

Abstract
Background
Acute lymphoblastic leukemia (ALL) is the most common cancer in children and the incidence of ALL varies by ethnicity. Although accumulating evidence indicates inherited predisposition to ALL, the genetic basis of ALL susceptibility in diverse ancestry has not been comprehensively examined.
Methods
We performed a multiethnic genome-wide association study in 1605 children with ALL and 6661 control subjects after adjusting for population structure, with validation in three replication series of 845 case subjects and 4316 control subjects. Association was tested by two-sided logistic regression.
Results
A novel ALL susceptibility locus at 10p12.31-12.2 (BMI1-PIP4K2A, rs7088318, P = 1.1×10−11) was identified in the genome-wide association study, with independent replication in European Americans, African Americans, and Hispanic Americans (P = .001, .009, and .04, respectively). Association was also validated at four known ALL susceptibility loci: ARID5B, IKZF1, CEBPE, and CDKN2A/2B. Associations at ARID5B, IKZF1, and BMI1-PIP4K2A variants were consistent across ethnicity, with multiple independent signals at IKZF1 and BMI1-PIP4K2A loci. The frequency of ARID5B and BMI1-PIP4K2A variants differed by ethnicity, in parallel with ethnic differences in ALL incidence. Suggestive evidence for modifying effects of age on genetic predisposition to ALL was also observed. ARID5B, IKZF1, CEBPE, and BMI1-PIP4K2A variants cumulatively conferred strong predisposition to ALL, with children carrying six to eight copies of risk alleles at a ninefold (95% confidence interval = 6.9 to 11.8) higher ALL risk relative to those carrying zero to one risk allele at these four single nucleotide polymorphisms.
Conclusions
These findings indicate strong associations between inherited genetic variation and ALL susceptibility in children and shed new light on ALL molecular etiology in diverse ancestry.
Acute lymphoblastic leukemia (ALL) is the most common childhood malignancy and a leading cause of death due to disease in children (1,2). A genetic basis of ALL susceptibility is supported by its association with certain congenital abnormalities (3,4) and, more recently, by genome-wide association studies (GWASs) identifying common variants at ARID5B (10q21.2), IKZF1 (7p12.2), and CEBPE (14q11.2) influencing ALL risk in children of European descent (5–9). In fact, the disease risk associated with these common variants are among the strongest in cancer susceptibility variants identified through GWASs (10), consistent with a relatively large impact of inherited genetic factors on the pathogenesis of this childhood malignancy (11). However, the loci reported in ALL GWASs thus far cumulatively accounted for only 8% of genetic variation in ALL risk (11), suggesting additional susceptibility variants yet to be discovered in larger studies.
There is an extreme lack of population diversity in GWASs such that 96% of subjects studied in GWASs so far are individuals of European descent (12,13). This exclusive focus on selected few ethnic groups raises a number of critical questions: For example, are findings from European-only GWASs transferable to other populations (14, 15)? Can disease etiology be different among populations and thus characterized by distinct genetic risk factors (16)? What is the contribution of ancestry-related genetic variation to ethnic differences in cancer prevalence (17,18)? These issues are of particularly relevance to childhood ALL because the incidence of ALL varies substantially by ethnicity (14.8 per million person-years in African Americans, 35.6 per million person-years in European Americans, and 40.9 per million person-years in Hispanic Americans) (19,20), at least partly attributable to population differences in inherited genetic variations [eg, ARID5B (21,22)].
To identify novel ALL susceptibility loci and also to evaluate the associations of known susceptibility variants in diverse populations, we examined 709059 single nucleotide polymorphisms (SNPs) for association with childhood ALL in a multiethnic GWAS of 1605 case subjects and 6661 control subjects, followed by three independent replications in 845 case subjects and 4316 control subjects of European American, African American, and Hispanic American ethnicity.
Methods
Subjects and Genotyping
Two nonoverlapping series of childhood ALL case subjects and control subjects were included: the GWAS series and the replication series. In the GWAS series, we included 1605 B-precursor childhood ALL case subjects enrolled on the Children’s Oncology Group (COG) P9904/P9905 protocols (23), and 6661 unrelated subjects from the Multi-Ethnic Study of Atherosclerosis (MESA) (dbGAP phs000209.v9) were considered as non-ALL control subjects because the prevalence of adult survivors of childhood ALL is less than 1 in 10 000 in the United States (5). The replication study included three case–control series separately by ethnicity (Supplementary Data, available online): European Americans: 574 case subjects and 2601 control subjects (24,25); African Americans: 128 case subjects and 1075 control subjects (26); Hispanic Americans: 143 case subjects and 640 control subjects (27). ALL case subjects in the replication series were from the St. Jude Total Therapy XIIIB/XV and the COG P9906 protocol (5). Ethnicity was defined by genetic ancestry as described below. ALL molecular subtypes included MLL rearrangements, ETV6-RUNX1, TCF3-PBX1, or BCR-ABL1, and hyperdiploid. Patients include in the genetic association analyses represented 85.3% (n = 1605 of 1882) of total enrolled participants on the COG P9904/9905 treatment protocols and 83.1% (n = 854 of 1017) of participants of the COG P9906 and St. Jude Total Therapy XIIIB/XV protocols (Supplementary Figure 1, available online).
Genotyping of ALL case subjects was performed by using the Affymetrix (Santa Clara, CA) Human SNP Array 6.0 (COG P9904/P9905, the GWAS series) or the Affymetrix GeneChip Human Mapping 500K Array (St. Jude Total Therapy XIIIB/XV and COG P9906, the replication series). Non-ALL control subjects in both the GWAS and replication series were genotyped using Affymetrix Human SNP Array 6.0. Genotype calls (coded as 0, 1, and 2 for AA, AB, and BB genotypes) were determined by the Birdseed (Affymetrix Human SNP Array 6.0) (28) or BRLMM (Affymetrix GeneChip Human Mapping 500K Array) algorithms (29). Samples for which genotype was ascertained at less than 95% of SNPs on the array were deemed to have failed and were excluded from the analyses (Supplementary Figure 1, available online). SNP quality control procedures were performed on the basis of call rate, minor allele frequency, and Hardy–Weinberg equilibrium, and 197541 of 906600 SNPs were excluded during GWAS quality control (Supplementary Figure 2 and Supplementary Data, available online).
