

Abstract
Thrombotic microangiopathy (TMA) represents the clinical picture of thrombocytopenia and hemolytic anemia in the setting of small blood vessel thrombosis, accompanied by varying degrees of organ dysfunction. Well-known to both nephrologists and hematologists alike, among the most-common and best-studied TMAs are hemolytic-uremic syndrome (HUS) and thrombotic thrombocytopenic purpura (TTP). Despite sharing a strong clinical and historical relationship, these disorders represent distinct clinical and pathophysiological entities. Here will be reviewed recent progress into the pathogenesis of TTP and HUS, focusing on events taking place at the endothelial surface.
Keywords: HUS, TTP, VWF, ADAMTS13, verotoxin
TTP and HUS as classical thrombotic microangiopathies
Since its first description by Eli Moschowitz in 1925, TTP has been considered to be a systemic hematological disease.1 Although the clinical picture can be quite variable, this disorder typically presents abruptly with fever, thrombocytopenia, and microangiopathic hemolytic anemia, along with varying degrees of cardiac, neurological, renal dysfunction.1–3 These clinical findings most likely occur secondary to the deposition of platelet-rich thrombi in the vasculature of various organs, most notably the heart, brain, and kidneys. Historically referred to as “hyaline thrombi” owing to their glassy-pink appearance following hematoxylin and eosin staining, these lesions also underlie the TMA findings of thrombocytopenia (secondary to platelet consumption in the thrombi), and microangiopathic hemolytic anemia (thought to result from abnormally high levels of shear stress in the microcirculation causing mechanical destruction of erythrocytes).1–3
Although the kidney is a common target organ in TTP, hypertension and acute renal failure requiring dialysis are uncommon.4 In a manner not soon forgotten by both physicians and family members, TTP can progress rapidly with a shock-like clinical picture to coma and death; with untreated mortality greater than 90%. Even with plasma exchange (the current standard-of-care), mortality remains in the 10–20% range.5
First described by von Gasser in 1955, the hemolytic-uremic syndrome (HUS) is considered to be a renal disease with systemic complications. HUS is a leading cause of renal failure in children, and an increasing worldwide public health concern.1,4,6 Mortality from childhood HUS remains at approximately 5%, and can be significantly higher in older adults.7–9 Acute morbidity is also a serious concern with approximately 70% of patients requiring red blood cell (RBC) transfusions, 50% requiring renal replacement therapy, and 25% demonstrating neurological complications including stroke, seizures, and coma. Furthermore, up to 40% of childhood HUS survivors will develop long-term renal dysfunction ranging from chronic proteinuria and hypertension, to end stage renal disease requiring transplant.8,9
In a manner analogous to that which is thought to occur in TTP, thrombocytopenia and microangiopathic hemolytic anemia likely result from deposition of thrombi throughout the kidneys. Notably different however, HUS thromboses occur principally in the glomerular microcirculation, while those in TTP that occur in the kidneys are found in small renal arterioles.1,6 Additionally, HUS thrombi typically are more developed than those found in TTP, containing platelets, VWF, fibrin, and inflammatory cells, while those found in TTP typically comprise only platelets and VWF.6,10,11
Owning perhaps to these differences in thrombi composition, renal damage typically is more severe in HUS than TTP, although systemic thromboses may not be as widespread.1,4,7 Nevertheless, neurological findings are common, and when present, can greatly complicate the distinction from TTP. Despite the fact that HUS typically is considered to be a renal disease, it should be kept in mind that on another level, it must also be a disorder of the hemostatic system.
Although both diseases share a strong clinical relationship, much historical and recent investigation into HUS and TTP have followed independent and seemingly unrelated directions.12 It is important to stress that renal biopsy is uncommon in these disorders because of the associated thrombocytopenia and risk of biopsy-associated bleeding complications. TTP research has focused on regulation of the blood coagulation protein von Willebrand factor (VWF), whereas much effort in HUS research has been concentrated on understanding the mechanisms of injury to the renal endothelium. However, as will be discussed below, accumulating evidence suggests that these TMA disorders may share more of a common mechanistic link than originally thought, and that this link may center on the interactions of various blood cells and plasma proteins with the endothelium.
TTP pathophysiology and the roles of VWF and ADAMTS13
The role of abnormal VWF homeostasis recently has become the most widely supported hypothesis in TTP pathogenesis.1,2 VWF is an abundant plasma glycoprotein that plays a critical role in hemostasis and thrombosis by providing the initial adhesive link between circulating blood platelets and sites of vascular injury. Also importantly, VWF functions as a carrier for coagulation Factor VIII in circulation, significantly increasing its half-life.
