

Abstract
While a number of methods exist for the production of N-methyl amino acid derivatives, the methods for the production of N-methyl cysteine (MeCys) derivatives are suboptimal as they either have low yields or lead to significant sulfhydryl deprotection during the synthetic protocol. This paper focuses on the generation of MeCys and its subsequent use in Fmoc solid-phase peptide synthesis for the generation of N-methyl cystine containing peptides. Various methods for amino methylation of cysteine, in the presence of acid labile or acid stable sulfhydryl protecting groups, are compared and contrasted. Production of MeCys is best attained through formation of an oxazolidinone precursor obtained via cyclization of Fmoc-Cys(StBu)-OH. Following oxazolidinone ring opening, iminium ion reduction generates Fmoc-MeCys(StBu)-OH with an overall yield of 91%. The key to this procedure is using an electronically neutral Cys-derivative, as other polar Cys-derivatives gave poor results using the oxazolidinone procedure. Subsequently, the Fmoc-MeCys(StBu)-OH building block was used to replace a Cys residue with a MeCys residue in two peptide fragments that correspond to the active sites of glutaredoxin and thioredoxin reductase. The examples used here highlight the use of a MeCys(StBu) derivative, which allows for facile on-resin conversion to a MeCys(5-Npys) residue that can be subsequently used for intramolecular disulfide bond formation with concomitant cleavage of the peptide from the solid support.
Introduction
The construction of N-methyl amino acid building blocks has received much attention in the areas of medicinal chemistry and peptide chemistry due to their presence in an array of natural bioactive molecules [1-3] and the unique chemical and physical properties that N-methylation confers to the peptide [4-6], especially perturbations in backbone geometry [7,8]. As a result of their limited commercial availability [9], a number of synthetic strategies exist for their production which include; 1) reduction of oxazolidinones, oxazinanes, and thiazolidines, 2) direct methylation mediated by silver oxide, sodium hydride, sterically hindered organic bases, diazomethane, or potassium carbonate, 3) reductive amination involving O'Donnell's, or other Schiff bases, 4) reductive elimination of N-methylols, 5) amino substitution of α-bromo carboxylic acids, 6) Mitsunobu alkylation, and 7) retro aza-Diels-Alder reactions [10,11]. While as a whole these methods are well suited for the production of a number of N-methyl amino acid derivatives, they are less suitable for the generation of N-methyl cysteine (MeCys 2, Figure 1) [10,11]. The procedures that do exist for the production of MeCys are fraught with low overall yields, difficult reaction conditions, or multiple step procedures that often require the use of amino and/or sulfhydryl protecting groups that are not easily amendable to Fmoc solid-phase peptide synthesis. Our interest in MeCys lies in the development of small molecule mimics of cyclocystine, a vicinal cysteine motif that upon oxidation forms an eight-membered disulfide-containing ring [12]. This paper will focus on our efforts to generate MeCys and its use in the construction of peptide fragments that corresponds to the active sites of glutaredoxin and thioredoxin reductase with an added MeCys as part of the cystine ring structure (3 and 4 respectively, Figure 1).
Figure 1.

Retrosynthetic analysis for construction of glutaredoxin and thioredoxin reductase peptide fragments containing MeCys as part of a half-cystinyl residue.
Materials and Methods
Materials
Resins for solid-phase synthesis were purchased from Applied Biosystems (Foster City, CA) and Novabiochem (San Diego, CA). N-α-Fmoc amino acids and HBTU were purchased from Synpep Corp (Dublin, CA). All other chemicals were purchased from either Sigma-Aldrich (Milwaukee, WI) or Fisher Scientific (Pittsburgh, PA) and were used as received or purified by standard procedures [13].