This study was approved by the respective institutional review boards, and informed consent was obtained from parents, guardians, or patients, as appropriate.
Ethnicity Classification
European, African, Asian, and Native American genetic ancestry was determined by using STRUCTURE (version 2.2.3) (30,31) with HapMap CEU, YRI, CHB/JPT, and indigenous Native Americans (32) as reference populations, respectively. European Americans, African Americans, and Asian Americans were defined as having more than 95% European genetic ancestry, more than 70% African ancestry, and more than 90% Asian ancestry, respectively. Hispanic Americans were individuals for whom Native American ancestry was more than 10% and greater than African ancestry; the remaining subjects were grouped as “Others” (Supplementary Figure 3, available online).
Statistical Analyses
In the GWAS, the association between genotypes at each of 709059 SNPs and ALL susceptibility was tested by comparing the genotype frequency between ALL case subjects and control subjects in the logistic regression model, after adjusting for the top four principal components inferred by EIGENSTRAT (33) to control for population stratification (Supplementary Figure 4, available online). To validate associations at four susceptibility loci previously identified in populations of European descent [ARID5B (5,6), IKZF1 (5,6), CEBPE (6), and CDKN2A/2B (8)], we focused on variants within 600kb of the top SNP at each locus and applied statistical significance threshold that corrected for the number of SNPs tested at each locus (n = 174, 241, 104, and 145, respectively). To agnostically search for novel susceptibility variants by GWAS, we applied a genome-wide statistical significance cutoff of P less than or equal to 5×10−8 and sought to verify SNPs meeting this threshold in independent replication series.
In the replication studies, we tested six SNPs at the BMI1-PIP4K2A locus separately in European Americans, African Americans, and Hispanic Americans by logistic regression test with genetic ancestries as covariates. Those with P less than .05 in replication series were considered as validated.
Logistic regression model was also used to determine the independent association of multiple SNPs within the same locus, to examine the cumulative effects of multiple susceptibility variants, and to evaluate the effects of susceptibility variants in different age groups. Association between PIP4K2A SNP genotype and gene expression was assessed by a linear regression model in HapMap CEU lymphoblastoid cell lines [GSE7851 (34)] and in diagnostic blasts from European American children with ALL from St. Jude Total Therapy XIIIB/XV protocols (35,36).
R (version 2.15.1) statistical software was used for all analyses unless indicated otherwise, and a detailed description of statistical procedures is provided in the Supplementary Data (available online). All statistical tests were two-sided.
Results
To comprehensively examine germline ALL susceptibility variants, we performed GWAS in 1605 children with newly diagnosed B-precursor ALL and 6661 unrelated non-ALL control subjects of diverse ancestry (ie, European, African, Asian, and Native American genetic ancestry) (Supplementary Figures 3 and 4, available online). Controlling for population structures, we evaluated 709059 germ line SNPs for differences in genotype frequency between ALL case subjects and control subjects.
We first focused on three susceptibility loci previously identified by GWAS in populations of European descent (5,6)—ARID5B at 10q21.2, IKZF1 at 7p12.2, and CEBPE at 14q11.2—to compare the association signals among populations, particularly in those of non-European descent (Table 1; Supplementary Table 1, available online). At the ARID5B locus (ie, rs10821936), the association with ALL was consistent in all three genetically defined ethnicities: European Americans (P = 6.9×10−30; n = 972 case subjects and 1386 control subjects); African Americans (P = .004; n = 89 case subjects and 1363 control subjects); Hispanics (P = 3.8×10−11; n = 305 case subjects and 1008 control subjects); and the multiethnic group (P = 5.9×10−46; n = 1605 case subjects and 6661 control subjects). The frequency of the ALL risk allele (C) at rs10821936 increased in the order of African Americans, European Americans, and Hispanic Americans, consistent with the ethnic differences in ALL incidence (21). Multivariable analyses adjusting for rs10821936 did not identify any additional independent association signal at this locus (Supplementary Figure 5, available online). The top SNP in IKZF1 (ie, rs11978267; P = 5.3×10−24 in the multiethnic group) was also associated with ALL risk across ethnicities. Interestingly, another cluster of SNPs further upstream of rs11978267 were statistically significant even after controlling for rs11978267 (ie, rs10235226; P = 1.4×10−5 in the multiethnic group) (Supplementary Figure 5 and Supplementary Data, available online). Association at CEBPE SNPs was validated in the multiethnic group (ie, rs4982731; P = 9×10−12) (Table 1), but multivariable model conditioning on the top SNP (rs4982731) did not support additional independent associations in this region (Supplementary Figure 5, available online). Another previously reported ALL risk locus at 9p21.3 (8) was also validated in our GWAS series (ie, rs1775631 at CDKN2A/2B; P = 1.4×10−5 in the multiethnic group). In total, of 664 SNPs at these four loci, 79 remained statistically significant after correcting for multiple testing (Supplementary Table 1, available online).
Table 1.