The crucial importance of VWF in maintaining hemostasis is illustrated clinically in patients with Von Willebrand Disease (VWD). Although type 1 VWD is associated with modestly decreased levels of circulating VWF with mild bruising and bleeding, type 3 VWD (severe VWF deficiency) clinically mimics hemophilia in severity.13,14
VWF is synthesized in endothelial cells and megakaryocytes where it is processed from an initial propeptide monomer into considerably larger multimeric forms.15–18 In endothelial cells, VWF multimers become especially large, and can exceed 20,000 kDa in size (>60 monomers in length). Endothelial cell VWF is released in both a constitutive and an inducible manner.19 VWF that is not released constitutively is stored in specialized organelles termed Weibel-Palade bodies.20,21 Upon endothelial cell stimulation or physical vascular injury, the VWF stored in Weibel-Palade bodies is released in its largest multimeric form, which is termed ultra-large VWF (UL-VWF).19,22
Following release, newly-secreted VWF becomes transiently anchored to the endothelial surface through an interaction involving (at least in part) integrin αvβ3 and P-selectin on the endothelial surface.23,24 Endothelial-associated UL-VWF subsequently is cleaved into smaller multimers by the plasma metalloprotease ADAMTS13, and therefore typically is not detected in normal human plasma.25,26 Endothelial-associated VWF can bind circulating platelets and leukocytes, and therefore can be considered to be an effector of endothelial activation.27,28
Identification of the crucial link between TTP pathogenesis and VWF was made in 1982 when Moake and colleagues demonstrated the presence of UL-VWF in the plasma of patients with an inherited familial form of TTP.29 Thus it was hypothesized that deficiency of a VWF “depolymerase” was the underlying cause of TTP. This VWF “depolymerase” was ultimately identified as the metalloprotease ADAMTS13, which cleaves VWF between amino acid residues tyrosine 1605 and methionine 1606.30–36 The activity of ADAMTS13 against VWF in vitro is dependent on conformational changes of VWF brought about by applied shear stress, or denaturants such as guanidine or urea.35,36 Thus it speculated that cleavage of VWF by ADAMTS13 in vivo may be dependent on a conformation change of VWF induced by factors such as fluid shear stress, in combination with binding to platelets, the endothelium, or possibly other molecules.37–39 Deficiency of ADAMTS13 can be genetic (the rare Upshaw-Schulman syndrome), or more commonly acquired, resulting from autoimmune production of inhibitory anti-ADAMTS13 antibodies.2,5,40,41
Lack of endothelial-associated VWF cleavage by ADAMTS13 is now thought to be the primary defect underlying TTP pathogenesis. However, it is clear from both human and animal studies that deficiency of ADAMTS13 in-and-of-itself is not sufficient for TTP pathogenesis, which likely requires additional genetic and environmental factors.40,42 Although the nature of the stimuli that can trigger TTP pathogenesis remains unknown, systemic factors causing endothelial activation and release of VWF seem more plausible than widespread physical vascular injury. Clinically, infections, pregnancy, malignancy, collagen vascular disease, and various medications have all been associated with the onset of TTP.1,2 As will be discussed below, recent studies with a murine model of TTP have provided insight into possible infectious triggers of TTP, which interestingly, also uncovered a potential link between TTP and HUS pathogenesis.
HUS pathophysiology and the roles of shigatoxin and complement
In contrast to TTP where much of the latest work has been directed to understanding the roles of VWF and ADAMTS13, recent effort in HUS has been focused on the mechanisms of damage to the renal endothelium. In the case of the more common diarrhea-associated HUS (D+HUS), renal endothelial damage is thought to be mediated in large part by verotoxins ([VTs], also referred to as shiga toxins [Stxs]), which are actually a family of bacterial toxins elaborated by distinct strains of E. coli and S. dysenteriae (in North America principally shigatoxigenic E. coli [STEC] strain O157:H7 is responsible for D+HUS pathogenesis).8,9,43 Once ingested, STEC infection localizes to the gastrointestinal mucosa, and is followed by release of VT and its subsequent translocation through the epithelium and into the circulation.8,9,43 By mechanisms and routes that remain unclear, VT then circulates throughout the body, but may preferentially localize to the renal endothelium due to the high concentration of its receptor, globotriaosylceramide, on these surfaces.8,9,43 Based primarily on its actions in cell culture, VT is thought to exert its effects in renal endothelial cells by cleaving a specific N-glycosidic bond in the the 28S rRNA subunit, inhibiting the elongation step of protein synthesis, resulting ultimately in death of the cell.8,9,43
Additionally however, it is becoming clear that that the mechanisms by which VT induces the thrombotic state of HUS involve more than the direct toxic effects on protein synthesis described above. For example, VT induces expression of pro-inflammatory genes in endothelial cells and leukocytes, up-regulates expression of adhesion molecules, and can directly activate platelets.43–45 Additionally, following STEC infection, prothrombotic coagulation abnormalities can be detected which precede clinical evidence of renal damage.46 Thus, the end result of these, and likely other mechanisms, is a shift in the hemostatic balance of the renal circulation towards thrombosis and inflammation, and the subsequent development of renal injury and clinical HUS.