Methods
Reactions employed oven-dried glassware under argon unless otherwise noted. Argon was passed through a column of anhydrous CaSO4 before use. Reactions were monitored by thin layer chromatography (TLC) using glass 0.25 mm silica gel plates with UV indicator. Flash chromatography was performed using columns packed with 230-400 mesh silica gel as a slurry in the elution solvent, unless otherwise noted. Gradient flash chromatography was conducted by adsorption of product mixture onto silica gel, packing onto a fresh silica bed as a slurry in minimal hexanes and eluting with a continuous gradient as noted in parentheses. Reverse-phase high performance liquid chromatography (RP-HPLC) was executed using a Shimadzu VP system with a Symmetry® C18-5 μm column from Waters (4.6 × 150 mm) for analytical analysis and a SymmetryPrep™ C18-7μm column from Waters (19 × 150 mm) for prepatory separation. Melting points were determined on a Meltemp apparatus and are uncorrected. Proton and carbon NMR data were obtained with a Varian or Bruker ARX 500 spectrometer. Chemical shifts for 1H-NMR and 13C-NMR are reported in parts per million (ppm) relative to tetramethylsilane (δ = 0.00 ppm) for 1H-NMR or chloroform-d (δ = 77.0 ppm) for 13C-NMR. Infrared spectra were recorded with a Perkin-Elmer 2000 FT-IR spectrophotometer. Low-resolution mass spectra were obtained with a Hewlett Packard 5988 GCMS. High-resolution mass spectra were performed by the University of South Carolina Mass Spectrometry Laboratory. A Voyager-DE™ PRO Workstation (Applied Biosystems) was used for mass spectral analysis of peptide samples. Peptides for this study were synthesized on a Symphony™ Multiple peptide synthesizer (Protein Technologies Inc., Tuscon, AZ) via Fmoc protocol, utilizing 2-chlorotrityl chloride or PAL-PEG-Fmoc resin as the solid support. Double coupling using standard HBTU activation was employed for peptide elongation. A typical on-resin coupling procedure is as follows: 20% piperidine/DMF (2 × 10 min); DMF washes (6 × 30 sec); 5 eq. Fmoc amino acid and HBTU in 0.4 M NMM/DMF (2 × 30 min); DMF washes (3 × 30 sec). Cleavage of peptides from the solid support was accomplished through treatment of the dried resin with 96:2:2 TFA/TIS/H2O for 1 h. Following filtration and washing of the resin (2 × 3 mL DCM), the cleavage supernatant was evaporated to one fifth its original volume in a stream of nitrogen, followed by precipitation into cold anhydrous diethyl ether.
Synthesis of Oxazolidinone (28)
To a room temperature solution of 10.0 g (23.1 mmol) Fmoc-Cys(StBu)-OH in 230 mL PhH under natural atmosphere was added 3.59 g (155.1 g:1 mmol) p-formaldehyde and 0.37 g (7 mol%) camphorsulfonic acid. The reaction was heated to reflux (oil-bath=100 °C) in a Dean-Stark apparatus and stirred for 14 h. The reaction was concentrated to provide a clear oil. After impregnation onto flash silica the oxazolidinone was purified via gradient flash-chromatography (hexanes; 10:1; 4:1; 1:1; EtOAc) to provide 9.63 g (94%) 28 as a white viscous gum. (28): Rf=0.71 (EtOAc); = −13.6 (c 0.23, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.76 (d, J = 7.6 Hz, 2 H), 7.55 (d, J = 7.6 Hz, 2 H), 7.40 (t, J = 7.6 Hz, 2 H), 7.31 (m, 2 H), 5.5-5.2 (br m's, 2 H), 4.75-4.35 (br m's, 2 H), 4.24 (br s, 1 H), 4.01 (br s, 1 H), 3.53 (br s, 0.5 H), 3.20 (br s, 0.5 H), 2.97 (br s, 0.5 H), 2.66 (br s, 0.5 H), 1.26 (s, 9 H); 13C NMR (125 MHz, CDCl3) δ 170.8 (C), 152.2 (C), 143.4 (C), 141.4 (C), 127.9 (CH), 127.2 (CH), 124.6 (CH), 120.0 (CH), 78.4 (CH2), 73.9 (CH2), 67.6 (CH), 55.3 (C), 48.2 (CH), 47.2 (CH2), 29.6 (CH3); IR (neat) 1800 (s), 1714 (s), 1417 (s), 1288 (m), 1129 (m), 1052 (m), 910 (w), 737 (m) cm−1; HRMS (EI) m/z 446.1457 [(M+), calcd. for C23H27NO4S2: 446.1460].