Associations at four known acute lymphoblastic leukemia susceptibility loci: 7p12.2, 9p21.3, 10q21.2, and 14q11.2*
| SNP | Chr | Position† | Alleles‡ | Gene(s) | European American (n = 972/n = 1386)§ | African American (n = 89/n = 1363) § | Hispanic American (n = 305/n = 1008)§ | All ethnicities (n = 1605/ n = 6661)§ |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| RAF (case/control) | P | ORǁ (95% CI) | RAF (case/control) | P | ORǁ (95% CI) | RAF (case/control) | P | ORǁ (95% CI) | P | ORǁ (95% CI) | |||||
| rs11978267 | 7 | 50433798 | A/G | IKZF1 | 0.39/0.28 | 8.36×10−19 | 1.67 (1.49 to 1.87) | 0.27/0.19 | .005 | 1.59 (1.15 to 2.18) | 0.31/0.26 | .01 | 1.31 (1.07 to 1.61) | 5.29×10−24 | 1.59 (1.45 to 1.74) |
| rs17756311 | 9 | 22043895 | C/T | CDKN2A/B | 0.13/0.09 | 3.25×10−5 | 1.43 (1.21 to 1.69) | 0.11/0.1 | .62 | 1.12 (0.71 to 1.76) | 0.08/0.06 | .10 | 1.36 (0.94 to 1.97) | 1.37×10−5 | 1.36 (1.18 to 1.56) |
| rs10821936 | 10 | 63393583 | C/T | ARID5B | 0.48/0.33 | 6.93×10−30 | 1.88 (1.68 to 2.10) | 0.33/0.24 | .004 | 1.52 (1.14 to 2.02) | 0.63/0.47 | 3.78×10−11 | 1.95 (1.60 to 2.38) | 5.88×10−46 | 1.86 (1.71 to 2.03) |
| rs4982731 | 14 | 22655173 | C/T | CEBPE | 0.34/0.28 | 9.05×10−6 | 1.29 (1.15 to 1.45) | 0.41/0.38 | .41 | 1.13 (0.85 to 1.50) | 0.5/0.39 | 2.32×10−6 | 1.58 (1.31 to 1.91) | 8.97×10−12 | 1.36 (1.24 to 1.48) |
* Association of variants at these four loci was tested in the genome-wide association study series and shown are the top single nucleotide polymorphisms at each locus. Chr = chromosome; CI = confidence interval; OR = odds ratio; RAF = risk allele frequency.
† Chromosomal locations are based on hg18.
‡ Bold denotes the allele that had a statistically significantly higher frequency in children with acute lymphoblastic leukemia than in the non–acute lymphoblastic leukemia control subjects (ie, risk allele for acute lymphoblastic leukemia).
§ Ethnicity was defined by single nucleotide polymorphism genotype-based European, African, East Asian, and Native American genetic ancestry (see Methods), and numbers of acute lymphoblastic leukemia patients vs non–acute lymphoblastic leukemia control subjects are indicated.
ǁ Odds ratio represents the increase in the risk of developing acute lymphoblastic leukemia for each copy of the risk allele compared with subjects who do not carry the risk allele. P values and odds ratios were estimated by the logistic regression test (two-sided).
Importantly, our multiethnic GWAS also discovered a novel ALL susceptibility locus at 10p12.31-12.2 that was not identified by previous studies in populations of European descent. As shown in Figure 1, Figure 2, and Table 2, six variants in the BMI1-PIP4K2A region exhibited genome-wide statistically significant associations with ALL. Four SNPs were clustered within the intronic region of the PIP4K2A gene; the other two were upstream of the COMMD3 and BMI1 genes and further distal to the centromere (Figure 2). The SNPs with the strongest association in each region were rs7088318 (PIP4K2A; P = 1.1×10−11) and rs4748793 (COMMD3/BMI1; P = 8.4×10−9), respectively (Table 2). Although both SNPs conferred a similar degree of increase in ALL risk (odds ratio [OR] = 1.4), they were independently associated with disease susceptibility (P < .0001) in a multivariable model adjusting for each other (Supplementary Figure 5, available online) and were separated by distinct linkage disequilibrium (LD) blocks in all ethnic groups (Figure 2). Also, the frequency of the ALL risk allele at rs7088318 was highest in Hispanic Americans, followed by European Americans, and lowest in African Americans, in parallel with ALL incidence in these populations (20) (Table 2). To explore possible functional consequences of this PIP4K2A variation, we investigated the relationship between rs7088318 genotype and PIP4K2A mRNA expression. In lymphoblastoid cell lines derived from the HapMap CEU samples, the ALL risk allele (A) at rs7088318 was linked to higher PIP4K2A expression (P = .001; n = 55) (Figure 3). Consistently, the number of A allele at this SNP was also positively associated with PIP4K2A expression in diagnostic blasts from children with ALL (P = .02; n = 228) (Figure 3), indicative of a cis-acting expression quantitative trait locus.
Figure 1.
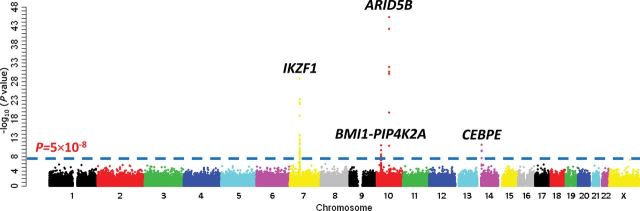
Genome-wide association study (GWAS) results of acute lymphoblastic leukemia (ALL) susceptibility in multiethnic populations. Association between genotype and ALL was evaluated using a logistic regression model (two-sided) for 709509 single nucleotide polymorphisms (SNPs) in 1605 ALL case subjects and 6661 non-ALL control subjects. P values (-log10 P, y axis) were plotted against respective chromosomal position of each SNP (x axis). Gene symbols were indicated for four loci achieving genome-wide significance threshold (P < 5×10−8; dashed blue line): ARID5B (10q21.2), IKZF1 (7p12.2), CEBPE (14q11.2), and BMI1-PIP4K2A (10p12.31-12.2).
Figure 2.

Association results and linkage disequilibrium (LD) at the 10p12.31-10p12.2 locus. A) The negative logarithm of the GWAS P values is plotted for each single nucleotide polymorphism (SNP) in a 600-kb window. Highlighted in red are six SNPs associated with ALL susceptibility with P < 5×10–8 (dashed orange line). Refseq genes are indicated below chromosomal position, which is based on hg18. B) LD at this locus is depicted based on r 2 in HapMap CEU (European), YRI (African), and MEX (Hispanic) samples, and the plots were constructed using the HaploView software. Of six genome-wide statistically significant SNPs at this locus, four are in the intronic region of the PIP4K2A gene and are within a single LD block across ethnic groups. The other two SNPs are located in a region upstream of the COMMD3 and BMI1 genes. P value was calculated by two-sided logistic regression test.
