioscicnce, Minneapolis, MN) (5 µM to 2.54 nM, 1:3 titration at 10 concentration levels) were mixed with 10 nM GST-hFXR-LBD, 1.5 nM Tb-anti-GST antibody (Invitrogen, Carlsbad, CA), 10 nM DY246, and TR-FRET assay buffer (Invitrogen, Carlsbad, CA) in a reaction volume of 20 µl/well. The final DMSO concentration was 0.1% for all wells. The DMSO group and GW4064 (5 µM) group served as negative and positive controls, respectively. The plate was spun down after a brief shake and incubated under room temperature. The TR-FRET signal for each well was then collected, using an Envision plate reader with excitation wavelength of 340 nm and emission wavelengths of 520 nm and 490 nm at 15, 20 and 30 min. The 520 nm/490 nm ratio from each well was calculated. The Signal/Background ratio was defined as the ratio of negative control group (DMSO) against positive control group (5 µM GW4064). The Z’-factor values were calculated using the following equation:20
where σ+ is the standard deviation of the negative control (DMSO) group; σ− is the standard deviation of the positive control (5 µM GW4064) group; Mean+ is the mean of the negative control (DMSO) group; and Mean− is the mean of the positive control (5 µM GW4064) group. The 520 nm/490 nm ratios for dilutions of GW4064 were fit into a one-site competitive binding equation in a dose-dependent manner to derive the EC50 value for GW4064. Experiments were repeated in triplicate.
4.2.2. TR-FRET for detection of FXR ligand-binding activity
A TR-FRET kit (LanthaScreen™) from Invitorgen was used as per the vendor’s instructions. Terbium chelate (GST-FXR + terbium -anti-GST antibody) donor species was mixed with a fluorescein-tagged SRC2 derived peptide (LKEKHKILHRLLQDSSSPV) acceptor species. The TR-FRET value was determined as a ratio of the FRET-specific signal measured with a 520 nm filter to that of the signal measured with the terbium-specific 495 nm filter. The fluorescent signals were read in a time-resolved manner to reduce assay interference and increase data quality. The dose-response curve yielded an EC50 of ~4 µM for CDCA which is consistent with measurements form other assays.
4.2.3. Cell-Based Co-transfection Reporter Assay1
CV-1 cells were grown in Dulbecco’s modified Eagle’s medium supplemented with 10% resin charcoal–stripped fetal bovine serum, 50 U/mL penicillin G, and 50 µg/mL streptomycin sulfate (DMEM-FBS) at 37°C in 5% CO2. One day prior to transfection, cells were plated to 50%–80% confluence using phenol red free DMEM-FBS. Cells were transiently transfected as described.1 Reporter constructs (300 ng/105 cells) and cytomegalovirus-driven expression vectors (20–50 ng/105 cells) were added as indicated along with CMX-β-gal (500 ng/105 cells) as an internal control. After 2 h, the liposomes were removed and cells were treated for approximately 45 h with phenol red free DMEM-FBS containing either vehicle control (DMSO or the indicated compounds. After exposure to ligandindicated compound, the cells were harvested and assayed for luciferase and β-galactosidase activity. Results are expressed as the mean ± standard deviation of at least three independent experiments, and error bars indicate the standard deviation.
4.2.4. TR-FRET-Based hFXR HTS
In the primary TR-FRET hFXR HTS, 30 nl of 10 mM testing chemical was transferred with a pintool into 15 µl of a mixture of GST-hFXR and Tb-anti-GST in a 384-well black plate, and then 5 µl of 40 nM DY246 was dispensed to give a final volume of 20 µl/well with 10 nM GST-hFXR, 1.5 nM Tb-anti-GST, 15 µM testing chemical and 10 nM DY246. In addition, selected wells containing 5 µM GW4064 or DMSO were used as positive and negative controls, respectively. The final DMSO concentration was 0.15% in all wells. The plates were then spun down after a brief shake and incubated for 20 min at room temperature. The TR-FRET signal was then collected for each well with an Envison plate reader using an excitation wavelength of 340 nm and emission wavelengths of 520 nm and 490 nm. The 520 nm/490 nm ratio from each well was calculated and employed for the %Inhibition calculation. The %Inhibition for each well was calculated using the following equation:
In the dose response TR-FRET hFXR HTS, the general protocol used for the primary screening was followed with minor modification. Briefly, 70 nl of titrated testing chemical was transferred with a pintool into 20 µl of a mixture of GST-hFXR and Tb-anti-GST in a 384-well black plate, and then 10 µl of 30 nM DY246 was dispensed to give a final volume of 30 µl/well with 10 nM GST-hFXR, 1.5 nM Tb-anti-GST, 1-to-3 titrated testing chemical from 23.3 µM to 1.2 nM and 10 nM DY246. Selected wells containing 5 µM GW4064 or DMSO were again used as positive and negative controls, respectively. The final DMSO concentration was 0.23% in all wells. The plates were then spun down after a brief shake and incubated for 20 min at room temperature.