Synthesis of Fmoc-MeCys(StBu)-OH (29)
To a room temperature stirring solution of 9.03 g (20.3 mmol) 28 in 101 mL CHCl3 under natural atmosphere was added 32.8 mL (203 mmol) TES followed by the rapid addition of 50 mL TFA. The reaction was stirred at room temperature for 16 h and then concentrated. The oil was dissolved in 150 mL DCM and concentrated. This was repeated three consecutive times. After impregnation onto flash silica, MeCys 29 was purified via gradient flash-chromatography (hexanes; 10:1; 4:1; 2:1; 1:1; EtOAc) to provide 8.05 g (89%) 29 as a white solid in a 1.5:1.0 conformer ratio. (29): Rf=0.32 (EtOAc); = −30.4 (c 0.33, CHCl3); 1H NMR (500 MHz, CDCl3) δ Major: 10.36 (br s, 1 H), 7.82 (m, 2 H), 7.69 (m, 2 H), 7.46 (m, 2 H), 7.39 (m, 2 H), 4.91 (dd, J = 10.5, 4.3 Hz, 1 H), 4.52 (m, 2 H), 4.36 (t, J = 7.09 Hz, 1 H), 3.47 (dd, J = 13.9, 4.3 Hz, 1 H), 3.25 (dd, J = 13.9, 10.7 Hz, 1 H), 3.13 (s, 3 H), 1.43 (s, 9 H); Minor: 10.36 (br s, 1 H), 7.81 (m, 2 H), 7.68 (m, 2 H), 7.46 (m, 2 H), 7.39 (m, 2 H), 4.86 (m, 1 H), 4.63 (dd, J = 10.5, 6.4 Hz, 1 H), 4.57 (m, 1 H), 4.32 (t, J = 6.0 Hz, 1 H), 3.33 (dd, J = 12.2, 5.5 Hz, 0.5 H), 3.23 (dd, J = 13.9, 4.7 Hz, 1 H), 3.06 (s, 3 H), 2.84 (dd, J = 13.7, 10.5 Hz, 0.5 H), 1.41 (s, 9 H); 13C NMR (125 MHz, CDCl3) δ Major: 174.4 (C), 156.5 (C), 143.5 (C), 141.0 (C), 127.4 (CH), 126.8 (CH), 124.8 (CH), 124.6 (CH), 67.9 (CH2), 59.5 (CH), 48.0 (C), 46.8 (CH), 38.8 (CH2), 33.4 (CH3), 29.7 (CH3); Minor: 174.3 (C), 156.1 (C), 143.6 (C), 141.0 (C), 127.2 (CH), 126.9 (CH), 124.9 (CH), 124.7 (CH), 67.8 (CH2), 58.5 (CH), 47.9 (C), 46.7 (CH), 39.0 (CH2), 32.7 (CH3), 29.7 (CH3); IR (neat) 3353 (w), 1702 (s), 1449 (s), 1399 (m), 1317 (s), 1165 (s), 1129 (m), 976 (w), 738 (s) cm−1; HRMS (EI) m/z 444.1308 [(M+), calcd. for C23H25NO4S2: 444.1303].
Synthesis of Heptamer Fragment of Glutaredoxin (3)
The peptide Ac-GC(Trt)PY[MeCys(StBu)]VR- NH2 was synthesized via standard Fmoc protocol from PAL-PEG-Fmoc resin (0.21 mmol/g). All amino acid residues were attached using the previously described standard Fmoc protocol with the following exceptions: Fmoc-[MeCys(StBu)]-OH was coupled using HATU activation (3 eq. HATU & amino acid) for 1 h. The following residue, Fmoc-Tyr(tBu)-OH, required double HATU coupling as described above. The additional cysteine residue in the sequence was protected as the triphenylmethyl (Trt) derivative. Following peptide elongation, α-Fmoc protection was removed via treatment of the resin with 20% piperidine/DMF and N-terminal acetylation was accomplished with Ac2O (1 mL) in 5 mL 0.1 M NMM/DMF for 40 min.
Synthesis of Octamer Fragment of Thioredoxin Reductase (4)
The peptide H-PTVTGC(Mob)[MeCys(StBu)]G-OH was synthesized via standard Fmoc protocol from 2-chlorotrityl chloride resin (1.30 mmol/g), in which a ∼5-fold excess of resin was treated with 40 μmol of Fmoc-Gly-OH in 0.1M NMM/DCM for 40 min. Following this initial loading step, the remaining attachment sites on the resin were capped via treatment with 1:0.5:8.5 MeOH/NMM/DCM for an additional 40 min. The remaining amino acid residues were attached using the previously described standard Fmoc protocol with the following exception: Fmoc-[MeCys(StBu)]-OH was coupled using HATU activation (3 eq. HATU and amino acid) for 1 h. The following residue, Fmoc-Cys(Mob)-OH, required double HATU coupling as described above. Following peptide elongation, α-Fmoc protection was converted to Boc via treatment of the resin with 20% piperidine/DMF, followed by incubation with Boc2O (10 eq.) in 5 mL 0.1 M NMM in 1:1 DMF/DCM for 40 min.