While ~90% of HUS cases result from infection with STEC and are therefore mediated by VT, considerable insight into the pathogenesis of HUS has been gained from investigation into the pathogenesis of diarrhea-negative, or “atypical” HUS (aHUS), which accounts for ~5% of HUS cases.47 It is now clear that ~55% of patients with aHUS have a mutation in one of several genes encoding proteins which regulate complement activity (complement factor H, complement factor I, complement factor B, membrane cofactor protein, and thrombomodulin).47–49 These proteins function to control complement activation on self plasma membranes, and therefore to limit complement-mediated damage to host tissues. A decrease in complement regulatory activity therefore results in the loss of ability to suppress complement activation throughout the body.
However, similar to D+HUS in which the renal endothelium seems to be the primary target of globally-circulating VT (and for reasons that likewise are not well understood), glomerular endothelial cells are especially sensitive to loss of complement regulation, and microvascular damage in aHUS is restricted principally to the renal microcirculation.
Similar to VT-mediated endothelial damage and the subsequent development of TMA, the mechanisms following complement activation that lead to TMA and aHUS also are not yet well understood. However, among multiple actions, complement activation on the glomerular endothelium likely recruits inflammatory cells, which results in endothelial injury, platelet aggregation, activation of the coagulation cascade, and the subsequent development of TMA and clinical HUS.47
It has not escaped notice by a number of investigators that the nearly identical clinical phenotypes of D+HUS and aHUS can caused by the seemingly disparate triggers of VT, and complement activation, respectively. These observations suggest that VT and complement activation share a common mechanism of action at some point during their function in the pathogenesis of HUS, either before, during, or after interaction with glomerular endothelial cells.
VWF as a mediator of VT action
While it is now clear that ADAMTS13 deficiency and VWF play a major role in TTP pathogenesis, the role of these molecules in HUS has remained more uncertain. Recently however, several groups have demonstrated that potentially physiological concentrations of VT can stimulate the release of UL-VWF from cultured human umbilical vein endothelial cells (HUVECs), and human glomerular microvascular endothelial cells (HGMECs), revealing a possible additional mechanism of action for VT in mediating the pathogenesis of HUS.50,51 Interestingly, this effect occurred within several minutes of VT application, suggesting that VT-induced VWF release was occurring via rapid plasma membrane-mediated signaling events, rather than through the toxic effects of VT on protein synthesis discussed above (which would be expected to take considerably longer).
As each VT molecule comprises a single enzymatically-active A subunit (thought to be responsible for toxicity), surrounded by five identical B subunits (which mediate cell surface binding), it was hypothesized that the isolated B subunits of VT could mediated VWF secretion in the absence of the catalytic A subunit. Interestingly, this hypothesis turned out to be correct, and that this effect requires binding and clustering of the VT receptor globotriaosylceramide, and depends on plasma membrane cholesterol and caveolin-1.51 Thus these findings demonstrated the existence of a novel VT-induced lipid raft-dependent signaling pathway in endothelial cells which may be responsible for some of the biological effects attributed previously to the enzymatic A subunit of VT.
A pathogenic link between HUS and TTP?