The TR-FRET signal was then collected for each well using an Envison plate reader with excitation wavelength of 340 nm and emission wavelengths of 520 nm and 490 nm. The 520 nm/490 nm ratio from each well was calculated and used for the %Inhibition calculation. The %Inhibition for each well was calculated using the above mentioned equation. The %Inhibitions at different concentrations for each compound were then imported into GraphPad Prism and IC50 values for individual chemicals were subsequently derived.
4.2.5. PXR Transactivation assay
The PXR transactivation assay was performed as previously described23. HepG2 stably expressing PXR and the PXR-regulated CYP3A4-luc luciferase reporter constructs were maintained in Dulbecco’s Modified Eagle Medium (DMEM) (Invitrogen, Madison, WI) supplemented with 10% fetal bovine serum (Hyclone, Logan, UT) and 400 µg/mL of G418 (Invitrogen). Compound or diluted DMSO (10 µL/well) and cells (5,000/well in 20 µL), both in assay medium [phenol red-free DMEM (Invitrogen) supplemented with 5% charcoal/dextran-treated fetal bovine serum (Hyclone, Logan, UT), L-glutamine and sodium pyruvate (Invitrogen)] were mixed and seeded into white tissue culture-treated 384-well plates (PerkinElmer, Waltham, MA). Final DMSO concentration was 0.4%. Final compound concentrations of each titrated testing chemical range from 40 µM to 19.5 nM (1:2 titration, 12 concentrations). DMSO or 10 µM rifampicin was used as negative (0%) or positive (100%) control, respectively. Luciferase assay was performed using SteadyLite HTS reagent (PerkinElmer) after 16 h of incubation. Luminescence signals were collected using an Envision plate reader (PerkinElmer). The data were expressed as a percentage of activation [%Activation = 100% × (compound signal – DMSO signal)/(10 µM rifampicin signal – DMSO signal)]. For compound with maximum %Activation > 50%, EC50 value was determined by using GraphPad PRISM 6 (GraphPad Software, La Jolla, CA).
DMSO, rifampicin, nimodipine, L-thyroxine, JWH-015, clotrimazole, Bio, fulvestrant, 6-hydroxy-DL-DOPA, ivermectin, NBI 27914, palmitoyl-DL-carnitine chloride and lithocholic acid were obtained from Sigma (St. Louis, MO). Felodipine, flutrimazole, sulconazole nitrate, raloxifene hydrochloride, loratadine, (R,R)-cis-Diethyl tetrahydro-2,8-chrysenediol and Z-guggulsterone were obtained from Santa Cruz Biotechnology (Dallas, TX). GW7647 was obtained from Tocris bioscience (Minneapolis, MN).
Acknowledgments
We gratefully thank Dr. Arthur D. Riggs for helpful discussion, support, and encouragement. We also thank Ramani Ravirala for technical assistance and Keely Walker for editing the manuscript. This work was supported by the American Lebanese Syrian Associated Charities (ALSAC), St. Jude Children’s Research Hospital (St. Jude), National Institutes of Health National Institute of General Medical Sciences [Grant GM086415], and National Institutes of Health National Cancer Institute [Grant P30-CA21765].