Tandem Resin Cleavage and Disulfide-bond Formation for MeCys Containing Glutaredoxin (3) and Thioredoxin Reductase(4)
Removal of the StBu protecting group from MeCys was accomplished by treating the resin-bound peptides with 20% βME in DMF (buffered to 0.1M in NMM) over 12 h. The resin was washed thoroughly with DMF and DCM, and the deprotected MeCys residue was functionalized and activated through treatment with 2,2′-dithiobis(5-nitropyridine) (DTNP) in DCM (2 × 5 eq; 30 min each) to form the corresponding p-nitropyridylsulfenyl (5-Npys) conjugate. The resin was then thoroughly washed with DCM and DMF in order to remove any residual DTNP. Formation of intramolecular disulfides was carried out concurrent with cleavage/deprotection in 98:2 TFA/H2O for 1 h. Following ether precipitation of the peptides, preparatory HPLC purification was carried out. In the case of the glutaredoxin fragment, full conversion to the desired oxidized structure required incubation of the crude isolate in 0.1% aq. TFA for 1 h before injection into the preparative HPLC.
Results and Discussion
Attempts at N-Methylation of Cysteine using Established Methods
The initial development of a viable procedure for the production of MeCys hinged on its incorporation into a peptide as part of a half-cystinyl residue. The original strategy for cystine construction was to generate a disulfide bond between Cys(Trt) and MeCys(Trt). After acidic deprotection to afford the dithiol, air oxidation to produce a disulfide-bond could then occur. As a result, our efforts began with the construction of MeCys(Trt) by using Cys 5 (Table 1). Classic Schiff base conditions required the carboxyl to be protected. Methyl esterification to produce 6 was non-problematic [14], however amino condensation either provided starting material 6 or dimethylated 8 depending if the amino-protected 6 or free amine 7 was utilized (Entries 1 and 2 respectively, Table 1). While the Schiff-base method has been used in the construction of MeCys(Trt), repeated attempts to repeat Hojo and Nakahara's conditions only provided 8[15]. A complimentary approach utilized O'Donnell's ketimine 9, which was produced in good yield after trans-imination [16]. Reductive amination also proceeded to generate ester 10 in good yield (results not shown), however removal of the dibenzylmethine was impossible under standard palladium-mediated hydrogenation with only starting material being moderately recovered (Entry 3, Table 1) [17]. The inability to remove this group is presumably a direct result of sulfur poisoning the catalyst. Direct methylation mediated either by silver oxide or anhydrous potassium carbonate had similar difficulties. Both acid (5 and 11) and ester (6 and 12) substrates, whether the sulfhydryl was protected with a Trt- or Mob-substituent, were inert to methylation and were recovered under a variety of experimental conditions (Entries 4-8, Table 1) [18]. These disheartening results using classical reductive amination and direct N-methylation conditions led to the use of another reductive amination protocol which proceeds via an oxazolidinone precursor.
Table 1. Schiff Base and Direct N-Methylations.