The initial observations above that VT is able to rapidly stimulate release of VWF from cultured endothelial cells suggested this toxin as a potential trigger of TTP in the setting of ADAMTS13 deficiency. The availability of ADAMTS13-deficient mice allowed this hypothesis to be addressed experimentally. Similar to humans with familial ADAMTS13 deficiency, these animals develop TTP spontaneously.52 Also similar to humans, additional environmental triggers and genetic modifying factors are thought to contribute to disease pathogenesis in this murine model of TTP.40,52
Thus, in a search for potential environmental triggers, VT was administered to ADAMTS13-deficient mice intravenously. Interestingly, the ADAMTS13-deficient mice (but not their ADAMTS13-sufficient littermates) developed findings identical to spontaneous TTP, including: severe thrombocytopenia, hemolytic anemia with schistocytes and RBC fragments visible on peripheral blood smear; VWF-rich and fibrin-poor thrombi in the arterioles of multiple organs, including the heart, brain, and kidneys; and increased mortality.52,53 It should be noted that these findings were consistent with human TTP, rather than HUS. This observation was subsequently extended to include the isolated B subunit of VT, which similar to complete VT, also was found to able to induce findings consistent with TTP in ADAMTS13-deficient mice.51 Based on this last observation, it can be concluded that VT likely is triggering TTP in this system via endothelial cell activation and stimulation of VWF secretion, rather than through inhibition of protein synthesis and the other longer-term effects of VT described above.
These experimental observations thus demonstrated the existence of a pathogenic link between TTP and HUS, as a single agent (VT) is able to trigger both disorders. Additionally, the ability of VT to induce the systemic findings of TTP demonstrates that his molecule can indeed exert effects in different vascular beds throughout the body, as opposed to the localized activity of VT in HUS.
It is interesting to speculate regarding the mechanisms by which VT can trigger both TTP and HUS, and whether VT and similar stimuli play any role in TTP pathophysiology in humans. Perhaps the simplest explanation would be that the ADAMTS13 status of the animal determines the outcome of VT challenge. In the case of ADAMTS13 deficiency, VT (or similar endothelial-activating stimuli [e.g. histamine, cytokines, other bacterial toxins from otherwise sub-clinical infections]) would result in a systemic TTP-like picture. Conversely, the situation of ADAMTS13 sufficiency would result in the potential for HUS, which could conceivably require a higher and/or more sustained exposure to VT, along with effects mediated by the A subunit of VT, in addition to VWF release stimulated by the B subunit.
Likewise, although it is clear that VWF plays a central role in TTP pathophysiology, its potential role in the pathogenesis of HUS remains unclear. However, given that HUS also is a disorder of the blood coagulation system, a role for VWF in HUS potentially downstream of VT and/or complement activation should not be considered unwarranted.
Consistent with a potential role for VWF in HUS pathogenesis, a recent genetic association study of polymorphisms demonstrated that the T145M variant of the platelet VWF receptor, GPIb-alpha, was found with an increased frequency in HUS patients compared with controls (23% vs. 9%, P <0.001).54 This GPIb-alpha polymorphism confers increased affinity of platelets for VWF (and therefore the activated endothelial surface), and has been previously associated with other thrombotic diseases.55
Conclusions
Although considerable progress has been made regarding the pathogenesis of TTP and HUS (only a fraction of which was reviewed here), many important issues remain. The observations discussed above support the possibility that similar factors may trigger both TTP and HUS, and that these two disorders may share some degree of subsequent pathophysiology— and therefore the possibility of future therapeutic approaches as well. The interactions of platelets, leukocytes, and multiple plasma proteins with the endothelial surface clearly underlie the pathogenesis of both disorders. Further investigation into the potential function of these factors, including VWF and ADAMTS13 in HUS pathogenesis (and likewise complement regulation in TTP pathogenesis) likely will require additional genetic approaches, and refinement of the current animal models of these diseases.
Figure 1.
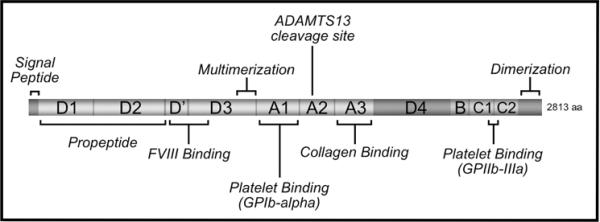
Schematic representation of VWF. Domains of VWF are indicated by lettering. Known functional elements are as labeled.
Figure 2.

Multimerization of VWF. Individual VWF monomers form dimers via disulfide bonds near the C-termini of each molecule. Much larger multimers form via N-terminal disulfide bonds between dimers. Multimers of VWF can comprise greater than 100 individual monomers, and exceed 20,000 kDa in molecular weight.
Figure 3.