Table 2.
Genome-wide statistically significant association and replication of novel acute lymphoblastic leukemia susceptibility variants at 10p12.31-12.2
| SNP | Chr | Position* | Alleles† | Gene(s) | GWAS series (case/control) | Replication series (case/control) | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| European American (n = 972/n = 1386) ‡ | African American (n = 89/n = 1363)‡ |
Hispanic American (n = 305/n = 1008)‡ |
All ethnicities (n = 1605/n = 6661)‡ | European American (n = 574/n = 2601)‡ | African American (n = 128/n = 1075)‡ | Hispanic American (n = 143/n = 640)‡ | |||||||||||||||
| RAF (Case/Control) | P | ORǁ (95% CI) | RAF (Case/Control) | P | ORǁ (95% CI) | RAF (Case/Control) | P | ORǁ (95% CI) | P | ORǁ (95% CI) | P | ORǁ (95% CI) | P | ORǁ (95% CI) | P | ORǁ (95% CI) | |||||
| rs4266962 | 10 | 22381580 | C/G | COMMD3/BMI1 | 0.83/0.76 | 4.35×10−8 | 1.41 (1.20 to 1.65) | 0.95/0.94 | .97 | 1.08 (0.49 to 2.38) | 0.8/0.78 | .48 | 1.03 (0.82 to 1.30) | 2.39×10−8 | 1.39 (1.25 to 1.56) | .0003 | 1.33 (1.13 to 1.57) | NA§ | NA§ | .17 | 1.20 (0.82 to 1.76) |
| rs4748793 | 10 | 22523017 | C/T | COMMD3/BMI1 | 0.83/0.78 | 4.60×10−6 | 1.35 (1.15 to 1.58) | 0.94/0.89 | .04 | 1.83 (0.95 to 3.53) | 0.83/0.78 | .04 | 1.23 (0.97 to 1.57) | 8.40×10−9 | 1.40 (1.26 to 1.57) | .0004 | 1.33 (1.13 to 1.58) | .46 | 1.03 (0.65 to 1.63) | .11 | 1.26 (0.86 to 1.83) |
| rs7901152 | 10 | 22873159 | C/T | PIP4K2A | 0.67/0.61 | 5.84×10−5 | 1.22 (1.07 to 1.39) | 0.8/0.7 | .006 | 1.61 (1.1 to 2.34) | 0.83/0.78 | .02 | 1.33 (1.04 to 1.70) | 1.89×10−8 | 1.33 (1.21 to 1.45) | .002 | 1.22 (1.06 to 1.40) | .14 | 1.18 (0.88 to 1.57) | .09 | 1.31 (0.80 to 1.94) |
| rs11013046 | 10 | 22880589 | A/G | PIP4K2A | 0.66/0.6 | 2.58×10−5 | 1.23 (1.08 to 1.40) | 0.48/0.39 | .01 | 1.47 (1.07 to 2.01) | 0.66/0.6 | .009 | 1.31 (1.07 to 1.59) | 2.92×10−9 | 1.32 (1.21 to 1.43) | .001 | 1.23 (1.08 to 1.42) | .03 | 1.28 (0.98 to 1.68) | .04 | 1.34 (0.97 to 1.86) |
| rs7088318 | 10 | 22892954 | A/C | PIP4K2A | 0.65/0.59 | 5.25×10−6 | 1.25 (1.10 to 1.42) | 0.5/0.39 | .001 | 1.65 (1.21 to 2.26) | 0.81/0.75 | .009 | 1.42 (1.12 to 1.80) | 1.13×10−11 | 1.40 (1.28 to 1.53) | .001 | 1.23 (1.07 to 1.41) | .009 | 1.38 (1.05 to 1.81) | .04 | 1.40 (0.95 to 2.05) |
| rs7075634 | 10 | 22893108 | C/T | PIP4K2A | 0.66/0.6 | 1.78×10−5 | 1.23 (1.08 to 1.40) | 0.51/0.4 | .003 | 1.62 (1.19 to 2.22) | 0.82/0.75 | .008 | 1.43 (1.12 to 1.82) | 2.06×10−10 | 1.38 (1.26 to 1.50) | .0001 | 1.29 (1.12 to 1.48) | .006 | 1.41 (1.08 to 1.84) | .07 | 1.34 (0.91 to 1.97) |
* Chromosomal locations are based on hg18. Chr = chromosome; CI = confidence interval; NA = not applicable; OR = odds ratio; RAF = risk allele frequency.
† Bold denotes the allele that had a statistically significantly higher frequency in children with acute lymphoblastic leukemia than in the non–acute lymphoblastic leukemia control subjects (ie, risk allele for acute lymphoblastic leukemia).
‡ Ethnicity was defined by single nucleotide polymorphism genotype-based European, African, East Asian, and Native American genetic ancestry (see Methods), and numbers of acute lymphoblastic leukemia patients vs non–acute lymphoblastic leukemia control subjects are indicated.
§ Not applicable (single nucleotide polymorphism call rate <95% in the respective replication series).
ǁ Odds ratio represents the increase in the risk of developing acute lymphoblastic leukemia for each copy of the risk allele compared with subjects who do not carry the risk allele. P values and odds ratios were estimated by the logistic regression test (two-sided).
Figure 3.