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Forman BM, Goode E, Chen J, Oro AE, Bradley DJ, Perlmann T, Noonan DJ, Burka LT, McMorris T, Lamph WW, Evans RM, Weinberger C. Cell. 1995;81:687–693. doi: 10.1016/0092-8674(95)90530-8. [DOI] [PubMed] [Google Scholar]
- 2.Wang H, Chen J, Hollister K, Sower LC, Forman BM. Mol. Cell. 1999;3:543–553. doi: 10.1016/s1097-2765(00)80348-2. [DOI] [PubMed] [Google Scholar]
- 3.Wang WD, Chen YD, Zhang L, Shiah S, Wang M, Yang F, Yu D, Forman BM, Huang W. Hepatology. 2010;51:953–962. doi: 10.1002/hep.23390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bishop-Bailey D, Walsh DT, Warner TD. Proc. Natl. Acad. Sci. U.S.A. 2004;101:3668–3673. doi: 10.1073/pnas.0400046101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fiorucci S, Mencarelli A, Palladino G, Cipriani S. Trends Pharmacol. Sci. 2009;30:570–580. doi: 10.1016/j.tips.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 6.Fiorucci S, Mencarelli A, Distrutti E, Palladino G, Cipriani S. Curr. Med. Chem. 2010;17:139–159. doi: 10.2174/092986710790112666. [DOI] [PubMed] [Google Scholar]
- 7.Yu D, Mattern DL, Forman BM. Steroids. 2012;77:1335–1338. doi: 10.1016/j.steroids.2012.09.002. [DOI] [PubMed] [Google Scholar]
- 8.Lambert G, Amar MJA, Guo G, Brewer HB, Jr., Gonzalez FJ, Sina CJ. J. Bio.l Chem. 2003;278:2563–2570. doi: 10.1074/jbc.M209525200. [DOI] [PubMed] [Google Scholar]
- 9.Kainuma M, Makishima M, Hashimoto Y, Miyachi H. Bioorganic & Medicinal Chemistry. 2007;15:2587–2600. doi: 10.1016/j.bmc.2007.01.046. [DOI] [PubMed] [Google Scholar]
- 10.Urizar NL, Moore DD. Annu. Rev. Nutr. 2003;23:303–313. doi: 10.1146/annurev.nutr.23.011702.073102. [DOI] [PubMed] [Google Scholar]
- 11.Pellicciari R, Costantino G, Fiorucci S. J. Med. Chem. 2005;48:5383–5403. doi: 10.1021/jm0582221. [DOI] [PubMed] [Google Scholar]
- 12.Cui J, Huang L, Zhao A, Lew JL, Sahoo S, Meinke PT. J. Biol. Chem. 2003;278:10214–10220. doi: 10.1074/jbc.M209323200. [DOI] [PubMed] [Google Scholar]
- 13.Sepe V, Bifulco G, Renga B, D’Amore C, Fiorucci S, Zampella A. J. Med. Chem. 2011;54:1314–1320. doi: 10.1021/jm101336m. [DOI] [PubMed] [Google Scholar]
- 14.Renga1 B, Mencarelli A, D’Amore C, Cipriani S, D’Auria MV, Sepe V, Chini MG, Monti MC, Bifulco G, Zampella A, Fiorucci S. PLoS ONE. 2012;7:e30443. doi: 10.1371/journal.pone.0030443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dussault I, Beard R, Lin M, Hollister K, Chen J, Xiao JH, Chandraratna R, Forman BM. J. Biol. Chem. 2003;278:7027–7033. doi: 10.1074/jbc.M209863200. [DOI] [PubMed] [Google Scholar]
- 16.Huang H, Yu Y, Gao Z, Zhang Y, Li C, Xu X, Jin H, Yan W, Ma R, Zhu J, Shen X, Jiang H, Chen L, Li J. J. Med. Chem. 2012;55:7037–7053. doi: 10.1021/jm3002718. [DOI] [PubMed] [Google Scholar]
- 17.Overington JP, Al-Lazikani B, Hopkins AL. Nat Rev Drug Discov. 2006;5:993–996. doi: 10.1038/nrd2199. [DOI] [PubMed] [Google Scholar]
- 18.Schulman IG, Heyman RA. Chem Biol. 2004;11:639–646. doi: 10.1016/j.chembiol.2003.12.021. [DOI] [PubMed] [Google Scholar]
- 19.Han K, Kim JH, Kim K, Kim EE, Seo J, Yang EG. Anal. Biochem. 2010;398:185–190. doi: 10.1016/j.ab.2009.11.008. [DOI] [PubMed] [Google Scholar]
- 20.Zhang JH, Chung TD, Oldenburg KR. J. Biomol. Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 21.Jeyakumar M, Webb P, Baxter JD, Scanlan TS, Katzenellenbogen JA. Biochemistry. 2008;47:7465–7476. doi: 10.1021/bi800393u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leonardi R, Zhang YM, Yun MK, Zhou R, Zeng FY, Lin W, Cui J, Chen T, Rock CO, White SW, Jackowski S. Chem. Biol. 2010;17:892–902. doi: 10.1016/j.chembiol.2010.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lin W, Wu J, Dong H, Bouck D, Zeng FY, Chen T. J. Biol. Chem. 2008;283:30650–30657. doi: 10.1074/jbc.M806132200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maloney PR, Parks DJ, Haffner CD, Fivush AM, Chandra G, Plunket KD, Greech KL, Moore PR, Wilson JG, Lewis MC, Jones SA, Willson TM. J. Med. Chem. 2000;43:2971–2974. doi: 10.1021/jm0002127. [DOI] [PubMed] [Google Scholar]