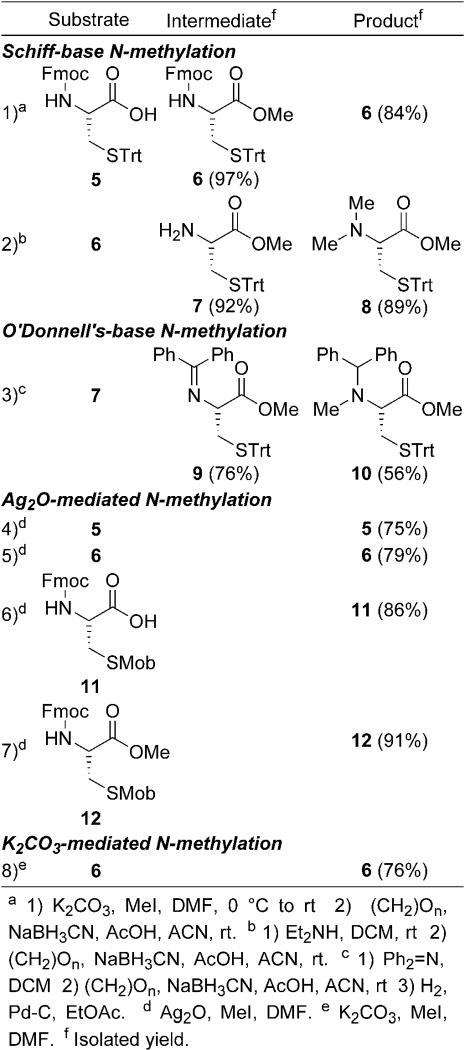
|
N-Methylations via Oxazolidinones
Originally developed by Ben-Ishai [19], and later improved upon by Freidinger (Table 2) [20], the oxazolidinone procedure seemed a suitable place to start anew due to the simple reaction conditions and ease of purification of the desired material from substrate and/or possible side-products. Due to its common place use in solid phase synthesis, Fmoc-Cys(Trt)-OH 5 was first subjected to N-methylation conditions via oxazolidinone 13. While formation of oxazolidinone 13 was non-problematic, the acidic conditions necessary for reductive installation of the N-methyl substituent provided only thiazolidine 14 (Entry 1, Table 2). Its production is a result of sulfur deprotection under the acidic reaction conditions with concomitant 5-endo-trig-cyclization upon iminium ion formation (Step 2, Scheme 1). With that in mind, a number of other cysteine derivatives, varying in sulfhydryl protection, were subjected to N-methylation via an oxazolidinone intermediate.
Table 2. N-Methylations via Oxazolidinone Intermediates.
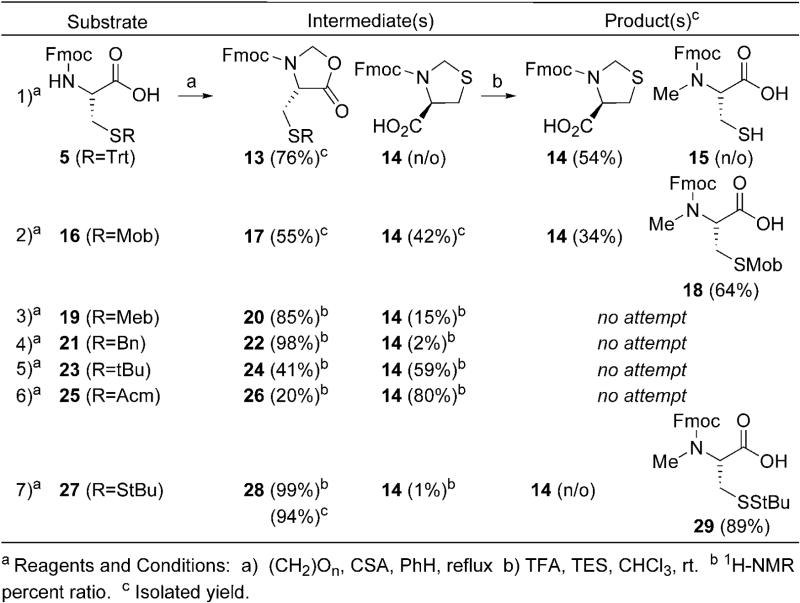
|
Scheme 1.

Proposed mechanism for MeCys formation.
The Mob-substituent was chosen first as a result of its ease of removal [21], as well as its stability to acidic conditions [22]. Unfortunately, upon oxazolidinone formation not only was the desired intermediate 17 formed but also thiazolidine 14 (Entry 2, Table 2). In the case of Cys(Trt) 5, thiazolidine 14 was not observed until acidic ring opening of oxazolidinone 13. However here the Mob-protected sulfur-atom captures the iminium ion prior to capture by the carboxyl during the oxazolidinone forming step (Step 1, Scheme 1). After isolation, treatment of oxazolidinone 17 with Freidinger's conditions did lead to the formation of Fmoc-MeCys(Mob)-OH 18 (Entry 2, Table 2). However, thiazolidine 14 was formed again. While the production of 18 was satisfactory, the generation of thiazolidine 14 during both steps diminished its overall appeal. This was coupled with difficulty storing 18 as well as using it in solid phase synthesis as a result of its propensity to decompose, which was also observed by Hughes [23].