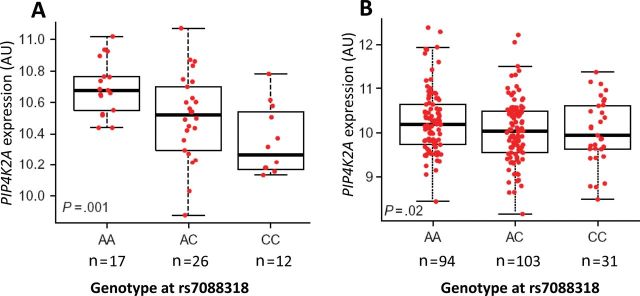
Expression quantitative loci (eQTL) analysis of PIP4K2A single nucleotide polymorphism rs7088318 in lymphoblastoid cell lines and primary acute lymphoblastic leukemia (ALL) blasts. PIP4K2A expression was determined in HapMap CEU (European) cell lines (A) and diagnostic blasts from children with ALL enrolled on St. Jude Total Therapy XIIIB/XV protocols (B). Genotype-expression association was evaluated using a linear regression model. AU = arbitrary unit.
We next sought to validate the association at the novel susceptibility locus BM1-PIP4K2A in three independent case–control series in an ethnicity-specific manner: European Americans (n = 574 case subjects and 2601 control subjects), African Americans (n = 128 case subjects and 1075 control subjects), and Hispanic Americans (n = 143 case subjects and 640 control subjects). The top PIP4K2A SNP, rs7088318, was statistically significantly associated with ALL susceptibility in all three ethnic groups: European Americans (P = .001); African Americans, (P = .009); and Hispanic Americans, (P = .04) (Table 2). The remaining five SNPs at this locus were all replicated in at least one ethnic group (Table 2) and so was the independent association at rs4748793 (P rs4748793 = 3.5×10−4, after adjusting for rs7088318 and genetic ancestry in replication series).
The genetic underpinning of childhood ALL susceptibility is likely to be complex, and current evidence strongly favors a polygenic model of ALL risk (11). We next examined the combined effects of four genome-wide statistically significant loci (Figure 1) on ALL susceptibility by multimarker analyses on the basis of genotype at top SNPs at each locus: rs10821936 at ARID5B, rs11978267 at IKZF1, rs7088318 at PIP4K2A, and rs4982731 at CEBPE. In the combined GWAS and replication series (n = 2450 case subjects and 10 977 control subjects), there was a positive correlation (P = 1.6×10−5; correlation coefficient = 0.39, 95% confidence interval [CI] = 0.33 to 0.45) between the number of risk alleles at these four SNPs and relative ALL risk (ie, odds ratio, relative to subjects carrying 0–1 copy of the risk alleles) (Figure 4). For example, subjects with six to eight copies of risk alleles (n = 252 case subjects and 314 control subjects) were at ninefold (95% CI = 6.9 to 11.8) higher risk of developing ALL than those with zero to one copy of the risk alleles (n = 153 case subjects and 1753 control subjects). Cumulative effects of these variants were also estimated separately in the GWAS and replication series (Supplementary Figure 6, available online).
Figure 4.

Cumulative effects of top variants at 7p12.2, 10p12.31-12.2, 10q21.2, and 14q11.2 on acute lymphoblastic leukemia (ALL) susceptibility. Odds ratio for ALL is plotted against the number of risk alleles at rs10821936 (ARID5B), rs11978267 (IKZF1), rs7088318 (PIP4K2A), and rs4982731 (CEBPE). Bars indicate 95% confidence intervals, and dotted line is odds ratio of 1. Odds ratio was estimated by logistical regression test combining genome-wide association study and replication series (n = 2450 ALL case subjects and n = 10 977 control subjects) after adjusting for genetic ancestry.
Finally, because the incidence of ALL is highly related to age with the majority of cases occurring in children aged 2 to 5 years (2), we examined the effects of ALL susceptibility variants by age. Combining GWAS and replication series, risk allele frequency at rs10821936 was higher in children who developed ALL before 10 years of age than in those diagnosed with ALL at ages older than 10 years (P = .02, .18, .007 in European Americans, African Americans, and Hispanic Americans, respectively) (Table 3), most evidently in hyperdiploid ALL (Table 3). Consistently, when we further classified children into age groups of those aged less than 5 years, those aged 5 to 10 years, and those aged greater than 10 years, there was a trend for decreasing allelic odds ratio (ie, relative risk of ALL conferred by each copy of the C allele at rs10821936) as age increased: 2.01 (95% CI = 1.85 to 2.19), 1.8 (95% CI = 1.6 to 2.02), and 1.48 (95% CI = 1.3 to 1.68), respectively (Supplementary Figure 7, available online). Similar results were observed when we restricted the analysis to hyperdiploid ALL (Supplementary Figure 7, available online). In contrast, the effects of IKZF1, CEBPE, and PIP4K2A variants did not differ between age groups (data not shown). Together, these results suggest possible modifying effects of age on genetic predisposition to ALL.
Table 3.
The frequency of acute lymphoblastic leukemia risk (ALL) allele at rs10821936 (ARID5B) by ALL status, age at diagnosis, ALL molecular subtype, and ethnicity
| European American* (RAF/No.)† | African American* (RAF/No.)† | Hispanic American* (RAF/No.)† | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Non-ALL control | Age at ALL diagnosis, years | Non-ALL control | Age at ALL diagnosis, years | Non-ALL control | Age at ALL diagnosis, years | |||||||
| <10 | ≥10 | All ages | <10 | ≥10 | All ages | <10 | ≥10 | All ages | ||||
| ALL (all subtypes) | 0.33/3987 | 0.49/1,219‡ | 0.44/327‡ | 0.48/1546 | 0.23/2438 | 0.34/156 | 0.27/61 | 0.32/217 | 0.46/1648 | 0.62/355‡ | 0.5/93‡ | 0.59/448 |
| Hyperdiploid | 0.57/351‡ | 0.45/65‡ | 0.55/416 | 0.4/29‡ | 0.13/8‡ | 0.34/37 | 0.67/98 | 0.6/15 | 0.66/113 | |||
| ETV6-RUNX1 | 0.42/339 | 0.37/23 | 0.41/362 | 0.35/50 | 0.25/2 | 0.34/52 | 0.56/85 | 0.25/4 | 0.54/89 | |||
* Ethnicity was defined by single nucleotide polymorphims genotype-based European, African, East Asian, and Native American genetic ancestry (see Methods). RAF = risk allele frequency.