- 25.Nicolaou KC, Evans RM, Roecker AJ, Hughes R, Downes M, Pfefferkorn JA. Org. Biomol. Chem. 2003;1:908–920. [PubMed] [Google Scholar]
- 26.Akwabi-Ameyaw A, Bass JY, Caldwell RD, Caravella JA, Chen L, Creech KL, Deaton DN, Jones SA, Kaldor I, Liu Y, Madauss KP, Marr HB, McFadyen RB, Miller AB, Navas III F, Parks DJ, Spearing PK, Todd D, Williams SP, Wisely GB. Bioorg. Med. Chem. Lett. 2008;18:4339–4343. doi: 10.1016/j.bmcl.2008.06.073. [DOI] [PubMed] [Google Scholar]
- 27.Bass JY, Caldwell RD, Caravella Justin A, Chen L, Creech KL, Deaton DN, Madauss KP, Marr HB, McFadyen RB, Miller AB, Parks DJ, Todd D, Williams SP, Wisely GB. Bioorg. Med. Chem. Lett. 2009;19:2969–2973. doi: 10.1016/j.bmcl.2009.04.047. [DOI] [PubMed] [Google Scholar]
- 28.Mi L-Z, Devarakonda S, Harp JM, Han Q, Pellicciari R, Willson TM, Khorasanizadeh S, Rastinejad F. Mol. Cell. 2003;11:1093. doi: 10.1016/s1097-2765(03)00112-6. [DOI] [PubMed] [Google Scholar]
- 29.Pellicciari R, Gioiello A, Costantino G, Sadeghpour BM, Rizzo G, Meyer U, Parks DJ, Entrena-Guadix A, Fiorucci S. J. Med. Chem. 2006;49:4208. doi: 10.1021/jm060294k. [DOI] [PubMed] [Google Scholar]
- 30.Bass JY, Caldwell RD, Caravella JA, Chen L, Creech KL, Deaton DN, Madauss KP, Marr HB, McFadyen RB, Miller AB, Parks DJ, Todd D, Williams SP, Wisely GB. Bioorg. Med. Chem. Lett. 2009;19:2969. doi: 10.1016/j.bmcl.2009.04.047. [DOI] [PubMed] [Google Scholar]
- 31.Abel U, Schlüter T, Schulz A, Hambruch E, Steeneck C, Hornberger M, Hoffmann T, Perović-Ottstadt S, Kinzel O, Burnet M, Deuschle U, Kremoser C. Bioorg. Med. Chem. Lett. 2010;16:4911–4917. doi: 10.1016/j.bmcl.2010.06.084. [DOI] [PubMed] [Google Scholar]
- 32.Kojima H, Urano Y, Kikuchi K, Higuchi T, Nagano T. Angew. Chem. Int. Ed. 1999;38:2899–2901. doi: 10.1002/(sici)1521-3773(19991004)38:19<2899::aid-anie2899>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 33.Alavijeh MS, Chishty M, Qaiser MZ, Palmer AM. NeuroRx. 2005;2:554–571. doi: 10.1602/neurorx.2.4.554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ding X, Staudinger JL. J. Pharmacol. Exp. Ther. 2005;314:120–127. doi: 10.1124/jpet.105.085225. [DOI] [PubMed] [Google Scholar]
- 35.Staudinger JL, Goodwin B, Jones SA, Hawkins-Brown D, MacKenzie KI, LaTour A, Liu Y, Klaassen CD, Brown KK, Reinhard J, Willson TM, Koller BH, Kliewer SA. Proc. Natl. Acad. Sci. U.S.A. 2001;98:3369–3374. doi: 10.1073/pnas.051551698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sepe V, Ummarino R, D'Auria MV, Chini MG, Bifulco G, Renga B, D'Amore C, Debitus C, Fiorucci S, Zampella A. J. Med. Chem. 2012;12:84–93. doi: 10.1021/jm201004p. [DOI] [PubMed] [Google Scholar]
- 37.Lehmann JM, McKee DD, Watson MA, Willson TM, Moore JT, Kliewer SA. J. Clin. Invest. 1998;102:1016–1023. doi: 10.1172/JCI3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pan Y, Li L, Kim G, Ekins S, Wang H, Swaan PW. Drug. Metab. Dispos. 2011;39:337–344. doi: 10.1124/dmd.110.035808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xiao L, Nickbarg E, Wang W, Thomas A, Ziebell M, Prosise WW, Lesburg CA, Taremi SS, Gerlach VL, Le HV, Cheng KC. Biochem. Pharmacol. 2011;81:669–679. doi: 10.1016/j.bcp.2010.12.003. [DOI] [PubMed] [Google Scholar]