Both Meb- and Bn-protected cysteines also formed thiazolidine 14, however in much lower amounts (Entries 3 and 4, Table 2). The degrees of thiazolidine 14 formation between Mob, Meb, and Bn are attributed to the electron-donating character of the para-substituent on the benzene ring. While both Meb and Bn did provide the desired oxazolidinones 20 and 22 respectively, the eventual difficult sulfhydryl deprotection of these groups led to investigating other sulfur-protected cysteines such as tBu 23 and Acm 25. Regrettably both Cys(tBu) and Cys(Acm) derivatives formed significant quantities of thiazolidine 14 (Entries 5 and 6, Table 1). In the case of the Cys(tBu) derivative the electron-donating tBu-substituent and 3°-cation produced upon tBu cleavage are responsible for thiazolidine formation. This was not observed with Trt-protected 5 due to the enhanced bulky character of the Trt-protecting group making sulfur-cyclization difficult. The pronounced formation of thiazolidine 14 with the Cys(Acm) derivative is a result of the Acm protecting group's lability to TFA in the presence of hydride donors such as was used in this procedure [24]. Clearly the electronics and acid lability of the sulfhydryl protecting group play a dominant role in the formation of undesired thiazolidine 14.
A suitable protecting group, one that is either electron-withdrawing or electronically neutral coupled with TFA stability and ease of deprotection post-synthesis, was still needed. This led to the investigation of StBu-protected cysteine 27 since this disulfide protecting group is primarily electronically neutral, stable to TFA, and can be removed easily by thiolysis. Oxazolidinone formation was superb and 28 was isolated in excellent yield (Entry 7, Table 2). Satisfyingly, acidic cleavage of oxazolidinone 28 provided MeCys(StBu) 29 in excellent isolated yield. Not only are the isolated (89%) and overall yields (91%) desirable, but also this two-step process is amenable to large-scale synthesis with Entry 7 being conducted on a 10 g scale (Table 2). Use of Cys(StBu) 27 for the generation of MeCys completes the study of Hughes and coworkers since thiazolidine formation is circumvented and the difficulties with decomposition observed in their study with sulfur-containing amino acids was not observed with this derivative[23]. Now with a viable large-scale synthesis of MeCys 29 elucidated, construction of MeCys substituted glutaredoxin and thioredoxin reductase peptide fragments 3 and 4, respectively, could be carried out.
Tandem Cleavage and Cyclization Protocol for Peptides Containing N-Methyl Cystine
The choice of the StBu protecting group for the production of MeCys is fortuitous since not only does the Cys(StBu) derivative give the best yields of oxazolidinone and the final protected derivative, but as illustrated in Scheme 2 on-resin thiolysis and conversion to a MeCys(5-Npys) residue facilitates facile intramolecular disulfide bond formation with concomitant cleavage of the peptide from the resin. Prior to cleavage from the solid support, the StBu group is removed upon treatment with β-mercaptoethanol and the resultant sulfhydryl is protected as a mixed disulfide with a 5-Npys group (32). During cleavage from the resin, the electron-withdrawing character of the 5-Npys-substituent facilitates disulfide-bond formation for production of the MeCys containing cystines 3 and 4. Mechanistically, cystine formation in the glutaredoxin peptide fragment proceeds after Trt-deprotection during the resin cleavage (33 to 34, Scheme 2). The unmasked sulfhydryl attacks the mixed disulfide bond of the MeCys(5-Npys) derivative, expelling 5-nitropyridinylthione and generating the desired cystine 3 after loss of a proton. A crude HPLC chromatogram of the resulting cystine-containing glutaredoxin fragment is shown in Figure 2A. As shown in the chromatogram, our chosen coupling conditions results in a peptide without any truncations evident and a largely pure peptide product. The major peak in the chromatogram corresponds to the peptide with an intramolecular disulfide-bond as revealed by MALDI-TOF analysis (Figure 2B). After purification by preparative HPLC and lyophilization the final yield of this peptide fragment was 36%.
Scheme 2.

Proposed mechanism for peptide resin cleavage with concomitant disulfide-bond formation. For the thioredoxin reductase fragment n = 0 and for the glutaredoxin peptide fragment n = 2.
Figure 2.

(A) HPLC chromatogram of the crude glutaredoxin peptide preparation containing MeCys. The major peak (labeled) is the desired oxidized peptide. The high purity of the product is evident from the chromatogram. The dark line is the absorbance at 214 nm and the grey dashed line is the absorbance at 254 nm. (B) MALDI-TOF mass spectral chromatogram of the desired oxidized peptide.