† Numbers indicate the C allele frequency at rs10821936 and total number of subjects in each category (eg, in 3987 EA non-ALL control subjects, the C allele frequency is 33%), combining the genome-wide association study and replication series.
‡ Differences in allele frequency were statistically significant between two age groups (ie, P < .05 as determined by logistical regression test after adjusting genetic ancestry).
Discussion
Non-European populations are indisputably underrepresented in GWASs (12,13). Recent GWASs in diverse populations reveal both similarities and differences in genetic architecture of disease susceptibility among ethnic groups. We reported here the first GWAS of ALL in multiethnic populations (including African Americans and Hispanic Americans), in which we discovered novel susceptibility variants at BMI1-PIP4K2A locus and comprehensively compared associations at known susceptibility loci (ARID5B, IKZF1, CEBPE, and CDKN2A/2B) in different ethnic groups.
The discovery of the BMI1-PIP4K2A susceptibility variants that were not detected by previous European-only GWASs of ALL (5–7) raises the question of improved power as a result of population diversity. When the disease variant is substantially more common in non-European populations, GWASs in these ethnic groups obviously heighten the power to discover such loci compared with GWASs in Europeans with the same sample size, as illustrated in the case of the KCNQ1 locus in type 2 diabetes (37) and by simulation using the 1000 Genome data (38). However, this is probably unlikely to explain the BMI1-PIP4K2A variants that are actually less frequent in African Americans than European Americans, although they are modestly more common in Hispanic Americans. Another plausible explanation is population differences in LD around the BMI1-PIP4K2A variants: if the causal variant is better tagged in African populations, including African Americans in the GWAS is likely to improve the sensitivity to detect the signal at the genome-wide threshold. At this locus, LD pattern is similar between European Americans and Hispanic Americans, but is much less extensive in African Americans, as expected (Figure 2). Lastly, population heterogeneity in effect size of the risk allele can also influence the sample size required in GWAS. In type 2 diabetes, the allelic risk at multiple disease variants is statistically significantly greater in the Japanese population than in the European population, although these variants are statistically significant in both ethnic groups (39). Interestingly, the per-allele odds ratio at rs7088318 was greater in African Americans and Hispanic Americans relative to European Americans (Table 2), consistent with possibly improved power when these non-European populations are included in GWASs of ALL.
The population diversity in our GWAS also offered a unique opportunity to examine the genetic basis of ethnic differences in ALL incidence (20). Variants at the ARID5B, IKZF1, and BMI1-PIP4K2A loci were associated with ALL susceptibility across ethnic groups (ie, the SNP with the strongest association at each locus was statistically significant in all three populations), suggesting common causal variants across ancestral backgrounds. In contrast, CEBPE SNPs were strongly related to ALL risk in European Americans, with variable effects in non-European populations. Such disparities might reflect existence of true population-specific disease variants but can also arise from population differences in genomic structure at these loci (differences in LD between tagging SNPs and causal variants). Further, the frequency of ALL risk variants at the ARID5B and PIP4K2A loci vary substantially by ethnicity in a pattern consistent with their possible contribution to ethnic differences in ALL incidence (21) (Tables 1 and 2).
The genetic basis of ALL is most likely to be polygenic (11). However, it should be noted that carrying ALL risk variants at merely four SNPs (ARID5B, IKZF1, CEBPE, and PIP4K2A) conferred a ninefold increase in disease susceptibility (Figure 4) and these GWAS signals are concentrated to genes directly related to hematopoietic differentiation and development [ARID5B (40), IKZF1 (41), and CEBPE (42)]. We hypothesize that genetic predisposition to ALL might be largely mediated by robust effects of a modest number of key genes rather than cumulative effects of tens of thousands of variants with small effects (OR = 1.1–1.2), as seen in GWASs of other common diseases (43,44). In fact, it is estimated that variants in ARID5B, IKZF1, CEBPE, and CDKN2A/2B account for approximately one-third of ALL risk conferred by common genetic polymorphisms (11). The effect of ALL susceptibility variants was particularly strong in younger children (Supplementary Figure 7, available online), suggesting possible variation in ALL genetic predisposition at different developmental stages. Interestingly, several of the GWAS hits are also frequently targeted by somatic aberrations in ALL cells [IKZF1 (45) and CEBPE (46)]. Susceptibility variants in ARID5B are also related to gross cytogenetic abnormalities in ALL blasts (ie, hyperdiploidy) (Table 3), consistent with prior reports from us and others (5,6,21,47). The C allele at rs10821936 confers a greater disease risk for this subtype of ALL (5), although the molecular mechanisms linking ARID5B to aneuploidy remain unclear. Nevertheless, these observations raise the possibility of interactions between inherited (germline) and acquired (somatic) genetic variations in the pathogenesis of ALL.
PIP4K2A is a member of the family of enzymes that catalyze phosphorylation of phosphatidylinositol-5-phosphate to form phosphatidylinositol-5,4-bisphosphate (PIP2), a precursor of the important second messenger molecule, PIP3. Upon B-cell receptor activation, PIP4K2A is directly recruited by BTK to the plasma membrane as a means of stimulating local PIP2 synthesis (48). Similarly, PIP5K enzymes also interact with the Rho-family small GTP-binding proteins (eg, Rac1) to regulate membrane PIP2 synthesis and PI3K and PLC signaling in B cells (49). Although these observations point to PIP4K2A as a plausible regulator of lymphoid cell differentiation, functional studies are warranted to determine the mechanisms linking PIP4K2A to leukemogenesis.
Our study was not without limitations. Further fine-mapping and/or resequencing of the causal variants will be required to completely characterize the contribution of BMI1-PIP4K2A variants to ALL etiology in the context of ethnicity. Future GWASs and/or admixture mapping with even larger samples of non-European populations are needed to comprehensively characterize genetic variants that predispose children to this most common childhood cancer and to fully understand the genetic basis of ethnic disparity in ALL. Nonetheless, we argue that a GWAS approach that includes multiethnic subjects is likely to be more effective in discovering ALL risk loci than analyses selectively procuring large samples in a single population, as suggested by observations from GWASs of other diseases (13,15,16,50).