The roles of ADAMTS13 and VWF in TTP pathogenesis. Panels A–C represent the normal state of ADAMTS13 sufficiency, while panels D–F represent the pathological state of ADAMTS13 deficiency. Black triangles represent ADAMTS13 in the circulation. VWF is stored in the Weibel-Palade bodies of resting/unactivated endothelial cells (A and D). Endothelial stimulation results in the rapid release of VWF, which remains adherent to the endothelial surface (B and E). When present, ADAMTS13 rapidly binds to and cleaves VWF (B), releasing it into the circulation (C). If ADAMTS13 is absent or non-functional, platelets bind to endothelial-associated VWF (E), leading to the formation of platelet/VWF thrombi throughout the circulation (F).
Figure 4.
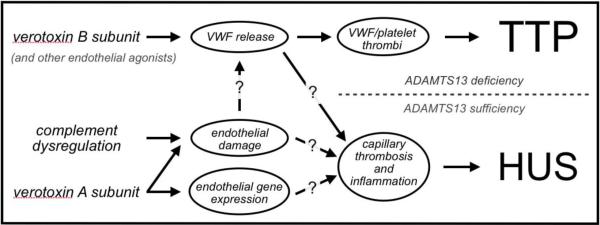
Mechanisms of pathogenesis in TTP and HUS. Question marks indicate either hypothetical links between events, or poorly-understood mechanisms of action. Please see text for detailed explanation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
No financial disclosures.
References
- 1.Moake JL. Thrombotic microangiopathies. New England Journal of Medicine. 2002;347:589–600. doi: 10.1056/NEJMra020528. [DOI] [PubMed] [Google Scholar]
- 2.Sadler JE, Moake JL, Miyata T, George JN. Recent advances in thrombotic thrombocytopenic purpura. Hematology Am Soc Hematol Educ Program. 2004:407–23. doi: 10.1182/asheducation-2004.1.407. [DOI] [PubMed] [Google Scholar]
- 3.Lammle B, George JN. Thrombotic thrombocytopenic purpura: advances in pathophysiology, diagnosis, and treatment--introduction. Semin Hematol. 2004;41:1–3. doi: 10.1053/j.seminhematol.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 4.Tsai HM. The molecular biology of thrombotic microangiopathy. Kidney Int. 2006;70:16–23. doi: 10.1038/sj.ki.5001535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Levy GG, Motto DG, Ginsburg D. ADAMTS13 turns 3. Blood. 2005;106:11–7. doi: 10.1182/blood-2004-10-4097. [DOI] [PubMed] [Google Scholar]
- 6.Moake JL. Thrombotic thrombocytopenic purpura and the hemolytic uremic syndrome. Arch Pathol Lab Med. 2002;126:1430–3. doi: 10.5858/2002-126-1430-TTPATH. [DOI] [PubMed] [Google Scholar]
- 7.Ruggenenti P, Noris M, Remuzzi G. Thrombotic microangiopathy, hemolytic uremic syndrome, and thrombotic thrombocytopenic purpura. Kidney Int. 2001;60:831–46. doi: 10.1046/j.1523-1755.2001.060003831.x. [DOI] [PubMed] [Google Scholar]
- 8.Tarr PI, Gordon CA, Chandler WL. Shiga-toxin-producing Escherichia coli and haemolytic uraemic syndrome. Lancet. 2005;365:1073–86. doi: 10.1016/S0140-6736(05)71144-2. [DOI] [PubMed] [Google Scholar]
- 9.Ray PE, Liu XH. Pathogenesis of Shiga toxin-induced hemolytic uremic syndrome. Pediatr Nephrol. 2001;16:823–39. doi: 10.1007/s004670100660. [DOI] [PubMed] [Google Scholar]
- 10.Asada Y, Sumiyoshi A. Pathological features of thrombotic thrombocytopenic purpura. In: Kaplan B, Trompeter R, Moake J, editors. Hemolytic Uremic Syndrome and Thrombotic Thrombocytopenic Purpura. Marcel Dekker, Inc.; New York: 1992. [Google Scholar]
- 11.Asada Y, Sumiyoshi A, Hayashi T, Suzumiya J, Kaketani K. Immunohistochemistry of vascular lesion in thrombotic thrombocytopenic purpura, with special reference to factor VIII related antigen. Thromb Res. 1985;38:469–79. doi: 10.1016/0049-3848(85)90180-x. [DOI] [PubMed] [Google Scholar]
- 12.Desch K, Motto D. Is there a shared pathophysiology for thrombotic thrombocytopenic purpura and hemolytic-uremic syndrome? J Am Soc Nephrol. 2007;18:2457–60. doi: 10.1681/ASN.2007010062. [DOI] [PubMed] [Google Scholar]