The MeCys(StBu) derivative can also be employed to isolate fully reduced peptide. In the case of the glutaredoxin fragment, the StBu group was removed by thiolysis on-resin as before, but the DTNP oxidation step was omitted. The yield of the fully reduced peptide was isolated after preparative HPLC and lyophilization was 50% (data not shown).
In the case of the thioredoxin reductase peptide fragment, it is the S-Mob thioether sulfur atom that attacks the mixed disulfide bond of the MeCys(5-Npys) group (instead of a naked sulfhydryl) to produce cystine 4 after the Mob-substituent is scavenged (Scheme 2) [21]. While disulfide bond formation via an activated Cys(5-Npys) residue and a free sulfhydryl is well established, the use of a Cys(Mob) thioether nucleophile for the formation of a disulfide-bond between vicinal cysteines has only recently been demonstrated by us [25]. The analytical HPLC chromatogram of the crude thioredoxin reductase peptide is shown in Figure 3A. While the major peak in the chromatogram corresponds to the intramolecular disulfide-containing peptide fragment as revealed by MALDI analysis (Figure 3B), the StBu group was somewhat resistant to thiolysis as shown by a significant amount of fully-protected peptide in the HPLC chromatogram. The vicinal context of the Cys(Mob) and MeCys(StBu) derivatives may be the cause of the reaction not going to completion as we have been able to carry out a similar reaction with vicinal Cys(Mob) and Cys(StBu) derivatives in higher yields previously [25]. The final yield of the isolated peptide fragment after preparative HPLC and lyophilization was 41%.
Figure 3.

(A) HPLC chromatogram of the crude thioredoxin reductase peptide preparation containing MeCys. There are two labeled peaks in the chromatogram with the largest peak corresponding to the desired oxidized vicinal disulfide containing peptide and the other significant peak corresponding to the fully protected peptide (both the Mob and StBu groups still attached to Cys and MeCys, respectively). The dark line is the absorbance at 214 nm and the grey dashed line is the absorbance at 254 nm. (B) MALDI-TOF mass spectral chromatogram of the desired oxidized peptide.
As discussed by Humphrey and Chamberlin, racemization during coupling of N-methyl amino acids can be a problem, especially if the α-nitrogem atom is protected by the Boc group [26]. In this study, the Fmoc protocol was employed and at least in the case of the thioredoxin reductase peptide fragment, racemization was not observed as evidenced by the single peak in the HPLC chromatogram (Figure 3A). It is possible that epimerization occurred during the synthesis of the glutaredoxin fragment since there are several small peaks evident in the chromatogram close to the main peak, however if epimerization has occurred it is only present to a small extent as observed in the chromatogram. It has been shown that long coupling times increase the likelihood of epimerization. In our case we adopted the methodology of Rich and coworkers [27], in which they relied on HOAt additives and a HATU uronium coupling agent for quantitative coupling efficiencies onto N-methyl amino acids without epimerization.
Conclusions
While a number of methods exist for the production of NMAs, none are optimal for the production of MeCys, and those that do are characterized by low yields, have cumbersome protocols, or utilize protecting groups that are not amenable to Fmoc solid phase peptide synthesis. Here we have demonstrated a viable multigram synthesis of Fmoc-MeCys(StBu)-OH 29 using oxazolidinone chemistry that can be easily used for the production of peptides via the Fmoc protocol. Production of StBu-protected MeCys has been highlighted by its subsequent use in generating unique disulfide bond containing peptide fragments of glutaredoxin and thioredoxin reductase containing MeCys as part of a half-cystine residue, replacing the normal cystine connectivity found in these peptide fragments. The presence of the MeCys residue did not impede oxidation to the disulfide using a standard method or our previously published procedure for vicinal disulfide bond formation [21,25]. Both examples used here highlight the use of a MeCys(StBu) derivative, which allows for facile on-resin conversion to a MeCys(5-Npys) residue that can be subsequently used for intramolecular disulfide bond formation with concomitant cleavage of the peptide from the solid support.
Supplementary Material
Acknowledgments
We wish to thank Ms. Katharine Harris for her previous contributions in our laboratory on disulfide bond forming reactions.
Footnotes
These studies were supported by National Institutes of Health Grants GM070742 to RJH.