Funding
This work was supported by the National Institutes of Health (grant numbers CA156449, CA21765, CA36401, CA98543, CA114766, CA98413, CA140729, and U01GM92666), in part by the Intramural Program of the National Cancer Institute, and by the American Lebanese Syrian Associated Charities (ALSAC).
Genome-wide genotyping of COG P9904/P9905 samples was performed by the Center for Molecular Medicine with the generous financial support from the Jeffrey Pride Foundation and the National Childhood Cancer Foundation. S.P. Hunger is the Ergen Family Chair in Pediatric Cancer, and J.J. Yang is supported by the American Society of Hematology Scholar Award, Alex Lemonade Stand Foundation for Childhood Cancer Young Investigator Grant, and by the Order of St. Francis Foundation.
H. Xu and W. Yang contributed equally to this work.
The study sponsors were not directly involved in the design of the study, the collection, analysis, and interpretation of the data, the writing of the manuscript, or the decision to submit the manuscript.
We thank the patients and parents who participated in the St. Jude and COG protocols included in this study, the clinicians and research staff at the St. Jude and COG institutions, and J. Pullen (University of Mississippi at Jackson) for assistance in classification of patients with ALL. We thank M. Shriver (Pennsylvania State University) for sharing SNP genotype data of the Native American references. Full acknowledgements of the dbGAP datasets are provided in the Supplementary Data (available online).
References
- 1. Pui CH, Evans WE. Treatment of acute lymphoblastic leukemia. N Engl J Med. 2006; 354(2):166–178 [DOI] [PubMed] [Google Scholar]
- 2. Greaves M. Infection, immune responses and the aetiology of childhood leukaemia. Nat Rev Cancer. 2006; 6(3):193–203 [DOI] [PubMed] [Google Scholar]
- 3. Hasle H, Clemmensen IH, Mikkelsen M. Risks of leukaemia and solid tumours in individuals with Down’s syndrome. Lancet. 2000; 355(9199):165–169 [DOI] [PubMed] [Google Scholar]
- 4. Morrell D, Cromartie E, Swift M. Mortality and cancer incidence in 263 patients with ataxia-telangiectasia. J Natl Cancer Inst. 1986; 77(1):89–92 [PubMed] [Google Scholar]
- 5. Trevino LR, Yang W, French D, et al. Germline genomic variants associated with childhood acute lymphoblastic leukemia. Nat Genet. 2009; 41(9):1001–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Papaemmanuil E, Hosking FJ, Vijayakrishnan J, et al. Loci on 7p12.2, 10q21.2 and 14q11.2 are associated with risk of childhood acute lymphoblastic leukemia. Nat Genet. 2009; 41(9):1006–1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ellinghaus E, Stanulla M, Richter G, et al. Identification of germline susceptibility loci in ETV6-RUNX1-rearranged childhood acute lymphoblastic leukemia. Leukemia. 2012; 26(5):902–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sherborne AL, Hosking FJ, Prasad RB, et al. Variation in CDKN2A at 9p21.3 influences childhood acute lymphoblastic leukemia risk. Nat Genet. 2010; 42(6):492–494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Orsi L, Rudant J, Bonaventure A, et al. Genetic polymorphisms and childhood acute lymphoblastic leukemia: GWAS of the ESCALE study (SFCE). Leukemia. . 2012 26(12):2561–2564 [DOI] [PubMed] [Google Scholar]
- 10. Fletcher O, Houlston RS. Architecture of inherited susceptibility to common cancer. Nat Rev Cancer. 2010; 10(5):353–361 [DOI] [PubMed] [Google Scholar]
- 11. Enciso-Mora V, Hosking FJ, Sheridan E, et al. Common genetic variation contributes significantly to the risk of childhood B-cell precursor acute lymphoblastic leukemia. Leukemia. 2012; 26(10):2212–2215 [DOI] [PubMed] [Google Scholar]
- 12. Bustamante CD, Burchard EG, De la Vega FM. Genomics for the world. Nature. 2011; 475(7355):163–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rosenberg NA, Huang L, Jewett EM, et al. Genome-wide association studies in diverse populations. Nat Rev Genet. 2010; 11(5):356–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ioannidis JP. Population-wide generalizability of genome-wide discovered associations. J Natl Cancer Inst. 2009; 101(19):1297–1299 [DOI] [PubMed] [Google Scholar]
- 15. Ntzani EE, Liberopoulos G, Manolio TA, et al. Consistency of genome-wide associations across major ancestral groups. Hum Genet. 2012; 131(7):1057–1071 [DOI] [PubMed] [Google Scholar]
- 16. Torgerson DG, Ampleford EJ, Chiu GY, et al. Meta-analysis of genome-wide association studies of asthma in ethnically diverse North American populations. Nat Genet. 2011; 43(9):887–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Postel-Vinay S, Veron AS, Tirode F, et al. Common variants near TARDBP and EGR2 are associated with susceptibility to Ewing sarcoma. Nat Genet. 2012; 44(3):323–327 [DOI] [PubMed] [Google Scholar]
- 18. Moonesinghe R, Ioannidis JP, Flanders WD, et al. Estimating the contribution of genetic variants to difference in incidence of disease between population groups. Eur J Hum Genet. 2012; 20(8):831–836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dores GM, Devesa SS, Curtis RE, et al. Acute leukemia incidence and patient survival among children and adults in the United States. 2001–2007 Blood. 2012; 119(1): 34–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Linabery AM, Ross JA. Trends in childhood cancer incidence in the U.S. (1992–2004). Cancer. 2008; 112(2): 416–432 [DOI] [PubMed] [Google Scholar]