- 13.Sadler JE. Biochemistry and genetics of von Willebrand factor. Annu Rev Biochem. 1998;67:395–424. doi: 10.1146/annurev.biochem.67.1.395. [DOI] [PubMed] [Google Scholar]
- 14.Sadler JE, Mannucci PM, Berntorp E, Bochkov N, Boulyjenkov V, Ginsburg D, Meyer D, Peake I, Rodeghiero F, Srivastava A. Impact, diagnosis and treatment of von Willebrand disease. Thromb Haemost. 2000;84:160–74. [PubMed] [Google Scholar]
- 15.Tsai HM, Nagel RL, Hatcher VB, Sussman II. Multimeric composition of endothelial cell-derived von Willebrand factor. Blood. 1989;73:2074–2076. [PubMed] [Google Scholar]
- 16.Ruggeri ZM, Zimmerman TS. The complex multimeric composition of Factor VIII/von Willebrand Factor. Blood. 1981;57:1140–1143. [PubMed] [Google Scholar]
- 17.Huang RH, Wang Y, Roth R, Yu X, Purvis AR, Heuser JE, Egelman EH, Sadler JE. Assembly of Weibel-Palade body-like tubules from N-terminal domains of von Willebrand factor. Proc Natl Acad Sci U S A. 2008;105:482–7. doi: 10.1073/pnas.0710079105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mayadas TN, Wagner DD. von Willebrand factor biosynthesis and processing. 1991;614:153–166. doi: 10.1111/j.1749-6632.1991.tb43700.x. [DOI] [PubMed] [Google Scholar]
- 19.Sporn LA, Marder VJ, Wagner DD. Inducible secretion of large, biologically potent von Willebrand factor multimers. Cell. 1986;46:185–190. doi: 10.1016/0092-8674(86)90735-x. [DOI] [PubMed] [Google Scholar]
- 20.Wagner DD. The Weibel-Palade body: the storage granule for von Willebrand factor and P-selectin. Thrombosis and Haemostasis. 1993;70:105–110. [PubMed] [Google Scholar]
- 21.Vischer UM, Wagner DD. von Willebrand factor proteolytic processing and multimerization precede the formation of Weibel-Palade bodies. Blood. 1994;83:3536–3544. [PubMed] [Google Scholar]
- 22.Arya M, Anvari B, Romo GM, Cruz MA, Dong JF, McIntire LV, Moake JL, Lopez JA. Ultralarge multimers of von Willebrand factor form spontaneous high-strength bonds with the platelet glycoprotein Ib-IX complex: studies using optical tweezers. Blood. 2002;99:3971–7. doi: 10.1182/blood-2001-11-0060. [DOI] [PubMed] [Google Scholar]
- 23.Padilla A, Moake JL, Bernardo A, Ball C, Wang Y, Arya M, Nolasco L, Turner N, Berndt MC, Anvari B, Lopez JA, Dong JF. P-selectin anchors newly released ultralarge von Willebrand factor multimers to the endothelial cell surface. Blood. 2004;103:2150–6. doi: 10.1182/blood-2003-08-2956. [DOI] [PubMed] [Google Scholar]
- 24.Huang J, Roth R, Heuser JE, Sadler JE. Integrin alpha(v)beta(3) on human endothelial cells binds von Willebrand factor strings under fluid shear stress. Blood. 2009;113:1589–97. doi: 10.1182/blood-2008-05-158584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dong JF, Moake JL, Nolasco L, Bernardo A, Arceneaux W, Shrimpton CN, Schade AJ, McIntire LV, Fujikawa K, Lopez JA. ADAMTS-13 rapidly cleaves newly secreted ultralarge von Willebrand factor multimers on the endothelial surface under flowing conditions. Blood. 2002;100:4033–4039. doi: 10.1182/blood-2002-05-1401. [DOI] [PubMed] [Google Scholar]
- 26.Lopez JA, Dong JF. Cleavage of von Willebrand factor by ADAMTS-13 on endothelial cells. Semin Hematol. 2004;41:15–23. doi: 10.1053/j.seminhematol.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 27.Frenette PS, Moyna C, Hartwell DW, Lowe JB, Hynes RO, Wagner DD. Platelet-endothelial interactions in inflamed mesenteric venules. Blood. 1998;91:1318–1324. [PubMed] [Google Scholar]
- 28.Bernardo A, Ball C, Nolasco L, Choi H, Moake JL, Dong JF. Platelets adhered to endothelial cell-bound ultra-large von Willebrand factor strings support leukocyte tethering and rolling under high shear stress. J Thromb Haemost. 2005;3:562–70. doi: 10.1111/j.1538-7836.2005.01122.x. [DOI] [PubMed] [Google Scholar]