References
- 1.DeGrado WF. Adv Protein Chem. 1988;39:51–124. doi: 10.1016/s0065-3233(08)60375-7. [DOI] [PubMed] [Google Scholar]
- 2.Nicolaou KC. J Med Chem. 2005;48:5613–5638. doi: 10.1021/jm050524f. [DOI] [PubMed] [Google Scholar]
- 3.Wilson RM, Danishefsky SJ. J Org Chem. 2006;71:8329–8351. doi: 10.1021/jo0610053. [DOI] [PubMed] [Google Scholar]
- 4.Fairlie DP, Abbenante G, March DR. Curr Med Chem. 1995;2:654–686. [Google Scholar]
- 5.Gordon DJ, Tappe R, Meredith SC. J Pept Res. 2002;60:37–55. doi: 10.1034/j.1399-3011.2002.11002.x. [DOI] [PubMed] [Google Scholar]
- 6.Kapurniotu A, Schmauder A, Tenidis K. J Mol Biol. 2002;315:339–350. doi: 10.1006/jmbi.2001.5244. [DOI] [PubMed] [Google Scholar]
- 7.Vitoux B, Aubry A, Cung MT, Marraud M. Int J Pept Protein Res. 1986;27:617–632. doi: 10.1111/j.1399-3011.1981.tb02016.x. [DOI] [PubMed] [Google Scholar]
- 8.Trzepałka E, Kowalczyk W, Lammek B. J Peptide Res. 2004;63:333–346. doi: 10.1111/j.1399-3011.2004.00103.x. [DOI] [PubMed] [Google Scholar]
- 9.Usually limited to glycine, alanine, leucine, iso-leucine, valine, and phenylalanine.
- 10.Aurelio L, Brownlee RTC, Hughes AB. Chem Rev. 2004;104:5823–5846. doi: 10.1021/cr030024z. [DOI] [PubMed] [Google Scholar]
- 11.Gilon C, Dechantsreiter MA, Burkhart F, Friedler A, Kessler H. In: Synthesis of Peptides and Peptidomimetics. Goodman M, Felix A, Moroder L, Toniolo C, editors. E22c, Chapter 10.1. Thieme; New York: 2004. pp. 215–271. [Google Scholar]
- 12.Ruggles EL, Hondal RJ. Tetrahedron Lett. 2006;47:4281–4284. doi: 10.1016/j.tetlet.2006.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Armarego WLF, Perrin DD. Purification of Laboratory Chemicals. 4th. Butterworth-Heinemann; Woburn, MA: 1998. [Google Scholar]
- 14.Garner P, Park JM. Organic Syntheses, Coll Vol 9. 1992;70:300–307. [Google Scholar]
- 15.Hojo H, Onuma Y, Akimoto Y, Nakahara Y, Nakahara Y. Tetrahedron Lett. 2007;48:25–28. [Google Scholar]
- 16.Polt R, Peterson MA, DeYoung L. J Org Chem. 1992;57:5469–5480. [Google Scholar]
- 17.Sames D, Polt R. J Org Chem. 1994;59:4596–4601. [Google Scholar]
- 18.Direct methylation using NaH/MeI is adequate for the construction of Boc-MeCys(Trt)-OH. See reference [10,11].
- 19.Ben-Ishai D. J Am Chem Soc. 1957;79:5736–538. [Google Scholar]
- 20.Freidinger RM, Hinkle JS, Perlow DS, Arison BH. J Org Chem. 1983;48:77–81. [Google Scholar]
- 21.Harris KM, Flemer S, Jr, Hondal RJ. J Pept Sci. 2007;13:81–93. doi: 10.1002/psc.795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wuts PGM, Greene TW. Greene's Protective Groups in Organic Synthesis. John Wiley & Sons, Inc.; New York: 2007. pp. 654–656. [Google Scholar]
- 23.Aurelio L, Box JS, Brownlee RTC, Hughes AB, Sleebs MM. J Org Chem. 2003;68:2652–2667. doi: 10.1021/jo026722l. [DOI] [PubMed] [Google Scholar]
- 24.Singh PR, Rajopadhye M, Clark SL, Williams NE. Tetrahedron Lett. 1996;37:4117–4120. [Google Scholar]
- 25.Flemer SJ, Lacey BM, Hondal RJ. J Pept Sci. 2007 doi: 10.1002/psc.961. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Humphrey JM, Chamberlin AR. Chem Rev. 1997;97:2243–2266. doi: 10.1021/cr950005s. [DOI] [PubMed] [Google Scholar]
- 27.Angell YM, Garcia-Echeverria C, Rich DH. Tetrahedron Lett. 1994;35:5981–5985. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.