- 21. Xu H, Cheng C, Devidas M, et al. ARID5B genetic polymorphisms contribute to racial disparities in the incidence and treatment outcome of childhood acute lymphoblastic leukemia. J Clin Oncol. 2012; 30(7):751–757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yang W, Trevino LR, Yang JJ, et al. ARID5B SNP rs10821936 is associated with risk of childhood acute lymphoblastic leukemia in blacks and contributes to racial differences in leukemia incidence. Leukemia. 2010; 24(4):894–896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Borowitz MJ, Devidas M, Hunger SP, et al. Clinical significance of minimal residual disease in childhood acute lymphoblastic leukemia and its relationship to other prognostic factors: a Children’s Oncology Group study. Blood. 2008; 111(12):5477–5485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shi J, Levinson DF, Duan J, et al. Common variants on chromosome 6p22.1 are associated with schizophrenia. Nature. 2009; 460(7256):753–757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Purcell SM, Wray NR, Stone JL, et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009; 460(7256):748–752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Troyer JL, Nelson GW, Lautenberger JA, et al. Genome-wide association study implicates PARD3B-based AIDS restriction. J Infect Dis. 2011; 203(10):1491–1502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Burchard EG, Avila PC, Nazario S, et al. Lower bronchodilator responsiveness in Puerto Rican than in Mexican subjects with asthma. Am J Respir Crit Care Med. 2004; 169(3):386–392 [DOI] [PubMed] [Google Scholar]
- 28. Korn JM, Kuruvilla FG, McCarroll SA, et al. Integrated genotype calling and association analysis of SNPs, common copy number polymorphisms and rare CNVs. Nat Genet. 2008; 40(10):1253–1260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rabbee N, Speed TP. A genotype calling algorithm for affymetrix SNP arrays. Bioinformatics. 2006; 22(1):7–12 [DOI] [PubMed] [Google Scholar]
- 30. Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000; 155(2):945–959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yang JJ, Cheng C, Devidas M, et al. Ancestry and pharmacogenomics of relapse in acute lymphoblastic leukemia. Nat Genet. 2011; 43(3):237–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mao X, Bigham AW, Mei R, et al. A genomewide admixture mapping panel for Hispanic/Latino populations. Am J Hum Genet. 2007; 80(6): 1171–1178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Patterson N, Price AL, Reich D. Population structure and eigenanalysis. PLoS Genet. 2006; 2(12):e190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhang W, Duan S, Kistner EO, et al. Evaluation of genetic variation contributing to differences in gene expression between populations. Am J Hum Genet. 2008; 82(3):631–640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. French D, Yang W, Cheng C, et al. Acquired variation outweighs inherited variation in whole genome analysis of methotrexate polyglutamate accumulation in leukemia. Blood. 2009; 113(19):4512–4520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pottier N, Yang W, Assem M, et al. The SWI/SNF chromatin-remodeling complex and glucocorticoid resistance in acute lymphoblastic leukemia. J Natl Cancer Inst. 2008; 100(24):1792–1803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Unoki H, Takahashi A, Kawaguchi T, et al. SNPs in KCNQ1 are associated with susceptibility to type 2 diabetes in East Asian and European populations. Nat Genet. 2008; 40(9):1098–1102 [DOI] [PubMed] [Google Scholar]
- 38. Pulit SL, Voight BF, de Bakker PI. Multiethnic genetic association studies improve power for locus discovery. PLoS One. 2010; 5(9):e12600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Waters KM, Stram DO, Hassanein MT, et al. Consistent association of type 2 diabetes risk variants found in Europeans in diverse racial and ethnic groups. PLoS Genet. 2010; 6(8):e1001078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lahoud MH, Ristevski S, Venter DJ, et al. Gene targeting of Desrt, a novel ARID class DNA-binding protein, causes growth retardation and abnormal development of reproductive organs. Genome Res. 2001; 11(8):1327–1334 [DOI] [PubMed] [Google Scholar]
- 41. Schmitt C, Tonnelle C, Dalloul A, et al. Aiolos and Ikaros: regulators of lymphocyte development, homeostasis and lymphoproliferation. Apoptosis. 2002; 7(3):277–284 [DOI] [PubMed] [Google Scholar]
- 42. Bedi R, Du J, Sharma AK, et al. Human C/EBP-epsilon activator and repressor isoforms differentially reprogram myeloid lineage commitment and differentiation. Blood. 2009; 113(2):317–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Eichler EE, Flint J, Gibson G, et al. Missing heritability and strategies for finding the underlying causes of complex disease. Nat Rev Genet. 2010; 11(6): 446–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Manolio TA, Collins FS, Cox NJ, et al. Finding the missing heritability of complex diseases. Nature. 2009; 461(7265):747–753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mullighan CG, Su X, Zhang J, et al. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N Engl J Med. 2009; 360(5):470–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Akasaka T, Balasas T, Russell LJ, et al. Five members of the CEBP transcription factor family are targeted by recurrent IGH translocations in B-cell precursor acute lymphoblastic leukemia (BCP-ALL). Blood. 2007; 109(8):3451–3461 [DOI] [PubMed] [Google Scholar]
- 47. Paulsson K, Forestier E, Lilljebjorn H, et al. Genetic landscape of high hyperdiploid childhood acute lymphoblastic leukemia. Proc Natl Acad Sci U S A. 2010; 107(50):21719–21724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Saito K, Tolias KF, Saci A, et al. BTK regulates PtdIns-4,5-P2 synthesis: importance for calcium signaling and PI3K activity. Immunity. 2003; 19(5):669–678 [DOI] [PubMed] [Google Scholar]
- 49. O’Rourke LM, Tooze R, Turner M, et al. CD19 as a membrane-anchored adaptor protein of B lymphocytes: costimulation of lipid and protein kinases by recruitment of Vav. Immunity. 1998; 8(5):635–645 [DOI] [PubMed] [Google Scholar]
- 50. Kurreeman FA, Stahl EA, Okada Y, et al. Use of a multiethnic approach to identify rheumatoid- arthritis-susceptibility loci, 1p36 and 17q12. Am J Hum Genet. 2012; 90(3):524–532 [DOI] [PMC free article] [PubMed] [Google Scholar]