- 29.Moake JL, Rudy CK, Troll JH, Weinstein MJ, Colannino NM, Azocar J, Seder RH, Hong SL, Deykin D. Unusually large plasma factor VIII:von Willebrand factor multimers in chronic relapsing thrombotic thrombocytopenic purpura. New England Journal of Medicine. 1982;307:1432–1435. doi: 10.1056/NEJM198212023072306. [DOI] [PubMed] [Google Scholar]
- 30.Zheng X, Chung D, Takayama TK, Majerus EM, Sadler JE, Fujikawa K. Structure of von Willebrand factor-cleaving protease (ADAMTS13), a metalloprotease involved in thrombotic thrombocytopenic purpura. J Biol Chem. 2001;276:41059–63. doi: 10.1074/jbc.C100515200. [DOI] [PubMed] [Google Scholar]
- 31.Levy GG, Nichols WC, Lian EC, Foroud T, McClintick J, McGee BM, Yang A, Siemieniak DR, Stark KR, Gruppo R, Sarode R, Shurin S, Chandrasekaran V, Stabler SP, Sabio H, Bouhassira EE, Upshaw JD, Ginsburg DT, H.-M. Mutations in a novel member of the ADAMTS gene family cause thrombotic thrombocytopenic purpura. Nature. 2001:488–494. doi: 10.1038/35097008. [DOI] [PubMed] [Google Scholar]
- 32.Gerritsen HE, Robles R, Lammle B, Furlan M. Partial amino acid sequence of purified von Willebrand factor-cleaving protease. Blood. 2001;98:1654–1661. doi: 10.1182/blood.v98.6.1654. [DOI] [PubMed] [Google Scholar]
- 33.Soejima K, Mimura N, Hirashima M, Maeda H, Hamamoto T, Nakagaki T, Nozaki C. A novel human metalloprotease synthesized in the liver and secreted into the blood: Possibly, the von Willebrand factor-cleaving protease? Journal of Biochemistry. 2001;130:475–480. doi: 10.1093/oxfordjournals.jbchem.a003009. [DOI] [PubMed] [Google Scholar]
- 34.Dent JA, Berkowitz SD, Ware J, Kasper CK, Ruggeri ZM. Identification of a cleavage site directing the immunochemical detection of molecular abnormalities in type IIA von Willebrand factor. Proc Natl Acad Sci U S A. 1990;87:6306–10. doi: 10.1073/pnas.87.16.6306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tsai HM. Physiologic cleavage of von Willebrand factor by a plasma protease is dependent on its conformation and requires calcium ion. Blood. 1996;87:4235–4244. [PubMed] [Google Scholar]
- 36.Furlan M, Robles R, Lamie B. Partial purification and characterization of a protease from human plasma cleaving von Willebrand factor to fragments produced by in vivo proteolysis. Blood. 1996;87:4223–34. [PubMed] [Google Scholar]
- 37.Nishio K, Anderson PJ, Zheng XL, Sadler JE. Binding of platelet glycoprotein Ibalpha to von Willebrand factor domain A1 stimulates the cleavage of the adjacent domain A2 by ADAMTS13. Proc Natl Acad Sci U S A. 2004;101:10578–83. doi: 10.1073/pnas.0402041101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shim K, Anderson PJ, Tuley EA, Wiswall E, Sadler JE. Platelet-VWF complexes are preferred substrates of ADAMTS13 under fluid shear stress. Blood. 2008;111:651–7. doi: 10.1182/blood-2007-05-093021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cao W, Krishnaswamy S, Camire RM, Lenting PJ, Zheng XL. Factor VIII accelerates proteolytic cleavage of von Willebrand factor by ADAMTS13. Proc Natl Acad Sci U S A. 2008;105:7416–21. doi: 10.1073/pnas.0801735105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Desch KC, Motto DG. Thrombotic thrombocytopenic purpura in humans and mice. Arterioscler Thromb Vasc Biol. 2007;27:1901–8. doi: 10.1161/ATVBAHA.107.145797. [DOI] [PubMed] [Google Scholar]
- 41.Moake JL. von Willebrand factor, ADAMTS-13, and thrombotic thrombocytopenic purpura. Semin Hematol. 2004;41:4–14. doi: 10.1053/j.seminhematol.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 42.Furlan M, Lammle B. Aetiology and pathogenesis of thrombotic thrombocytopenic purpura and haemolytic uraemic syndrome: the role of von Willebrand factor-cleaving protease. Best Pract Res Clin Haematol. 2001;14:437–54. doi: 10.1053/beha.2001.0142. [DOI] [PubMed] [Google Scholar]
- 43.Noris M, Remuzzi G. Hemolytic uremic syndrome. J Am Soc Nephrol. 2005;16:1035–50. doi: 10.1681/ASN.2004100861. [DOI] [PubMed] [Google Scholar]
- 44.Petruzziello TN, Mawji IA, Khan M, Marsden PA. Verotoxin biology: molecular events in vascular endothelial injury. Kidney Int Suppl. 2009:S17–9. doi: 10.1038/ki.2008.612. [DOI] [PubMed] [Google Scholar]
- 45.Karpman D, Papadopoulou D, Nilsson K, Sjögren A, Mikaelsson C, Lethagen S. Platelet activation by shiga toxin and circulatory factors as a pathogenic mechanism in the hemoytic uremic syndrome. Blood. 2001;97:3100–3108. doi: 10.1182/blood.v97.10.3100. [DOI] [PubMed] [Google Scholar]
- 46.Chandler WL, Jelacic S, Boster DR, Ciol MA, Williams GD, Watkins SL, Igarashi T, Tarr PI. Prothrombotic coagulation abnormalities preceding the hemolytic-uremic syndrome. N Engl J Med. 2002;346:23–32. doi: 10.1056/NEJMoa011033. [DOI] [PubMed] [Google Scholar]
- 47.Noris M, Remuzzi G. Atypical hemolytic-uremic syndrome. N Engl J Med. 2009;361:1676–87. doi: 10.1056/NEJMra0902814. [DOI] [PubMed] [Google Scholar]
- 48.Kavanagh D, Goodship TH, Richards A. Atypical haemolytic uraemic syndrome. Br Med Bull. 2006;77–78:5–22. doi: 10.1093/bmb/ldl004. [DOI] [PubMed] [Google Scholar]
- 49.Delvaeye M, Noris M, De Vriese A, Esmon CT, Esmon NL, Ferrell G, Del-Favero J, Plaisance S, Claes B, Lambrechts D, Zoja C, Remuzzi G, Conway EM. Thrombomodulin mutations in atypical hemolytic-uremic syndrome. N Engl J Med. 2009;361:345–57. doi: 10.1056/NEJMoa0810739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nolasco LH, Turner NA, Bernardo A, Tao Z, Cleary TG, Dong JF, Moake JL. Hemolytic uremic syndrome-associated Shiga toxins promote endothelial-cell secretion and impair ADAMTS13 cleavage of unusually large von Willebrand factor multimers. Blood. 2005;106:4199–209. doi: 10.1182/blood-2005-05-2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huang J, Motto DG, Bundle DR, Sadler JE. Shiga toxin B subunits induce VWF secretion by human endothelial cells and thrombotic microangiopathy in ADAMTS13-deficient mice. Blood. 2010 doi: 10.1182/blood-2010-02-271957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Motto DG, Chauhan AK, Zhu G, Homeister J, Lamb CB, Desch KC, Zhang W, Tsai HM, Wagner DD, Ginsburg D. Shigatoxin triggers thrombotic thrombocytopenic purpura in genetically susceptible ADAMTS13-deficient mice. J Clin Invest. 2005;115:2752–61. doi: 10.1172/JCI26007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chauhan AK, Walsh MT, Zhu G, Ginsburg D, Wagner DD, Motto DG. The combined roles of ADAMTS13 and VWF in murine models of TTP, endotoxemia, and thrombosis. Blood. 2008;111:3452–7. doi: 10.1182/blood-2007-08-108571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Taranta A, Gianviti A, Palma A, De Luca V, Mannucci L, Procaccino MA, Ghiggeri GM, Caridi G, Fruci D, Ferracuti S, Ferretti A, Pecoraro C, Gaido M, Penza R, Edefonti A, Murer L, Tozzi AE, Emma F. Genetic risk factors in typical haemolytic uraemic syndrome. Nephrol Dial Transplant. 2009;24:1851–7. doi: 10.1093/ndt/gfn720. [DOI] [PubMed] [Google Scholar]
- 55.Gonzalez-Conejero R, Lozano ML, Rivera J, Corral J, Iniesta JA, Moraleda JM, Vicente V. Polymorphisms of platelet membrane glycoprotein Ib associated with arterial thrombotic disease. Blood. 1998;92:2771–6. [PubMed] [Google Scholar]